
ABSTRACT
The human adenovirus (Ad) type 2/5 early region 4 (E4) ORF4 protein (E4orf4) exerts a remarkable tumor cell-selective killing activity in mammalian cells. This indicates that E4orf4 can target tumor cell-defining features and is a unique tool to probe cancer cell vulnerabilities. Recently, we found that E4orf4, through an interaction with the polarity protein PAR3, subverts nuclear envelope (NE) remodeling processes in a tumor cell-selective manner. In this Perspective, we outline mechanical signals that modify nuclear dynamics and tumor cell behavior to highlight potential mechanisms for E4orf4’s tumoricidal activity. Through an analysis of E4orf4’s cellular targets, we define a protein subnetwork that comprises phosphatase systems interconnected to polarity protein hubs, which could contribute to enhanced NE plasticity. We infer that elucidating E4orf4’s protein network at a functional level could uncover key mechanisms of NE remodeling that define the tumor cell phenotype.
Altered cell mechanics in cancer
Tumor progression is driven by a limited subset of genetic and epigenetic alterations that promote cell-autonomous capabilities and facilitating properties that form the various hallmarks of cancer[Citation1]. However, progression to a malignant cell phenotype is fueled by multiple microenvironmental cues, including mechanical signals that are largely generated by increased cell density and extracellular matrix rigidity [Citation2–4]. To cope with the highly selective pressures imposed by these microenvironment cues, tumor cells exploit cellular plasticity programs[Citation5]. This enables them to adapt to their microenvironment, in part, by modifying their mechanical properties. Epithelial-mesenchymal plasticity is a prototypal cellular plasticity program associated with tumor cell metastasis and high-grade carcinoma[Citation6]. Reversible remodeling of cell-cell junctions and cell-matrix signaling complexes enables tumor cells to spread (epithelial-to-mesenchymal transition, EMT) and to grow at distant sites (mesenchymal-to-epithelial transition, MET). Additionally, tumor cells typically display a deregulated mechanical response. While epithelial cell stiffness is linked to tissue rigidity, in rigid tumors, tumor cells appear “softer” than normal cells[Citation7]. Indeed, evidence indicates that in several tumor cell lines, cell stiffness is inversely correlated with the malignant phenotype [Citation8,Citation9]. However, tumor cells can exert higher traction forces on the microenvironment, due to increased cellular contractility [Citation4,Citation9]. Oncogenic signaling, which empowers cellular contractility, increases cellular mechanoperception and exacerbates cellular responses to subtle changes in the mechanical microenvironment[Citation10]. Thus, dynamic reciprocity of cell-tissue interactions, involving changes in tumor cell mechanical properties, has a crucial impact on tumor progression.
The mechanisms underlying these paradoxical changes in tumor cell mechanics are still poorly understood. However, actin network and nuclear envelope dynamics both play dominant roles in determining cell mechanical properties. Actomyosin structure remodeling, controlled by small GTPase signaling networks downstream of cell surface receptor activation, govern intracellular forces [Citation11,Citation12]. The spatial control of these contractile forces is crucial to determine cell behavior. The polarity protein signaling network acts to segregate actomyosin subnetworks, to control their attachment to membranes, and to locally restrict biochemical signaling that transduces intracellular forces[Citation13]. Since cancer cells typically lose some aspects of cell polarity, polarity proteins were assumed to function as tumor suppressors. However, it appears that they have a more complex relationship with cancer [Citation14,Citation15]. Changes in the polarity protein signaling network can promote new interactions of these proteins with the actin assembly machinery and enhance cell invasiveness and tumor progression[Citation16]. Moreover, these proteins show multifaceted interactions with components of the Hippo pathway, a pathway that controls epithelial cell proliferation and thereby influences intracellular forces[Citation15].
Mechanical signals also modify nuclear envelope (NE) dynamics. The nucleus is the largest and stiffest organelle limiting cell shape plasticity, particularly during cell migration in 3D environments [Citation17–19]. The nucleus can sense and trigger adaptive responses to mechanical forces that modify nuclear shape and deformability, along with cell polarization and migration [Citation20,Citation21]. Indeed, nuclear shape and stability are typically deregulated in tumor cells by multiple alterations of nuclear stiffness, notably, changes in lamin levels[Citation22–24]. These tumor cell alterations profoundly modify NE dynamics and correlate with increased metastatic potential. During metastatic migration, a reduction in nuclear stiffness is expected to facilitate cell shape adaptability in the face of physical constraints. However, enhanced nuclear plasticity is also correlated with nuclear fragility. In line with this, NE deformation and rupture are observed during migration in a restricted environment (confined cell migration) in vitro and in vivo [Citation25–27]. Moreover, tumor cells are prone to spontaneous NE ruptures, presumably because of NE component alterations[Citation28]. While most tumor cells survive to NE ruptures by NE repair, a transient loss of NE barrier function could promote innate immune signaling, genomic instability, tumor heterogeneity, and even resistance to first-line therapies [Citation29–31]. It is important to note that NE rupture is rare in most healthy cells during interphase [Citation28,Citation32–34]. Thus, NE rupture and repair are currently considered as novel hallmarks of cancer. Strategies to either inhibit NE rupture or NE repair could selectively target NE rupture-prone tumor cells. Impairing NE repair efficiency alone does not reduce tumor cell viability; however, simultaneously reducing both NE repair and DNA repair does increase cell death after NE rupture [Citation25,Citation26]. This suggests that NE rupture and repair could be exploited as synthetic lethal targets. Loss of function for p53 or RB tumor suppressors impairs genome stability and can also promote NE rupture via undefined mechanisms [Citation33,Citation35,Citation36]. Thus, targeting NE rupture and repair mechanisms could provide a novel entry point for combination therapies in a wide range of cancers bearing common alterations in p53 or RB pathways.
Figure 1. Migration-induced NE rupture and mechanisms of force transmission to the nucleus by different types of NE-associated actin cables
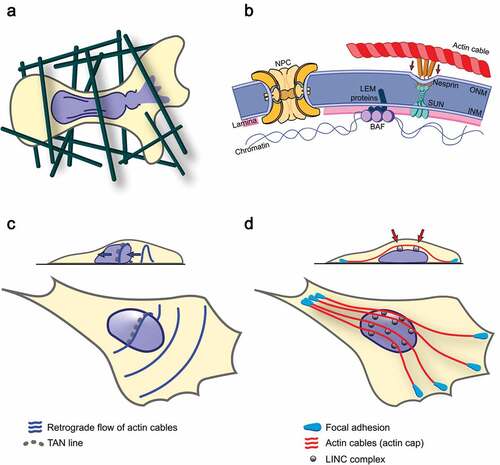
Despite important advances, studying these events at the molecular level is very challenging as they are stochastic. In this Perspective, we outline knowledge of mechanical stress-induced NE remodeling during interphase. We also cover relevant aspects of human adenovirus (Ad) type 2/5 early region 4 (E4) ORF4 protein (E4orf4) biology. In a novel paradigm, E4orf4 was found to exert tumor cell-selective killing by causing a high incidence of repetitive NE rupture-repair events.
Nuclear envelope remodeling during interphase
NE structure, nuclear mechanosensation, and nuclear mechanotransduction
The NE ensures nuclear compartmentalization and is composed of two specialized lipid bilayers. These include the outer nuclear membrane (ONM) contiguous with the endoplasmic reticulum and the inner nuclear membrane (INM)[Citation37]. NE membranes fuse at multiple sites to form donut-shaped channels (nuclear pore complexes, NPC) that control the bidirectional transport of macromolecules across the NE[Citation38]. In multicellular organisms, a dense fibrillar network of intermediate filaments (AC-type and B-type lamins), located between the INM and chromatin, provides a rigid scaffold that mechanically supports the NE and spatially organizes the genome by controlling heterochromatin domain organization[Citation39]. A-type lamins are believed to increase nuclear stiffness, while B-type lamins support nuclear elasticity [Citation40,Citation41]. The NE serves as an important signaling hub. This is due to interactions between intracellular signaling pathways and NE transmembrane (NET) proteins, including LEM (LAP2, Emerin, MAN1) domain proteins at the INM[Citation42]. Therefore, the NE functions as a scaffold for the regulation of cell cycle progression, mitosis, DNA repair, cell senescence, and cell migration. However, signal transduction across the NE, and the contribution of most NET proteins, remain poorly explored.
The NE undergoes multiple morphological changes [Citation43,Citation44]. In human cells, the most dramatic change is undoubtedly the rapid NE disassembly and reassembly during mitosis, a process that needs strict spatiotemporal coordination to prevent NE damages and aneuploidy. NE remodeling also occurs during interphase. For instance, NE remodeling is present in growing cells to increase NE surface and insert NPC. Additionally, NE remodeling occurs in response to force, for nuclear structure adaptation to mechanical stress, and to prepare cells for confined migration [Citation43–45]. Moreover, vesicular transport across the NE enables large RNP particles and viral particles to exit the nucleus via a NPC-independent process called nuclear budding, exemplifying unforeseen NE plasticity [Citation46,Citation47]. Therefore, NE remodeling uses conserved molecular mechanisms that may be exploited by tumor cells to increase NE plasticity and cell invasiveness.
Cytoplasmic mechanosensitive structures include ion channels, adhesion complexes, and intercellular junction complexes [Citation3,Citation48]. They also include cytoskeletal elements, and in particular, actin filaments. Forces are mainly sensed through the binding and conformation of proteins residing in mechanosensitive structures. These proteins have motifs that can change conformation over mechanical forces to expose cryptic sites and promote events, such as protein complex assembly and posttranslational modifications[Citation49]. These events, in turn, initiate biochemical responses that widely implicate focal adhesion kinase (FAK), oncogenic tyrosine kinase Src, mitogen-activated protein (MAP) kinases, and Rho family of small GTPases. Interestingly, all of these responses are generally upregulated in tumor cells and enable cells to adjust shape, contractility, and stiffness in response to extracellular matrix rigidity [Citation3,Citation48]. Upon sustained mechanical stress, tumor cells engage in cellular adaptive responses that have pro-proliferative and pro-migratory functions, such as Hippo pathway effectors YAP and TAZ[Citation50]. Downstream of these signaling cascades also, nuclear lamin A dephosphorylation can stabilize lamin filaments, remodel the NE, and adjust nuclear stiffness [Citation40,Citation41,Citation51]. However, lamin A/C can directly respond to mechanical stress since changes in lamin A/C epitope expression have been reported[Citation52].
Indeed, the forces generated at cell-surface mechanosensitive structures can be transmitted across the NE via the linker of nucleoskeleton and cytoskeleton (LINC) complex [Citation53–55]. This complex consists of KASH domain proteins (Nesprins) that span the ONM, and SUN domain proteins that span the INM [Citation56]. Interactions between the KASH and SUN domains in the perinuclear space form a physical bridge between the ONM and the INM. This bridge connects chromatin to the microenvironment through SUN protein binding to nuclear lamin A/C and Nesprin binding to elements of the cytoskeleton. Increased intracellular forces, through myosin-mediated stress fiber assembly, can remodel the LINC complex by reinforcing the coupling of LINC proteins to lamin A/C [Citation57,Citation58]. Various LINC complex connections act by anchoring actomyosin filament networks to the apical NE surface and regulating their spatial dynamics. For instance, LINC complex clusters, composed of Nesprin-2G and SUN2, that colocalize with actin cables were named transmembrane actin-associated nuclear (TAN) lines [Citation59]. TAN lines are functionally linked to nuclear positioning and cell migration. In addition, distinct actomyosin cables, which are aligned with the cell axis and connected to large peripheral focal adhesion sites, can form an actin cap at the apical cell surface [Citation60]. Actomyosin cables of the actin cap appear to provide a bridge for the rapid transmission to the nucleus of changes in external forces, such as in substrate compliance or fluid shear stress, which are relevant mechanical stimuli for circulating tumor cells [Citation57,Citation61]. By exerting both compressive forces to the NE and tension to the LINC complex, actin cap structures can also shape the cell’s nucleus. Thus, various types of actin-dependent cytoskeletal forces are generated in different cellular contexts and can remodel LINC complex connections in a regulated manner[Citation55].
Through these connections, LINC complex-mediated signal transduction could enable the cells to respond to strong forces, in part, by changing NE structure and chromatin organization [Citation20,Citation41,Citation55]. Applying local stresses to integrins can trigger, via the LINC complex, chromatin deformation and transcription[Citation62]. Through the LINC complex, forces associated with significant nuclear deformation can also alter NPC permeability and enhance the nuclear transport of large molecules, such as YAP[Citation63]. Moreover, applying direct force to the LINC complex stiffens isolated nuclei. This nuclear response is triggered by Src-induced phosphorylation of the INM protein Emerin/LEMD5[Citation64]. Remarkably, Emerin can mediate myosin II recruitment to the NE, perinuclear actin polymerization, cell polarization, confined cell migration, and YAP nuclear localization [Citation64,Citation65].
Thus, direct evidence supports a role for mechanical stress-induced NE remodeling in mechanotransduction. However, it is difficult to determine which nuclear event directly responds to forces or is downstream of cytoplasmic signaling cascades, since the pathways involved are tightly intertwined. Interestingly, NE rupture and repair is an extreme case of NE remodeling during interphase. Additionally, the mechanisms involved in NE rupture and repair may be particularly relevant for highly motile metastatic cancer cells.
Mechanisms of NE rupture and repair
When strong forces exceed nuclear mechanical resilience, NE deformation and remodeling can generate intranuclear pressure which causes the appearance of fragile NE sites. These fragile NE sites are believed to promote nuclear bleb formation and rupture [Citation28,Citation32,Citation66,Citation67]. NE rupture susceptibility, along with a tumor’s cell metastatic potential, depends on lamin levels. It was shown that NE rupture and confined cell migration are promoted by the downregulation of either A- and/or B-type lamin levels, whereas they are impeded by the upregulation of lamin A/C levels [Citation18,Citation28,Citation68,Citation69]. The primary mechanical forces that cause NE rupture are generated by actomyosin filaments[Citation66]. In line with this, loss of myosin phosphatase subunit MYPT1/PPP1R12A, which antagonizes actomyosin contractility, promotes cancer cell nuclear dysmorphia and NE rupture[Citation34]. Conversely, inhibition of myosin II or actin dynamics usually impedes NE rupture [Citation25,Citation66,Citation70,Citation71]. While forces applied to the nucleus during confined cell migration can be independent of the LINC complex[Citation72], they generally rely on an intact LINC complex to trigger efficient NE remodeling and rupture.
As discussed above, the LINC complex may provide a dynamic signal-responsive interface between the nucleus and the cytoplasm, similar to cellular adhesion complexes at the cell surface[Citation55]. Different LINC complexes appear to play specific roles in the organization of actin networks[Citation73]. However, little is known on the LINC complex-associated signal transduction mechanisms that have an impact on NE rupture events. Several proteins regulating TAN line assembly were identified, including Formin homology 2 domain-containing 1 FHOD1, the actin-bundling protein Fascin, the ATP-binding protein Torsin 1A, and Spindle-associated membrane protein 1 Samp1/TMEM201 [Citation74–79]. However, only Fascin has been tested for its ability to regulate nuclear deformation during confined cell migration and the role of other TAN line proteins in this process remains unclear[Citation76]. While TAN line assembly relies on the LINC complex protein SUN2, spontaneous NE rupture is specifically hindered by SUN1 depletion, suggesting different modes of NE-actomyosin filament tethering[Citation66]. Regarding actin cap assembly, an integrated signaling platform, using RhoA and Rac1 GTPases, appears to be essential [Citation60,Citation80,Citation81]. Intriguingly, SUN1 and SUN2 can have opposing effects on RhoA and actin organization[Citation80]. Therefore, future studies will be important to clarify LINC complex-associated signaling pathways that enhance NE rupture susceptibility in tumor cells.
Increased NE rupture can eventually cause cell death[Citation82]. However, NE rupture-susceptible tumor cells can survive by the rapid recruitment of molecular repair machinery[Citation83]. Studies have shown that NE repair after rupture has several molecular effectors in common with post-mitotic NE reassembly. Initial work on migration-associated NE rupture uncovered a role for components of the endosomal sorting complex required for transport III (ESCRT-III), which mirrored their function in post-mitotic NE reassembly and plasma membrane repair [Citation25,Citation26,Citation84–87]. Recently, the recruitment of cytosolic barrier to autointegration factor (BAF) was shown to be critical to initiate NE repair, as required for postmitotic NE reassembly [Citation88–91]. BAF is an early-acting factor that recruits LEM domain proteins and endoplasmic reticulum membranes to NE rupture sites. This effect is dependent on BAF’s ability to bind DNA, suggesting that NE sealing is initiated at the exposed chromatin surface [Citation89,Citation92,Citation93]. Finally, NE membrane composition and lipid synthesis also impact NE repair [Citation82,Citation83].
However, the factors that determine NE repair efficiency (e.g. whether the repaired NE site might be more susceptible to future rupture events) remain unknown. In this regard, it will be important to determine the role of the phosphatases that control post-mitotic NE reassembly[Citation94]. Another key question that remains is whether an integrated signaling interface exists between NE rupture and repair machinery to ensure a tight spatiotemporal coupling of these important events. In the following sections, evidence supporting the value of exploiting the adenoviral protein E4orf4 to address these issues in tumor cell-defining mechanics will be discussed.
Targeting tumor cell mechanics with the adenoviral protein E4orf4
E4orf4, perinuclear actomyosin dynamics, repetitive NE rupture, and tumor cell-selective killing
Adenoviral type 2/5 early region 4 (E4) open reading frame 4 (ORF4) protein (E4orf4) is an early viral gene product which, although not essential, plays several functions to facilitate viral replication. A recent comprehensive review on E4orf4 is available[Citation95]. The interest in the biology of E4orf4 stems from the discovery of its unusual intrinsic toxicity in a variety of tumor cells, which is typified by unique changes in actomyosin structure organization [Citation96–100]. E4orf4’s cell-killing ability is highly correlated with the assembly of robust actomyosin fibers that wrap around the nucleus at the apical cell surface and connects to enlarged focal adhesions at the basal cell surface [Citation101,Citation102]. The induction of polarized cell blebbing generally follows the assembly of the perinuclear contractile structure and is correlated with intense nuclear deformations and chromatin condensation. Mechanistically, E4orf4 deregulates focal adhesions stability and several components of mechanoresponsive signaling pathways, including tyrosine kinase Src, MAP kinases ERKs and JNKs, and Paxillin [Citation99,Citation102–104]. The E4orf4 phenotype also depends on the small GTPases RhoA, Rac1, Cdc42, and Rab11, along with myosin II [Citation101,Citation105,Citation106]. Importantly, the downregulation of myosin II motor activity inhibits E4orf4’s phenotypic hallmarks and impedes tumor cell killing [Citation101,Citation107]. Most intriguingly, myosin II inhibition also hinders E4orf4 nuclear exit, thereby inhibiting actomyosin filament remodeling. These E4orf4 hallmarks argue that it functions by deregulating tumor cell mechanics.
Figure 2. E4orf4's tumoricidal hallmark features
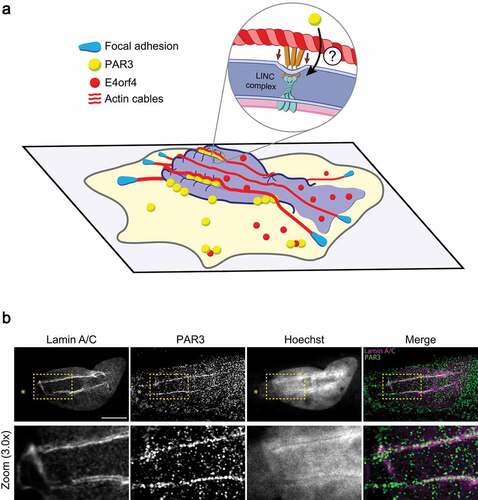
Recently, the deregulation of cell mechanics by E4orf4 was shown to be tumor cell-selective, since various nontumorigenic epithelial cell lines resist to E4orf4-induced cytoskeletal remodeling and killing[Citation107]. While these findings corroborate a longstanding premise that E4orf4 targets tumor cell-defining features, they provide a first hint on how E4orf4’s actions are dictated [Citation107,Citation108]. E4orf4-sensitive cells display striking nuclear lamina disorganization that forms folds at sites of NE-actomyosin filaments tethering, as typically observed when mechanical stress is applied to the nucleus[Citation109]. Live-cell imaging revealed that E4orf4 induces a high frequency of nuclear blebs that often burst, thereby provoking a sudden efflux of E4orf4 into the cytoplasm . Nuclear bleb formation is dependent on myosin II motor activity. Nuclear bleb formation and bursting were also shown to reflect repetitive NE rupture and repair events, often at related NE sites, suggesting that NE repair is not efficient. Ultimately, these events are associated with a sustained loss of nuclear compartmentalization that correlates with the onset of cell blebbing, nuclear condensation, and cellular integrity loss[Citation107].
Overall, these results argue that the disruption of NE mechanical regulation is a primary lesion induced by E4orf4 in tumorigenic cells. E4orf4-induced NE rupture appears to establish a vicious lethal cycle empowered by E4orf4 nuclear efflux. Cytoplasmic E4orf4, by promoting actomyosin filament assembly, may exacerbate the mechanical stress applied to the nucleus and exceed the cell’s NE repair capacity. E4orf4’s potential effects on DNA repair efficiency could also contribute to cell death in this context, considering that NE rupture and transient nuclear integrity loss can promote DNA damage due to the uncoordinated exchange of nuclear and cytoplasmic material [Citation29,Citation110]. In line with this notion, E4orf4 causes the accumulation of DNA damage foci in tumor cells showing NE rupture increases.Footnote1 However, E4orf4’s effects on DNA repair appear to be complex, as the protein can either inhibit or promote DNA damage signaling in the context of virally infected cells[Citation95]. In cells challenged with DNA-damaging compounds, E4orf4 can inhibit DNA damage signaling and presumably DNA repair. It will be informative to clarify E4orf4’s impact on both NE repair efficiency and DNA damage signaling in the context of NE rupture-associated tumor cell-selective killing, in the absence of externally inflicted DNA damage.
Nonetheless, our recent findings indicate that by identifying E4orf4’s targets in tumor cells, it will be possible to decipher conserved elements of molecular NE remodeling mechanisms underlying the tumor cell phenotype. Since it was shown that E4orf4 inhibits the development of highly aggressive tumors in Drosophila without causing significant damage to healthy tissues, the mechanisms identified will be relevant to the understanding of tumorigenesis[Citation111].
Deciphering E4orf4’s protein network
E4orf4, like many viral proteins, exerts its cellular activities by disrupting normal protein complex functions in cells. Studies have sought to exploit large scale affinity purification followed by mass spectrometry (AP-MS) to identify E4orf4’s protein targets. Three main E4orf4 proteomic datasets have been obtained using diverse experimental approaches and model cell lines [Citation107,Citation112,Citation113]. We combined these proteomic datasets to perform a comprehensive bioinformatic analysis and defined a set of 90 putative E4orf4’s cellular partners . Strong enrichments were found for subunits of two major phosphatase systems (PP1 and PP2), and proteins related to intercellular junctions and polarity signaling, mainly PAR3/PARD3 (2/3 studies; , asterisks). PP1 and PP2A are believed to account for ~90% of all phosphoserine/phosphothreonine phosphatase activity in mammalian cells [Citation114,Citation115]. They mainly consist of heterodimers and heterotrimers, which contain a catalytic subunit (C), a structural subunit (A, PP2A), and a variable regulatory subunit (R, usually designated B subunit for PP2A) that dictates substrate specificity[Citation115]. Remarkably, E4orf4 shows highly selective binding to a limited subset of PP1- and PP2A-regulatory proteins and . PAR3/PARD3 is a signaling scaffold that functions within the conserved polarity PAR complex, consisting of PAR3, PAR6, and atypical PKC (aPKC) (PKCɩ/PRKCi, ) [Citation116,Citation117]. Interestingly, PAR3 has both tumor suppressor and oncogenic activities and interacts with the Hippo signaling pathway[Citation15]. In the following sections, we discuss the role of PAR3 in E4orf4-induced NE remodeling and explore potential contributions for PP1- and PP2A-regulatory proteins.
Figure 3. E4orf4’s protein network identified by AP-MS analyses
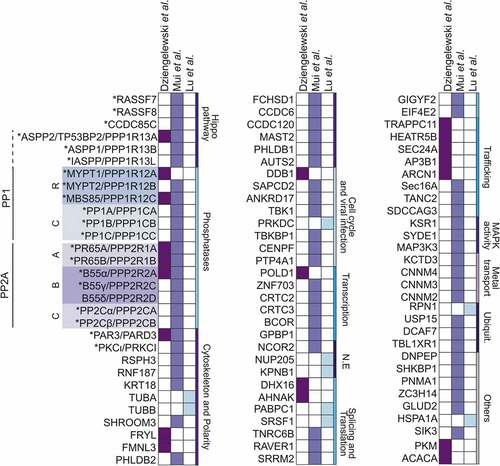
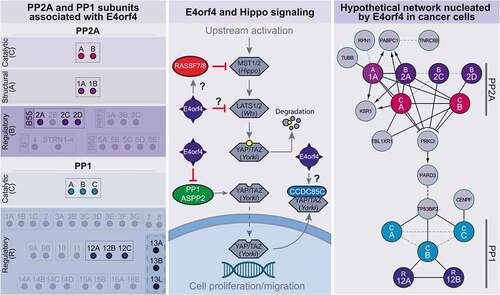
Figure 4. Schematics of E4orf4-associated PPP-regulatory proteins, and their relationships to the Hippo pathway and PAR complex network
PAR3 and the E4orf4-dependent induction of NE rupture
The PAR complex regulates epithelial polarity, apical domain formation, and cell junction remodeling [Citation117,Citation118]. PAR3 also plays a crucial role in the segregation of active Cdc42/aPKC to facilitate the spatial control of actomyosin filament organization [Citation119,Citation120]. Therefore, we postulated that E4orf4 subverts PAR3’s scaffolding function to deregulate perinuclear actomyosin filament dynamics and NE mechanical stability.
Our studies revealed that E4orf4’s main tumoricidal phenotypes, including NE rupture, are regulated by PAR3[Citation107]. Functional dissection of the E4orf4-PAR3 interaction using integrated protein biochemistry and cell imaging approaches indicates that these phenotypes are highly correlated with a proximal interaction between E4orf4 and PAR3 at perinuclear sites. Moreover, E4orf4 can associate with other members of the PAR complex (PAR6/PARD6 and PKCɩ), in a PAR3-dependent manner. While the E4orf4-PAR3 interaction is observed also in nontumorigenic cells, the functional outcome is different. Rather than the appearance of tumoricidal phenotypes, E4orf4 promotes the assembly of cell-cell junctions in nontumorigenic cells, suggesting that E4orf4 reinforces polarity signaling. These results argue that differences in cell mechanics enable E4orf4 to exploit an undescribed PAR complex activity in tumor cells. Mechanistically, E4orf4 induces PAR3 clustering at the NE in a tumor cell-selective manner, thereby triggering the recruitment of actin fibers, nuclear deformation, and transient NE rupture . High-resolution confocal microscopy revealed that the PAR3 clusters are recruited to sites of nuclear indentation, showing striking alignment with NE folds . Since apical actin cables also localize along nuclear surface invaginations or NE folds of lamin A/C in E4orf4-expressing cells, PAR3 clusters may define sites of actin-dependent mechanical stress at the NE[Citation107]. Importantly, disrupting either myosin II, PAR3, or the LINC complex, can all recapitulate the same inhibition of E4orf4-induced NE rupture, cell blebbing, and nuclear condensation[Citation107]. These results suggest that E4orf4 acts by corrupting PAR3’s scaffolding function within the NE, which may control actomyosin filament tethering and thereby, force transmission to the nucleus. Since we observed that PAR3 regulates a retrograde flowing of actin fibers towards the nucleus, the PAR complex could provide the spatial information to polarize actomyosin filaments and nucleo-cytoskeletal coupling. However, future work is needed to determine whether PAR3 is engaged with the NE via an interaction with the LINC complex or the nuclear lamina.
Based on current knowledge, PAR proteins could also affect nuclear stiffness via the phosphorylation of lamina proteins, as reported during NE remodeling. This enables the nuclear exit of large RNP particles via NPC-independent nuclear budding in Drosophila[Citation46]. By acting on both lamina proteins and perinuclear actin organization, PAR complex proteins could represent key regulators of nucleo-cytoskeletal connections in response to mechanical forces.
PP1-regulatory proteins targeted by E4orf4 in cellular mechanotransduction
PP1, through its catalytic subunits, interacts with hundreds of putative regulatory proteins that control substrate access and activity on different pathways[Citation115]. Proteomic datasets indicate that E4orf4 interacts with the three PP1 catalytic subunits (PPP1C) but only a few PP1-regulatory proteins (PPP1R; and ) [Citation101,Citation112]. Remarkably, these PP1-regulatory proteins show functional relationships with cellular contractility networks and polarity protein signaling.
Three PP1-regulatory proteins, MYPT1, MYPT2, and MBS85, are myosin phosphatase targeting subunit (MYPT) family members (, PPP1R12A-C). These subunits share homology and function in targeting PP1 catalytic activity to dephosphorylate myosin light chain, thereby inhibiting cellular contractile forces [Citation121,Citation122]. These interactions are likely to be relevant for E4orf4’s tumoricidal action, as myosin phosphatase complexes act to safeguard nuclear integrity in cancer cells. Indeed, the loss of either MYPT1 or PPP1CB subunits was shown to cause nuclear dysmorphia, NE rupture, and genome instability in cancer cells[Citation34]. Notably, the presence of actin filaments at NE rupture sites was correlated with nuclear damage. Thus, it is reasonable to speculate that E4orf4 may inhibit MYPT-PP1 catalytic activity to enhance NE-actomyosin filament tethering. In line with this, E4orf4 was found to stimulate MYPT1 phosphorylation on residues that inhibit myosin phosphatase complex activity, a process that is mainly catalyzed by ROCK [Citation101,Citation121]. Moreover, E4orf4 recruits the Rho-ROCK signaling axis to the perinuclear region, promoting actomyosin filament assembly [Citation101,Citation102]. Of note, E4orf4-interacting MYPT proteins share the ROCK-targeted residue[Citation121]. Additionally, myosin phosphatase complexes are suggested to play other non-contractile functions[Citation122]. Among potentially relevant substrates, MYPT1 can interact with ezrin-radixin-moesin (ERM) proteins that organize specialized actin filament-membrane domains in a phosphorylation-dependent manner[Citation123]. The MYPT1-PP1 complex also appears to modulate the tumor suppressor RB and could, in principle, affect its ill-defined function in NE rupture [Citation33,Citation124].
Intriguingly, other E4orf4- and PP1-associated proteins are members of the apoptosis stimulating proteins of p53 (ASPP) family, notably ASPP1 and ASPP2, and inhibitor of ASPP protein (iASPP) ( and ; PPP1R13A, 13B, 13 L). ASPP proteins were initially characterized as modulators of apoptosis mediated by the p53 family of tumor suppressors[Citation125]. However, E4orf4-induced cell killing is p53-independent [Citation96,Citation98,Citation108]. Therefore, E4orf4 might target other functions of ASPP proteins.
Current knowledge of PP1 regulation by ASPP proteins is limited. Interestingly, ASPP1/2-PP1 complexes were shown to dephosphorylate and activate the Hippo pathway transcriptional coactivator YAP [Citation126,Citation127]. As aforementioned, the Hippo pathway is deregulated in cancer and YAP regulates cytoskeletal remodeling and cell proliferation in response to high density or mechanosensory stimulation[Citation50]. It is important to note that E4orf4-induced cell killing was correlated with YAP phosphorylation and downregulation[Citation112]. Since YAP silencing can enhance cell death in response to E4orf4 expression, this further argues that E4orf4 susceptibility is regulated by cell mechanics. Moreover, several additional proteins linked to Hippo signaling were found in E4orf4 complexes and . This includes Ras-associated domain family proteins 7 and 8 (RASSF7/8) and coiled-coil domain-containing protein 85 C (CCDC85C), which were shown to differentially affect Hippo signaling [Citation128–131]. It will be important to dissect the exact impact of E4orf4 on these Hippo pathway components and PP1-catalytic activity. Since enhanced YAP phosphorylation seems independent of the Hippo pathway Large tumor suppressor kinase 1/2 (LATS1/2), YAP inhibition could rather be a result of E4orf4-dependent cellular mechanotransduction deregulation[Citation112]. Of note, we observed that NE rupture correlates with YAP nuclear exit in response to E4orf4 expression.Footnote2
Finally, ASPP2 function in cellular junction remodeling is particularly interesting for mechanical signal transduction to the nucleus. As mentioned earlier, intercellular adhesions, like cell-matrix adhesions, are connected to the nuclear lamina via the LINC complex that transmits actomyosin forces to the NE [Citation54,Citation55]. ASPP2, by restraining Src kinase activity and recruiting the polarity protein PAR3 and RASSF7/8 to cell junctions, positively regulates apical domain formation and epithelial cell polarity [Citation132–134]. Furthermore, ASPP2 was shown to act as a molecular switch in epithelial plasticity, favoring a mesenchymal-to-epithelial transition (MET) in tumors via a pathway that implicates cell junction remodeling[Citation135]. These findings suggest that an ASPP2 function in intercellular force transmission is relevant for tumorigenesis. A role for PP1 catalytic activity was not explored in this specific context. However, recent work indicates that Drosophila ASPP protein shares most of its biological functions with PP1 catalytic activity and the PP1-associated proteins RASFF7/8 and CCDC85C[Citation136]. It is tempting to speculate that E4orf4 could hijack components of the ASPP2-PP1 complex to exploit their function in the control of mechanical forces and segregation of specialized actomyosin filament-membrane domains. Recent finding for a dynamic between E4orf4 and PAR3, another component of epithelial cell junction that interacts with ASPP2, provides support to this hypothesis [Citation107,Citation132,Citation133].
E4orf4-associated PP2A targeting subunits in nuclear dynamics
PP2A phosphatase is the first identified E4orf4 target with important roles in both viral replication and cell killing [Citation95,Citation137], While there is still no consensus regarding how E4orf4 impacts PP2A catalytic activity, it is interesting to speculate on a potential function for PP2A in E4orf4-induced NE remodeling. Strong evidence supports a crucial role for B55α, PP2A regulatory subunit, in mediating efficient cell killing by E4orf4 in mammalian cell lines and Drosophila [Citation97,Citation111,Citation138]. Initial work revealed that E4orf4 selectively associates with the trimeric PP2A holoenzyme via interaction with B55α, consistent with proteomic datasets showing strong enrichment for B55α, but also for B55γ and δ and [Citation137–139]. Since the E4orf4-PP2A complex is associated with PP2A-like phosphatase activity, it is proposed that E4orf4 can target the B55-PP2A complexes to dephosphorylate a subset of substrates required for cell death. In the context of virally infected cells, E4orf4 can target PP2A to dephosphorylate serine/arginine-rich proteins (SR proteins) along with the NPC protein NUP205 [Citation113,Citation140,Citation141]. Nevertheless, in the context of tumor cell killing, no substrate for the B55-PP2A complexes has been formally identified.
Dissecting the role of B55 subunits in E4orf4-induced NE remodeling could help uncover relevant substrates. It is interesting to note that PP2A is a major regulator of cell junctions remodeling and polarity signaling, which could, therefore, affect force transmission to the NE[Citation142]. Moreover, B55α was identified as the main regulatory subunit to target PP2A to dephosphorylate BAF during post-mitotic NE reassembly [Citation143–146]. We confirmed that BAF is recruited to NE rupture sites in E4orf4-expressing cells.Footnote3 This points to the possibility that molecular mechanisms are induced to couple both NE rupture and repair in response to E4orf4 expression. Therefore, the role of B55α-PP2A complex in these events merits continued investigation.
Presently, it is not known if E4orf4 affects NE stiffness directly and decreases nuclear mechanical resilience to enhanced perinuclear actomyosin contractility. Chromatin compaction, which is linked to the nuclear lamina, is considered as a potential source of intranuclear forces that influence nuclear rigidity [Citation147,Citation148]. Since E4orf4 promotes PP2A targeting to the chromatin remodeler complex ACF, it could theoretically alter chromatin structure and thereby affect nuclear stiffness [Citation149–151]. Also, the ability of E4orf4 to modulate B55-PP2A-mediated dephosphorylation of NPC components, notably NUP205, could impact NE stability, as several connections between the nucleopore complex, lamina, and chromatin have been identified [Citation21,Citation113]. Finally, diversion of B55-PP2A complexes could indirectly alter the phosphorylation of NE components and affect NE mechanical stability. For instance, lamin A phosphorylation by constitutive kinases can stimulate its degradation, and, theoretically, reduce nuclear stiffness [Citation40,Citation51,Citation152]. If B55-PP2A complexes contribute to E4orf4-induced NE remodeling, the identification of relevant B55-PP2A complex interactions will have broad significance for cell biology.
Deciphering bona fide regulators of NE plasticity using the adenoviral protein E4orf4
The work using E4orf4 to model mechanical stress-induced NE rupture suggests the existence of unappreciated relationships between polarity protein signaling and NE remodeling in tumor cells. Since several of E4orf4’s targets share a function in cell junction remodeling, they could contribute to LINC complex-associated signal transduction mechanisms that may be rewired in tumor cells to promote NE plasticity. This hypothesis is in line with our finding that PAR3 can regulate spontaneous NE rupture in susceptible tumor cells, in the absence of E4orf4. Indeed, PAR3 depletion was shown to hinder spontaneous NE rupture facilitated by the downregulation of nuclear lamins[Citation107]. These results suggest that PAR3 is a bona fide regulator of the actin-dependent forces that remodel the NE in tumor cells that are harnessed by E4orf4. Cell-cell adhesion and polarity signaling mechanisms are typically deregulated in tumor cells [Citation3,Citation13,Citation15]. This can trigger ectopic signaling by these proteins in other cell compartments, which might render these networks prone to be hijacked by E4orf4 [Citation153,Citation154].
We suggest that E4orf4 subverts bona fide regulators of NE rupture and repair that enable tumor cells to cope with physical constraints during metastatic migration. Dissecting the biology of E4orf4’s targets will be crucial. Intriguingly, our analysis of proteomic datasets revealed that the B55-PP2A and MYPT-PP1 complexes can potentially interact with each other via the polarity signaling proteins aPKC-PAR3, and ASPP2, respectively [Citation155]. There is precedent for a PP1-PP2A phosphatase relay in yeast, and the molecular mechanisms appear to be conserved in mammalian cells[Citation156]. In this paradigm, PP1 is recruited to B55-PP2A at the mitotic exit to coordinate the sequential re-activation of these phosphatase systems that contribute to post-mitotic NE reassembly[Citation94]. It is tempting to speculate that polarity signaling hubs could couple actomyosin filament tethering at the NE and the recruitment of the NE repair machinery during NE remodeling, to rapidly cope with possible NE damage.
In conclusion, our understanding of E4orf4’s protein network, as it relates to NE rupture and repair is rather incomplete. Identifying highly dynamic and short-lived interactions that are likely to involve cytoskeletal and membrane-bound insoluble proteins will require using alternative methods. Since NE rupture and repair are tightly coupled, the rapid biotinylation of proximal proteins could capture bona fide molecular effectors that are near to E4orf4 as it leaks out via local NE rupturing. Future work using the adenovirus protein E4orf4 will undoubtedly decipher NE plasticity mechanisms that broadly define tumor cells and identify novel entry points for combination therapies.
Acknowledgments
We apologize to those whose work was not discussed, due to space limitations. The authors wish to acknowledge the crucial contribution of Claire Dziengelewski for the initial discovery of a role for PAR3 in NE rupture. This work was funded by the Cancer Research Society Inc (CRS) under Operating Grant to J.N.L. (NIP: 23395), and by the Natural Sciences and Engineering Research Council (NSERC) under Discovery Grant to J.N.L. (RGPIN/05849). M-A Rodrigue is supported by a doctoral award from the Fonds de la Recherche en Santé du Québec (FRQS).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Notes
1. Jacquet K. and Lavoie J.N., unpublished observations.
2. Dziengelewski C. and Lavoie J.N., unpublished observations.
3. Jacquet K. and Lavoie J.N., unpublished observations.
References
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
- Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO reports. 2014;15(12):1243–1253.
- Mohammadi H, Sahai E. Mechanisms and impact of altered tumour mechanics. Nat Cell Biol. 2018;20(7):766–774.
- Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9(2):108–122.
- Yuan S, Norgard RJ, Stanger BZ. Cellular plasticity in cancer. Cancer Discov. 2019;9(7):837–851.
- Williams ED, Gao D, Redfern A, et al. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer. 2019;19(12):716–732.
- Alibert C, Goud B, Manneville JB. Are cancer cells really softer than normal cells? Biol Cell. 2017;109(5):167–189.
- Coughlin MF, Bielenberg DR, Lenormand G, et al. Cytoskeletal stiffness, friction, and fluidity of cancer cell lines with different metastatic potential. Clin Exp Metastasis. 2013;30(3):237–250.
- Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular traction stresses increase with increasing metastatic potential. PloS One. 2012;7(2):e32572.
- Panciera T, Citron A, Di Biagio D, et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat Mater. 2020;19(7):797.
- Clayton NS, Ridley AJ. Targeting Rho GTPase signaling networks in cancer. Front Cell Dev Biol. 2020;8:222.
- Ohashi K, Fujiwara S, Mizuno K. Roles of the cytoskeleton, cell adhesion and rho signalling in mechanosensing and mechanotransduction. J Biochem. 2017;161(3):245–254.
- Halaoui R, McCaffrey L. Rewiring cell polarity signaling in cancer. Oncogene. 2015;34(8):939–950.
- Saito Y, Desai RR, Muthuswamy SK. Reinterpreting polarity and cancer: the changing landscape from tumor suppression to tumor promotion. Biochim Biophys Acta Rev Cancer. 2018;1869(2):103–116.
- Fomicheva M, Tross EM, Macara IG. Polarity proteins in oncogenesis. Curr Opin Cell Biol. 2020;62:26–30.
- Gandalovičová A, Vomastek T, Rosel, T, et al. Cell polarity signaling in the plasticity of cancer cell invasiveness. Oncotarget. 2016;7(18):25022-49.
- Wolf K, Te Lindert M, Krause M, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201(7):1069–1084.
- Davidson PM, Denais C, Bakshi MC, et al. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell Mol Bioeng. 2014;7(3):293–306.
- Calero-Cuenca FJ, Janota CS, Gomes ER. Dealing with the nucleus during cell migration. Curr Opin Cell Biol. 2018;50:35–41.
- Kirby TJ, Lammerding J. Emerging views of the nucleus as a cellular mechanosensor. Nat Cell Biol. 2018;20(4):373–381.
- Janota CS, Calero-Cuenca FJ, Gomes ER. The role of the cell nucleus in mechanotransduction. Curr Opin Cell Biol. 2020;63:204–211.
- Bell ES, Lammerding J. Causes and consequences of nuclear envelope alterations in tumour progression. Eur J Cell Biol. 2016;95(11):449–464.
- Denais C, Lammerding J. Nuclear mechanics in cancer. Adv Exp Med Biol. 2014;773:435–470.
- Chow KH, Factor RE, Ullman KS. The nuclear envelope environment and its cancer connections. Nat Rev Cancer. 2012;12(3):196–209.
- Denais CM, Gilbert RM, Isermann P, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352(6283):353–358.
- Raab M, Gentili M, de Belly H, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352(6283):359–362.
- Xia Y, Ivanovska IL, Zhu K, et al. Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J Cell Biol. 2018;217(11):3796–3808.
- Vargas JD, Hatch EM, Anderson DJ, et al. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus. 2012;3(1):88–100.
- Pfeifer CR, Vashisth M, Xia Y, et al. Nuclear failure, DNA damage, and cell cycle disruption after migration through small pores: a brief review. Essays Biochem. 2019;63(5):569–577.
- Shah P, Wolf K, Lammerding J. Bursting the bubble - nuclear envelope rupture as a path to genomic instability? Trends Cell Biol. 2017;27(8):546–555.
- Maciejowski J, Hatch EM. Nuclear membrane rupture and its consequences. Annu Rev Cell Dev Biol. 2020;36:85-114.
- De Vos WH, Houben F, Kamps M, et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet. 2011;20(21):4175–4186.
- Yang Z, Maciejowski J, de Lange T. Nuclear envelope rupture is enhanced by loss of p53 or Rb. Mol Cancer Res. 2017;15(11):1579–1586.
- Takaki T, Montagner M, Serres MP, et al. Actomyosin drives cancer cell nuclear dysmorphia and threatens genome stability. Nat Commun. 2017;8(1):16013. .
- Dick FA, Goodrich DW, Sage J, et al. Non-canonical functions of the RB protein in cancer. Nat Rev Cancer. 2018;18(7):442–451.
- Eischen CM. Genome Stability Requires p53. Cold Spring Harb Perspect Med. 2016;6(6): a026096
- Agrawal A, Lele TP. Mechanics of nuclear membranes. J Cell Sci. 2019;132(14): jcs229245.
- Knockenhauer KE, Schwartz TU. The nuclear pore complex as a flexible and dynamic gate. Cell. 2016;164(6):1162–1171.
- Osmanagic-Myers S, Dechat T, Foisner R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015;29(3):225–237.
- Swift J, Ivanovska IL, Buxboim A, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104.
- Cho S, Irianto J, Discher DE. Mechanosensing by the nucleus: from pathways to scaling relationships. J Cell Biol. 2017;216:305–315.
- Alvarado-Kristensson M, Rossello CA. The biology of the nuclear envelope and its implications in cancer biology. Int J Mol Sci. 2019;20(10):2586.
- Ungricht R, Kutay U. Mechanisms and functions of nuclear envelope remodelling. Nat Rev Mol Cell Biol. 2017;18(4):229–245.
- De Magistris P, Antonin W. The dynamic nature of the nuclear envelope. Curr Biol. 2018;28(8):R487–R97.
- Gundersen GG, Worman HJ. Nuclear positioning. Cell. 2013;152(6):1376–1389.
- Speese SD, Ashley J, Jokhi V, et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell. 2012;149(4):832–846.
- Hellberg T, Passvogel L, Schulz KS, et al. Nuclear egress of herpesviruses: the prototypic vesicular nucleocytoplasmic transport. Adv Virus Res. 2016;94:81–140.
- Martino F, Perestrelo AR, Vinarsky V, et al. Cellular Mechanotransduction: from Tension to Function. Front Physiol. 2018;9:824.
- Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. 2006;7:265–275.
- Panciera T, Azzolin L, Cordenonsi M, et al. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. 2017;18(12):758–770.
- Buxboim A, Swift J, Irianto J, et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr Biol. 2014;24:1909–1917.
- Ihalainen TO, Aires L, Herzog FA, et al. Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat Mater. 2015;14(12):1252–1261.
- Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc Natl Acad Sci U S A. 1997;94(3):849–854.
- Schwager SC, Taufalele PV, Reinhart-King CA. Cell-cell mechanical communication in cancer. Cell Mol Bioeng. 2019;12:1–14.
- Aureille J, Belaadi N, Guilluy C. Mechanotransduction via the nuclear envelope: a distant reflection of the cell surface. Curr Opin Cell Biol. 2017;44:59–67.
- Sosa BA, Kutay U, Schwartz TU. Structural insights into LINC complexes. Curr Opin Struct Biol. 2013;23(2):285–291.
- Chambliss AB, Khatau SB, Erdenberger N, et al. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci Rep. 2013;3(1):1087.
- Versaevel M, Braquenier JB, Riaz M, et al. Super-resolution microscopy reveals LINC complex recruitment at nuclear indentation sites. Sci Rep. 2014;4:7362.
- Luxton GW, Gomes ER, Folker ES, et al. Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science. 2010;329(5994):956–959.
- Khatau SB, Hale CM, Stewart-Hutchinson PJ, et al. A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci U S A. 2009;106(45):19017–19022.
- Kim DH, Chambliss AB, Wirtz D. The multi-faceted role of the actin cap in cellular mechanosensation and mechanotransduction. Soft Matter. 2013;9:5516–5523.
- Tajik A, Zhang Y, Wei F, et al. Transcription upregulation via force-induced direct stretching of chromatin. Nat Mater. 2016;15(12):1287–1296.
- Elosegui-Artola A, Andreu I, Beedle AEM, et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell. 2017;171:1397–410 e14.
- Guilluy C, Osborne LD, Van Landeghem L, et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol. 2014;16(4):376–381.
- Le HQ, Ghatak S, Yeung CY, et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat Cell Biol. 2016;18:864–875.
- Hatch EM, Hetzer MW. Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol. 2016;215(1):27–36.
- Zhang Q, Tamashunas AC, Agrawal A, et al. Local, transient tensile stress on the nuclear membrane causes membrane rupture. Mol Biol Cell. 2019;30(7):899–906.
- Harada T, Swift J, Irianto J, et al. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J Cell Biol. 2014;204:669–682.
- Rowat AC, Jaalouk DE, Zwerger M, et al. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J Biol Chem. 2013;288(12):8610–8618.
- Wiggan O, Schroder B, Krapf D, et al. Cofilin regulates nuclear architecture through a myosin-II dependent mechanotransduction module. Sci Rep. 2017;7(1):40953.
- Robijns J, Molenberghs F, Sieprath T, et al. In silico synchronization reveals regulators of nuclear ruptures in lamin A/C deficient model cells. Sci Rep. 2016;6(1):30325.
- Thiam HR, Vargas P, Carpi N, et al. Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat Commun. 2016;7:10997.
- HiedaHM. Signal Transduction across the nuclear envelope: role of the LINC complex in bidirectional signaling. Cells. 2019;8 (2): 124.
- Kutscheidt S, Zhu R, Antoku S, et al. FHOD1 interaction with nesprin-2G mediates TAN line formation and nuclear movement. Nat Cell Biol. 2014;16(7):708–715.
- Antoku S, Zhu R, Kutscheidt S, et al. Reinforcing the LINC complex connection to actin filaments: the role of FHOD1 in TAN line formation and nuclear movement. Cell Cycle. 2015;14(14):2200–2205.
- Jayo A, Malboubi M, Antoku S, et al. Fascin regulates nuclear movement and deformation in migrating cells. Dev Cell. 2016;38(4):371–383. DOI:10.1016/j.devcel.2016.07.021.
- Saunders CA, Luxton GW. LINCing defective nuclear-cytoskeletal coupling and DYT1 dystonia. Cell Mol Bioeng. 2016;9(2):207–216.
- Saunders CA, Harris NJ, Willey PT, et al. TorsinA controls TAN line assembly and the retrograde flow of dorsal perinuclear actin cables during rearward nuclear movement. J Cell Biol. 2017;216 (3):657–674.
- Borrego-Pinto J, Jegou T, Osorio DS, et al. Samp1 is a component of TAN lines and is required for nuclear movement. J Cell Sci. 2012;125(5):1099–1105.
- Thakar K, May CK, Rogers A, et al. Opposing roles for distinct LINC complexes in regulation of the small GTPase RhoA. Mol Biol Cell. 2017;28 (1):182–191.
- Woroniuk A, Porter A, White G, et al. STEF/TIAM2-mediated Rac1 activity at the nuclear envelope regulates the perinuclear actin cap. Nat Commun. 2018;9(1):2124. DOI:10.1038/s41467-018-04404-4.
- Kinugasa Y, Hirano Y, Sawai M, et al. The very-long-chain fatty acid elongase Elo2 rescues lethal defects associated with loss of the nuclear barrier function in fission yeast cells. J Cell Sci. 2019;132 (10):jcs229021.
- Lusk CP, Ader NR. CHMPions of repair: emerging perspectives on sensing and repairing the nuclear envelope barrier. Curr Opin Cell Biol. 2020;64:25–33.
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, et al. ESCRT machinery is required for plasma membrane repair. Science. 2014;343(6174):1247136.
- Scheffer LL, Sreetama SC, Sharma N, et al. Mechanism of Ca(2)(+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. 2014;5:5646.
- Olmos Y, Hodgson L, Mantell J, et al. ESCRT-III controls nuclear envelope reformation. Nature. 2015;522(7555):236–239.
- Olmos Y, Perdrix-Rosell A, Carlton JG. Membrane binding by CHMP7 coordinates ESCRT-III-dependent nuclear envelope reformation. Curr Biol. 2016;26(19):2635–2641.
- Young AM, Gunn AL, Hatch EM. BAF facilitates interphase nuclear membrane repair through recruitment of nuclear transmembrane proteins. Mol Biol Cell. 2020;31(15):1551–1560.
- Halfmann CT, Sears RM, Katiyar A, et al. Repair of nuclear ruptures requires barrier-to-autointegration factor. J Cell Biol. 2019;218(7):2136–2149.
- Gorjanacz M, Klerkx EP, Galy V, et al. Caenorhabditis elegans BAF-1 and its kinase VRK-1 participate directly in post-mitotic nuclear envelope assembly. Embo J. 2007;26:132–143.
- Haraguchi T, Koujin T, Segura-Totten M, et al. BAF is required for emerin assembly into the reforming nuclear envelope. J Cell Sci. 2001;114:4575–4585.
- Haraguchi T, Kojidani T, Koujin T, et al. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. J Cell Sci. 2008;121(15):2540–2554.
- Samwer M, Schneider MWG, Hoefler R, Schmalhorst PS, Jude JG, Zuber J, Gerlich DW. DNA Cross-Bridging. Shapes a Single Nucleus from a Set of Mitotic Chromosomes. Cell. 2017;170:956–72 e23.
- Huguet F, Flynn S, Vagnarelli P. The role of phosphatases in nuclear envelope disassembly and reassembly and their relevance to pathologies. Cells. 2019;8(7):687.
- Kleinberger T. Biology of the adenovirus E4orf4 protein: from virus infection to cancer cell death. FEBS Lett. 2020;594(12):1891–1917.
- Lavoie JN, Nguyen M, Marcellus RC, et al. E4orf4, a novel adenovirus death factor that induces p53-independent apoptosis by a pathway that is not inhibited by zVAD-fmk. J Cell Biol. 1998;140(3):637–645.
- Shtrichman R, Sharf R, Barr H, et al. Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc Natl Acad Sci U S A. 1999;96(18):10080–10085.
- Marcellus RC, Lavoie JN, Boivin D, et al. The early region 4 orf4 protein of human adenovirus type 5 induces p53- independent cell death by apoptosis. J Virol. 1998;72(9):7144–7153.
- Lavoie JN, Champagne C, Gingras MC, et al. Adenovirus E4 open reading frame 4-induced apoptosis involves dysregulation of src family kinases. J Cell Biol. 2000;150(5):1037–1056.
- Lavoie JN, Landry MC, Faure RL, et al. Src-family kinase signaling, actin-mediated membrane trafficking and organellar dynamics in the control of cell fate: lessons to be learned from the adenovirus E4orf4 death factor. Cell Signal. 2010;22(11):1604–1614.
- Robert A, Smadja-Lamere N, Landry MC, et al. Adenovirus E4orf4 hijacks rho GTPase-dependent actin dynamics to kill cells: a role for endosome-associated actin assembly. Mol Biol Cell. 2006;17(7):3329–3344.
- Smadja-Lamere N, Boulanger MC, Champagne C, et al. JNK-mediated phosphorylation of paxillin in adhesion assembly and tension-induced cell death by the adenovirus death factor E4orf4. J Biol Chem. 2008;283(49):34352–34364.
- Gingras M-C, Champagne C, Roy M, et al. Cytoplasmic death signal triggered by src-mediated phosphorylation of the Adenovirus E4orf4 protein. Mol Cell Biol. 2002;22(1):41–56.
- Champagne C, Landry MC, Gingras MC, et al. Activation of adenovirus type 2 early region 4 ORF4 cytoplasmic death function by direct binding to Src kinase domain. J Biol Chem. 2004;279(24):25905–25915.
- Landry MC, Sicotte A, Champagne C, et al. Regulation of cell death by recycling endosomes and golgi membrane dynamics via a pathway involving Src-family kinases, Cdc42 and Rab11a. Mol Biol Cell. 2009;20(18):4091–4106.
- Landry MC, Champagne C, Boulanger MC, et al. A functional interplay between the small GTPase Rab11a and mitochondria-shaping proteins regulates mitochondrial positioning and polarization of the actin cytoskeleton downstream of src family kinases. J Biol Chem. 2014;289:2230–2249.
- Dziengelewski C, Rodrigue MA, Caillier A, et al. Adenoviral protein E4orf4 interacts with the polarity protein Par3 to induce nuclear rupture and tumor cell death. J Cell Biol. 2020;219(4):e201805122.
- Shtrichman R, Sharf R, Barr H, et al. Induction of apoptosis by adenovirus E4orf4 protein is specific to transformed cells and requires an interaction with protein phosphatase 2A. Proc Natl Acad Sci U S A. 1999;96(18):10080–10085.
- Lele TP, Dickinson RB, Gundersen GG. Mechanical principles of nuclear shaping and positioning. J Cell Biol. 2018;217(10):3330–3342.
- Irianto J, Xia Y, Pfeifer CR, et al. DNA damage follows repair factor depletion and portends genome variation in cancer cells after pore migration. Curr Biol. 2017;27(2):210–223.
- Rosen H, Sharf R, Pechkovsky A, et al. Selective elimination of cancer cells by the adenovirus E4orf4 protein in a Drosophila cancer model: a new paradigm for cancer therapy. Cell Death Dis. 2019;10(6):455.
- Mui MZ, Zhou Y, Blanchette P, et al. The Human Adenovirus Type 5 E4orf4 Protein Targets Two Phosphatase Regulators of the Hippo Signaling Pathway. J Virol. 2015;89(17):8855–8870.
- Lu Y, Kucharski TJ, Gamache I, et al. Interaction of adenovirus type 5 E4orf4 with the nuclear pore subunit Nup205 is required for proper viral gene expression. J Virol. 2014;88(22):13249–13259.
- Ferrigno P, Langan TA, Cohen P. Protein phosphatase 2A1 is the major enzyme in vertebrate cell extracts that dephosphorylates several physiological substrates for cyclin-dependent protein kinases. Mol Biol Cell. 1993;4(7):669–677.
- Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33(5):537–545.
- Lang CF, Munro E. The PAR proteins: from molecular circuits to dynamic self-stabilizing cell polarity. Development. 2017;144(19):3405–3416.
- Goldstein B, Macara IG. The PAR proteins: fundamental players in animal cell polarization. Dev Cell. 2007;13(5):609–622.
- Nance J, Zallen JA. Elaborating polarity: PAR proteins and the cytoskeleton. Development. 2011;138(5):799–809.
- Mack NA, Georgiou M. The interdependence of the Rho GTPases and apicobasal cell polarity. Small GTPases. 2014;5(2):10.
- Munro E. Protein Clustering Shapes Polarity Protein Gradients. Dev Cell. 2017;42(4):309–311.
- Grassie ME, Moffat LD, Walsh MP, et al. The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch Biochem Biophys. 2011;510:147–159.
- Kiss A, Erdodi F, Lontay B. Myosin phosphatase: unexpected functions of a long-known enzyme. Biochim Biophys Acta Mol Cell Res. 2019;1866(1):2–15.
- Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–287.
- Kiss A, Lontay B, Becsi B, et al. Myosin phosphatase interacts with and dephosphorylates the retinoblastoma protein in THP-1 leukemic cells: its inhibition is involved in the attenuation of daunorubicin-induced cell death by calyculin-A. Cell Signal. 2008;20:2059–2070.
- Bergamaschi D, Samuels Y, Jin B, et al. ASPP1 and ASPP2: common activators of p53 family members. Mol Cell Biol. 2004;24(3):1341–1350.
- Liu CY, Lv X, Li T, et al. PP1 cooperates with ASPP2 to dephosphorylate and activate TAZ. J Biol Chem. 2011;286:5558–5566.
- Royer C, Koch S, Qin X, et al. ASPP2 links the apical lateral polarity complex to the regulation of YAP activity in epithelial cells. PloS One. 2014;9:e111384.
- Wang W, Li X, Huang J, et al. Defining the protein-protein interaction network of the human hippo pathway. Mol Cell Proteomics. 2014;13(1):119–131.
- Moya IM, Halder G. Discovering the Hippo pathway protein-protein interactome. Cell Res. 2014;24(2):137–138.
- Zheng X, Dong Q, Zhang X, et al. The coiled-coil domain of oncogene RASSF 7 inhibits hippo signaling and promotes non-small cell lung cancer. Oncotarget. 2017;8:78734–78748.
- Couzens AL, Knight JD, Kean MJ, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013;6:rs15.
- Cong W, Hirose T, Harita Y, et al. ASPP2 regulates epithelial cell polarity through the PAR complex. Curr Biol. 2010;20(15):1408–1414.
- Sottocornola R, Royer C, Vives V, et al. ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev Cell. 2010;19(1):126–137. DOI:10.1016/j.devcel.2010.06.003.
- Langton PF, Colombani J, Chan EH, et al. The dASPP-dRASSF8 complex regulates cell-cell adhesion during Drosophila retinal morphogenesis. Curr Biol. 2009;19(23):1969–1978.
- Wang Y, Bu F, Royer C, et al. ASPP2 controls epithelial plasticity and inhibits metastasis through beta-catenin-dependent regulation of ZEB1. Nat Cell Biol. 2014;16(11):1092–1104.
- Bertran MT, Mouilleron S, Zhou Y, et al. ASPP proteins discriminate between PP1 catalytic subunits through their SH3 domain and the PP1 C-tail. Nat Commun. 2019;10(1):771.
- Kleinberger T, Shenk T. Adenovirus E4orf4 protein binds to protein phosphatase 2A, and the complex down regulates E1A-enhanced junB transcription. J Virol. 1993;67(12):7556–7560.
- Marcellus RC, Chan H, Paquette D, et al. Induction of p53-independent apoptosis by the adenovirus E4orf4 protein requires binding to the Balpha subunit of protein phosphatase 2A. J Virol. 2000;74(17):7869–7877.
- Roopchand DE, Lee JM, Shahinian S, et al. Toxicity of human adenovirus E4orf4 protein in Saccharomyces cerevisiae results from interactions with the Cdc55 regulatory B subunit of PP2A. Oncogene. 2001;20(38):5279–5290.
- Kanopka A, Muhlemann O, Petersen-Mahrt S, et al. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature. 1998;393(6681):185–187.
- Estmer Nilsson C, Petersen-Mahrt S, Durot C, et al. The adenovirus E4-ORF4 splicing enhancer protein interacts with a subset of phosphorylated SR proteins. EMBO J. 2001;20(4):864–871.
- Schuhmacher D, Sontag JM, Sontag E. Protein Phosphatase 2A: more Than a Passenger in the Regulation of Epithelial Cell-Cell Junctions. Front Cell Dev Biol. 2019;7:30.
- Schmitz MH, Held M, Janssens V, et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol. 2010;12(9):886–893.
- Snyers L, Erhart R, Laffer S, et al. LEM4/ANKLE-2 deficiency impairs post-mitotic re-localization of BAF, LAP2alpha and LaminA to the nucleus, causes nuclear envelope instability in telophase and leads to hyperploidy in HeLa cells. Eur J Cell Biol. 2018;97:63–74.
- Mehsen H, Boudreau V, Garrido D, et al. PP2A-B55 promotes nuclear envelope reformation after mitosis in Drosophila. J Cell Biol. 2018;217(12):4106–4123.
- Asencio C, Davidson IF, Santarella-Mellwig R, et al. Coordination of kinase and phosphatase activities by Lem4 enables nuclear envelope reassembly during mitosis. Cell. 2012;150(1):122–135.
- Mazumder A, Roopa T, Basu A, et al. Dynamics of chromatin decondensation reveals the structural integrity of a mechanically prestressed nucleus. Biophys J. 2008;95(6):3028–3035.
- Krause M, Yang FW, Te Lindert M, et al. Cell migration through three-dimensional confining pores: speed accelerations by deformation and recoil of the nucleus. Philos Trans R Soc London, Ser B. 2019;374:20180225.
- Brestovitsky A, Sharf R, Mittelman K, et al. The adenovirus E4orf4 protein targets PP2A to the ACF chromatin-remodeling factor and induces cell death through regulation of SNF2h-containing complexes. Nucleic Acids Res. 2011;39(15):6414–6427.
- Fyodorov DV, Blower MD, Karpen GH, et al. Acf1 confers unique activities to ACF/CHRAC and promotes the formation rather than disruption of chromatin in vivo. Genes Dev. 2004;18(2):170–183.
- Machida S, Takizawa Y, Ishimaru M, et al. Structural Basis of Heterochromatin Formation by Human HP1. Mol Cell. 2018;69(3):385–397 e8.
- Bertacchini J, Beretti F, Cenni V, et al. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 2013;27(6):2145–2155.
- Loie E, Charrier LE, Sollier K, et al. CRB3A Controls the morphology and cohesion of cancer cells through Ehm2/p114RhoGEF-dependent signaling. Mol Cell Biol. 2015;35(19):3423–3435.
- Hirano M, Hashimoto S, Yonemura S, et al. EPB41L5 functions to post-transcriptionally regulate cadherin and integrin during epithelial-mesenchymal transition. J Cell Biol. 2008;182(6):1217–1230.
- Fabregat A, Sidiropoulos K, Viteri G, et al. Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics. 2017;18(1):142.
- Grallert A, Boke E, Hagting A, et al. A PP1-PP2A phosphatase relay controls mitotic progression. Nature. 2015;517:94–98.
- Jamin A, Wiebe MS. Barrier to Autointegration Factor (BANF1): interwoven roles in nuclear structure, genome integrity, innate immunity, stress responses and progeria. Curr Opin Cell Biol. 2015;34:61–68.
- Luxton GW, Gomes ER, Folker ES, et al. TAN lines: a novel nuclear envelope structure involved in nuclear positioning. Nucleus. 2011;2(3):173–181.
- Kim DH, Khatau SB, Feng Y, et al. Actin cap associated focal adhesions and their distinct role in cellular mechanosensing. Sci Rep. 2012;2:555.
- Choi H, Larsen B, Lin ZY, et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods. 2011;8:70–73.
- Choi H, Liu G, Mellacheruvu D, et al. Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr Protoc Bioinformatics. 2012;Chapter 8: Unit8.15.
- The UniProt C. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D69.
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504.
- Jassal B, Matthews L, Viteri G, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503.