
ABSTRACT
During muscle regeneration, extracellular signal-regulated kinase (ERK) promotes both proliferation and migration. However, the relationship between proliferation and migration is poorly understood in this context. To elucidate this complex relationship on a physiological level, we established an intravital imaging system for measuring ERK activity, migration speed, and cell-cycle phases in mouse muscle satellite cell-derived myogenic cells. We found that in vivo, ERK is maximally activated in myogenic cells two days after injury, and this is then followed by increases in cell number and motility. With limited effects of ERK activity on migration on an acute timescale, we hypothesized that ERK increases migration speed in the later phase by promoting cell-cycle progression. Our cell-cycle analysis further revealed that in myogenic cells, ERK activity is critical for G1/S transition, and cells migrate more rapidly in S/G2 phase 3 days after injury. Finally, migration speed of myogenic cells was suppressed after CDK1/2—but not CDK1—inhibitor treatment, demonstrating a critical role of CDK2 in myogenic cell migration. Overall, our study demonstrates that in myogenic cells, the ERK-CDK2 axis promotes not only G1/S transition but also migration, thus providing a novel mechanism for efficient muscle regeneration.
Introduction
To efficiently regenerate skeletal muscles, the right cells need to be at the right place at the right time. This coordinated process is dependent on muscle stem cells, or so-called satellite cells, that remain quiescent in uninjured muscles [Citation1–3]. Upon injury, activated satellite cells proliferate and differentiate into myoblasts. Myoblasts proliferate, migrate to the site of injury, and then differentiate into myofibers, completing the regeneration process. A subpopulation of satellite cells undergoes self-renewal to restore the pool of quiescent satellite cells. Recent studies have indicated that satellite cell dysfunction can contribute to age-associated muscle diseases and influence genetic disorders such as Duchenne muscular dystrophy (DMD) [Citation4–6].
Several myogenic transcription factors are sequentially activated to restore muscle structure and function after injury. Satellite cells express the transcription factor paired box 7 (PAX7), which is essential for satellite cell survival and muscle regeneration [Citation7–9]. Satellite cell activation is characterized by the expression of myogenic determination protein (MYOD) and myogenic factor 5 (MYF5). The differentiation of myoblasts involves the downregulation of PAX7 and the expression of myogenin (MYOG) [Citation1,Citation2].
The relationship between proliferation and migration is complex and context-dependent, and has been mostly studied in tumorigenesis and development. Historically, cancer cell proliferation and migration were considered to be mutually exclusive in time and space (i.e., the “go or grow” hypothesis) [Citation10–12]. This hypothesis is corroborated by reports showing that tumor cells in G0/G1 phase are more mobile than those in S/G2/M phase [Citation13,Citation14]. However, there are several lines of evidence to the contrary. For example, others have reported that tumor cells migrate faster in S/G2/M phase than in G0/G1 phase [Citation15,Citation16]. Furthermore, in development, neural crest cells in fish and avian embryo migrate faster in S phase [Citation17,Citation18]. Similarly, during mouse cerebral cortex development, migrating nuclei of neural progenitors in the ventricular zone show greater motility in S/G2/M phase than in G1 [Citation19]. It is thus likely that the relationship between proliferation and migration depends on the cells, tissues, and the surrounding environment, and much remains unknown in other physiological context such as muscle regeneration.
Extracellular signal-regulated kinase (ERK) signaling pathway has been suggested to play crucial roles in muscle regeneration. Previous studies have shown that ERK1/2 promotes myoblast proliferation and migration in vitro [Citation20,Citation21]. In addition, ERK1/2 has also been reported to be important for muscle differentiation in vitro [Citation22–24] and in vivo [Citation25]. Erk1-/- mutant mice have 40% less quiescent satellite cells compared to control [Citation26], further emphasizing the importance of ERK signaling in satellite cells. In many of these reports, fibroblast growth factor (FGF) acts upstream of the ERK signaling pathway. The significance of FGF is highlighted by a muscle regeneration defect in FGF6-/- mutant mice [Citation27], severe muscular dystrophy in FGF2-/-/FGF6-/-/mdx mutant mice [Citation28], and enhanced wound repair by the delivery of FGF2 [Citation29]. However, the contribution of ERK activity to muscle regeneration remains to be elucidated with spatio-temporal resolution in vivo.
Intravital imaging by two-photon microscopy is becoming a powerful technique to study the complexity of biological events in living tissues including skeletal muscle [Citation30,Citation31]. For example, Webster et al. developed an intravital imaging technique to observe cells labeled with Pax7-CreERT2 in living mice [Citation32]. They demonstrated that extracellular matrix (ECM) remnants guide the direction of migration and orient the division plane. Another intravital two-photon microscopy technique developed by Mercier et al. revealed that single fibers contraction occurs spontaneously and independently of neighboring fibers within the same muscle [Citation33]. More recently, Hotta et al. characterized alterations in microvascular permeability following eccentric contraction-induced skeletal muscle injury [Citation34]. Thus, intravital imaging has provided novel insights into fundamental processes such as cell division, cell migration, myofiber contraction, and vascular permeability at a level of detail previously inaccessible to researchers.
To further understand the role of ERK signaling and how cell migration is affected by cell-cycle modulations during muscle regeneration in vivo, we established an intravital imaging technique to observe live mouse muscle regeneration. We incorporated in this imaging system a Förster/fluorescence resonance energy transfer (FRET) biosensor that measures ERK activity and a fluorescent reporter that indicates cell cycle phase. With this intravital imaging platform, we found that ERK promotes G1/S phase transition and that satellite cell-derived myogenic cells migrate faster in S/G2 phase. Moreover, our data suggest that CDK2 is responsible for increasing migration speed of myogenic cells. In summary, our study establishes that myogenic cell migration is cell cycle-dependent in vivo and may provide a novel mechanism of efficient tissue regeneration.
Results
ERK is activated in myogenic cells during muscle regeneration
Satellite cell-derived myogenic cell proliferation and migration have been reported to be essential for muscle regeneration. To investigate the relationship between myogenic cell proliferation and migration, we focused on ERK, which has been reported to promote both myoblast proliferation [Citation20] and migration [Citation21] in vitro. To study ERK activity in living tissues, we used a previously developed R26R-EKAREV mice strain that ubiquitously expresses a floxed FRET biosensor for monitoring ERK activity, EKAREV [Citation35]. We crossed the R26R-EKAREV mice with Pax7-CreERT2 mice [Citation36] to generate R26R-EKAREV/Pax7-CreERT2 mice ()). Skeletal muscle damage was induced by cardiotoxin injection, and then live imaged under an upright microscope via imaging window (,c)) [Citation37]. After Cre-mediated recombination induced by tamoxifen, R26R-EKAREV/Pax7-CreERT2 mice express a FRET biosensor for ERK, in the nuclei of Pax7 lineage cells, hereafter referred to as “myogenic cells” (green cells and pseudo-colored cells in )). Cells that were not recombined, i.e. myofibers, expressed a large Stokes shift fluorescent protein, tdKeima. We confirmed that tdKeima was expressed ubiquitously in muscle fibers before injury (magenta cells in )). Marked reduction in the number of tdKeima-expressing cells was observed between 0 and 2 days post-injury (dpi) (Figure 1(d,e)). The nuclear density of myogenic cells was measured from the z-stack images of skeletal muscle, and assessed by a multiple contrast method, Scheffe’s F-test. The nuclear density was increased by 3.2 fold from 2 to 3 dpi, indicating the proliferation of myogenic cells.
Figure 1. ERK is activated in myogenic cells during muscle regeneration. (a) Scheme of R26R-EKAREV/Pax7-CreERT2 mice. (b) Experimental scheme of Cre-mediated recombination and in vivo imaging of skeletal muscle regeneration. (c) Layout for the in vivo imaging system. The muscle under the imaging window was observed with a two-photon microscope repetitively. (d) Representative images of regenerating muscles expressing EKAREV and tdKeima at 0, 1, 2, 3, and 5 days post-injury (dpi). The nuclei of Pax7 lineage cells (satellite cell-derived myogenic cells) express EKAREV are shown in green, while the myofibers that existed before injury express tdKeima and are shown in magenta (top panels). Note that the nuclei of Pax7 lineage cells include the myonuclei of myotubes or myofibers at 5 dpi. Biceps femoris muscles were imaged at indicated time points; maximum intensity projection images of 30 µm z-stack with 2 µm intervals are shown. EKAREV-NLS was used to monitor ERK activity in the nucleus. ERK activity (FRET/CFP) images of myogenic cells shown in the intensity-modulated display (IMD) mode (bottom panels). (e) Averaged nuclear density of myogenic cells calculated from the z-stack images (bars, SDs; N = 3 mice for each day; *p < 0.05). (f and g) Bee swarm plot (f) and histogram (g) of ERK activity (FRET/CFP) in myogenic cells. Each color represents data from a single mouse in (f) (bars, averages; N = 3 mice for each day; ***p < 0.001)
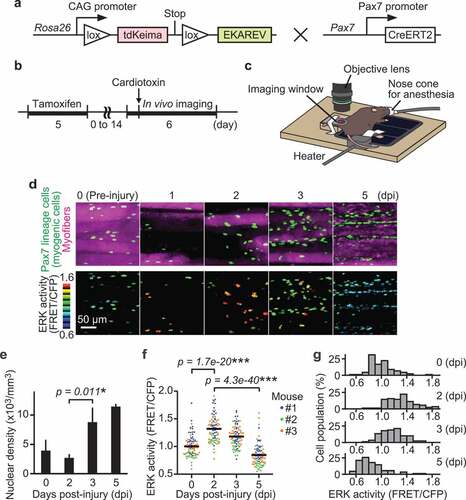
During muscle regeneration, ERK activity (FRET/CFP) in myogenic cells was maximally increased at 2 dpi and decreased below the basal level at 5 dpi (Figure 1(d,f,g)). The nuclei of Pax7 lineage cells include the myonuclei of myotubes or myofibers at 5 dpi. Statistically significant differences were found among every pair of days. We confirmed that in vitro ERK activity (FRET/CFP) is positively correlated with phospho-ERK/total-ERK, but not with total-ERK levels, supporting that ERK activation during muscle regeneration is not dependent on altered expression of total-ERK (Figure S1(a,b)). We also examined whether cells with high ERK activity at 3 dpi were undergoing apoptosis and found that only a small fraction of cells was TUNEL-positive at 3 and 5 dpi, negating the possibility (Figure S2(a,b)). Collectively, our results indicate that ERK activation precedes proliferation in myogenic cells.
Instantaneous ERK activity regulates migration in some but not all myogenic cells
Since ERK activity regulates both cell migration and proliferation [Citation20,Citation21], we first tested the relationship between ERK activity and cell migration speed ()). The migration speed of myogenic cells was calculated by dividing the displacement of nuclear centroids by elapsed time. The migration speed was significantly and maximally increased at 3 dpi and decreased at 5 dpi ()). Because ERK activity was already increased at 2 dpi ()), this observation indicates that ERK activation precedes the increase in migration speed as well as proliferation, in myogenic cells during muscle regeneration.
Figure 2. Instantaneous ERK activity regulates migration in some but not all myogenic cells. (a) Representative time-lapse images of myogenic cells (white dots) and their cell tracks (pseudo-colored lines) (left). FRET/CFP ratio images of myogenic cells (IMD mode dots) and their cell tracks (white lines) (right). (b) Migration speed of myogenic cells, which was calculated from the displacement of EKAREV-NLS centroids and divided by the time. Data for cells with more than one hour of tracking is shown. Each color represents data from a single mouse (bars, averages; N = 3 mice for each day; ***p < 0.001). (c) Representative cell tracks for 2 hours. X-axis corresponds to the long axis of myofibers. (d) Scatter plot of normalized ERK activity (FRET/CFP) against migration speed in migrating myogenic cells. Myogenic cells with a speed of more than 7 µm/hr were defined as “migrating” and taken into account. ERK activity was normalized by the averaged ERK activity of each mouse. Each color represents data from a single mouse (N = 3 mice). (e) Representative images of myogenic cells (white dots) and their cell tracks (two-colored lines). Yellow lines indicate cell tracks during the first two hours. Blue lines indicate cell tracks during the latter two hours after treatment with DMSO (1 mL/kg) or a MEK inhibitor (PD0325901, 5 mg/kg). (f) The difference in migration speed and ERK activity in myogenic cells, calculated by subtracting values before MEKi treatment from values after MEKi treatment. Gray dashed lines indicate the median of ERK activity and migration speed in DMSO group. Percentages of each cell groups after MEKi treatment are indicated in the scatter plot. Histograms of the difference in migration speed and ERK activity are shown at the top and right side of the figure, respectively (N = 4 mice for DMSO group; N = 3 mice for MEKi group)
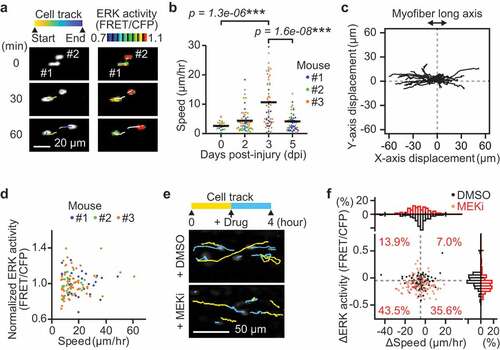
Interestingly, we found that myogenic cells migrate predominantly along the long axis of myofibers ()), consistent with the finding that extracellular matrix of the basal laminae around myofibers serves as a guide for myogenic cells to migrate [Citation32]. Moreover, the direction of myogenic cell migration was not biased toward either of the ends along the long axis of myofibers ()). This result suggests that, at least in muscle regeneration at 3 dpi, myogenic cell migration is governed by random walk rather than by chemotaxis.
Although ERK has been reported to promote myoblast migration in vitro [Citation21], whether ERK activity is required for myogenic cell migration has not been demonstrated in vivo. Therefore, we examined the relationship between ERK activity and speed in myogenic cells. Unexpectedly, we failed to observe a strong correlation between migration speed and ERK activity at 3 dpi (), only myogenic cells with a speed of more than 7 µm/hr were defined as “migrating” and analyzed). This suggests that migration speed of myogenic cells is not determined by instantaneous ERK activity on an acute timescale. We further assessed the contribution of instantaneous ERK activity to myogenic cell migration by acutely inhibiting MEK, a kinase of ERK, at 3 dpi (,f)). A MEK inhibitor treatment only moderately decreased the speed in migrating myogenic cells (top histogram, )). Some myogenic cells (43.5%) decreased in migration speed and ERK activity (bottom left cell population in scatter plot, )). However, it is important to note that many other migrating myogenic cells (35.6%) did not alter their speed after MEK inhibitor treatment, even though ERK activity was significantly decreased (bottom right cell population in scatter plot, )). These results indicate that instantaneous ERK activity may regulate migration speed in some but not all myogenic cells.
ERK activation is required for G1/S transition in myogenic cells
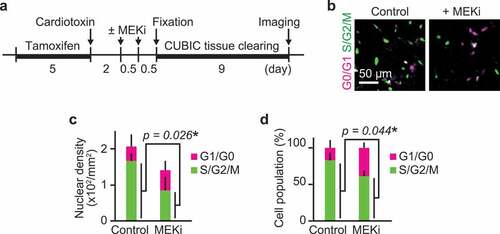
Due to the lack of correlation in instantaneous ERK activity and migration speed, we speculated that ERK promotes cell migration through its transcriptional targets (). This is consistent with our observation that there was a one-day gap between the peak of ERK activity and the peak of cell migration speed (). We thus focused on cell-cycle progression, a key long-term process that is linked to ERK-mediated transcription. First, to clarify the role of ERK in cell-cycle progression in vivo, we inhibited ERK activity in R26Fucci2aR/Pax7-CreERT2 mice that expressed a cell-cycle indicator, Fucci, in Pax7 lineage cells. Fucci2a is composed of two chimeric proteins, mCherry-hCdt1 and mVenus-hGeminin, which accumulate reciprocally in the nucleus of the cells during the cell cycle, labeling the nuclei of G0/G1 phase cells with mCherry and those of S/G2/M phase cells with mVenus. The proportion of cells expressing mCherry-hCdt1 and cells expressing mVenus-hGeminin was analyzed after ERK activity was suppressed by a MEK inhibitor, PD0325901 ()). Fixed muscle was cleared by CUBIC reagents to obtain the broad cross-sectional area of the tissue. MEK inhibitor treatment at 2 and 2.5 dpi decreased cell density as well as the percentage of cells expressing mVenus-hGeminin at 3 dpi (–d)), suggesting that myogenic cells were arrested at G1/S boundary. This result indicates that ERK activation is required for G1/S transition in vivo during muscle regeneration.
Figure 3. ERK activation is required for G1/S transition in myogenic cells. (a) Experimental scheme of cre-mediated recombination and tissue clearing. Mice were injected with a MEK inhibitor (PD0325901, 5 mg/kg) at 2 and 2.5 dpi. MEK inhibitor-injected mice and uninjected controls were fixed at 3 dpi. (b–d) Representative images (b), averaged nuclear density (c), percentage (d) of myogenic cells expressing Fucci in mouse muscle regenerating regions. Magenta and green colors represent cells in G0/G1 and S/G2/M phase, respectively. All data above were obtained from the same experiment described in (a) (bars, SDs; N = 3 mice for each group; *p < 0.05)
Migration speed of myogenic cells increases in S/G2 phase
From these data, we hypothesized that ERK promotes G1/S transition, which precedes the peak of cell migration speed. To further investigate the relationship between cell cycle and migration in myogenic cells, progression of cell cycle phase during muscle regeneration was examined using R26Fucci2aR/Pax7-CreERT2 mice. Again, Fucci2aR was expressed in satellite cells by tamoxifen injection. Then, skeletal muscle damage was induced by cardiotoxin injection. At 0 dpi, almost all of the cells were mCherry positive (i.e., in G0 phase) (,b)). The cells expressing mVenus-hGeminin increased at 2 to 3 dpi and decreased at 5 dpi. These data indicate myogenic cells mainly divide from 1 to 4 dpi.
Figure 4. Migration speed of myogenic cells increases in S/G2 phase. (a) Representative images of myogenic cells expressing Fucci at 0, 2, 3, and 5 dpi. Biceps femoris muscles were imaged as indicated time points, and shown in maximum intensity projection images of 100 µm z-stack with 2 µm intervals. Magenta and green dots indicate cells in G0/G1 phase and those in S/G2/M phase, respectively. (b) Averaged nuclear densities of myogenic cells expressing Fucci calculated from the z-stack images (bars, SDs; N = 3 mice for each day; ***p < 0.001). (c) Representative images of cell trajectories for 4 hours at 3 dpi. Magenta and green lines indicate the trajectories of cells in G0/G1 phase and those in S/G2/M phase, respectively. (d) Migration speed of myogenic cells expressing Fucci during each cell cycle phase at 3 dpi. Cells in the G0 and G1 phase were determined by the intensity of mCherry-hCdt1 (Figure S3(a,b)). Cells in the M phase was discriminated from cells in S/G2 phase by cytosolic distribution and subsequent disappearance of mVenus-hGeminin (Figure S3(c)). Each color represents data from a single mouse (bars, averages; N = 3 mice for each day; ***p < 0.001)
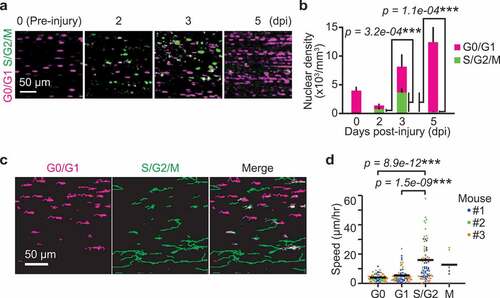
Next, we asked whether the migration speed varies depending on cell cycle phase. For this purpose, we focused on 3 dpi because myogenic cells are both in G1/S and G2 phases ()). We tracked mCherry-positive or mVenus-positive cells for 4 hours ()). Time-lapse imaging of the skeletal muscle of R26Fucci2aR/Pax7-CreERT2 mice revealed that S/G2/M cells expressing the mVenus-hGeminin migrate faster than G0/G1 cells expressing mCherry-hCdt1. The cells were classified into G0, G1, S/G2 and M phases to further examine whether migration speed is dependent on cell cycle phase. Cells that do not express Ki67 (and thus identified as G0 cells) have been previously reported to express higher levels of mKO2-hCdt1 [Citation38]. Thus, we classified cells expressing higher and lower levels of mCherry-hCdt1 as cells in G0 and G1 phase, respectively (Figure S3(a,b)). Cells in the M phase were discriminated from cells in S/G2 phase by nuclear membrane breakdown and subsequent disappearance of mVenus-hGeminin (Figure S3(c)). With these analyses, we found that the migration speed of myogenic cells was fastest in S/G2 phase and slower in M phase and G1 phase, while G0 phase cells showed the slowest migration speed ()).
CDK2 promotes myogenic cell migration during muscle regeneration
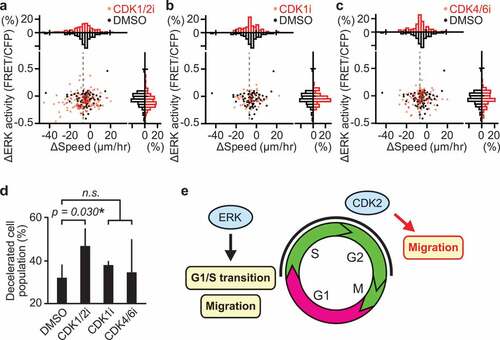
These results motivated us to search for a mechanism underlying cell cycle-dependent migration. To this end, we examined the contribution of CDKs, whose activities are tightly controlled according to cell cycle progression [Citation39]. To test the hypothesis that a downstream substrate of CDK directly regulates myogenic cell migration, CDK inhibitors were injected in mice during in vivo imaging at 3 dpi. The change in migration speed of each cell following inhibitor injection was plotted against the corresponding change in ERK activity (–c)). To clarify the effects of CDK inhibitors on cell migration, we focused on the migrating myogenic cells that decreased their speed more than 7 µm/hr after the inhibitor treatment. This population of cells was defined as “decelerated”. Gray dashed lines denote the threshold for classifying cells as “decelerated” (–c)). Interestingly, a CDK1/2 inhibitor, roscovitine, but not a CDK1 or CDK4/6 inhibitor increased the number of decelerated cells ()). From these results, we speculated that CDK2 promotes myogenic cell migration. CDK1 is not likely to contribute myogenic cell migration because it is most activated in M phase, when migration speed is lower than in S/G2 phase ()). Given that roscovitine is a kinase inhibitor, this result implies that phosphorylation of a CDK2 downstream substrate promotes myogenic cell migration in S/G2 phase during muscle regeneration.
Figure 5. CDK2 promotes myogenic cell migration during muscle regeneration. (a–c) Changes in migration speed and in ERK activity in myogenic cells, calculated by subtracting values before the drug treatment from values after the drug treatment. Gray dashed lines indicate -7 µm/hr, which was chosen as the threshold for classifying cells as decelerated. Histograms of the difference in migration speed and ERK activity are shown at the top and right side of the figure, respectively (N = 4 mice for DMSO group; N = 4 mice for CDK1/2i group; N = 3 mice for CDK1i group; N = 3 mice for CDK4/6i group). Mice expressing EKAREV-NLS in myogenic cells were injected intravenously with DMSO (1 mL/kg), CDK1/2 inhibitor (roscovitine, 0.05 mg/kg), CDK1 inhibitor (RO-3306, 1 mg/kg), or CDK4/6 inhibitor (palbociclib, 1 mg/kg) during in vivo imaging at 3 dpi. (d) Percentage of cells classified as decelerated after DMSO or CDK inhibitors. Migrating myogenic cells that decreased their speed more than 7 µm/hr are defined as “decelerated” and taken into account (bars, SDs; *p < 0.05; n.s., not significant). (e) Schematic model of cell cycle progression and cell migration by ERK and CDK2 in myogenic cells during muscle regeneration
Discussion
Despite a number of studies demonstrating that ERK is crucial to myogenic cell proliferation and migration in vitro [Citation20–24] and in vivo [Citation25,Citation26], the spatio-temporal dynamics of ERK activity in muscle regeneration in vivo have hardly been explored. Here we propose two phases of ERK action in myogenic cells during regeneration: in the early phase, ERK helps drive entry into the cell cycle in satellite cells and promotes cell migration; in the later phase, ERK promotes G1/S transition and cell migration through CDK2 activation ()).
We demonstrate, for the first time, that ERK is activated upon entry into the cell cycle in satellite cells during muscle regeneration in vivo. Our data suggest that ERK activation precedes other regulators of muscle regeneration. ERK activation peaks at 2 dpi, while the myogenic master transcription factor MyoD expression peaks at 3 to 4 dpi [Citation40] and p38, which is another mitogen-activated protein kinase, peaks at 7 to 14 dpi [Citation41]. This supports the notion that ERK is activated early in muscle regeneration, when satellite cells exit from quiescence in response to injury.
The varied effects of ERK on cell migration among myogenic cells in vivo could be caused by the difference in focal adhesion signaling. Pro-migratory functions of ERK and the responsible substrates have been characterized in numerous cell types. Among the identified substrates, two focal adhesion–associated proteins, FAK and paxillin are most likely to be involved in ERK-induced cell migration in myogenic cells. ERK is suggested to interact with FAK/paxillin and promote cell migration by enhancing focal adhesion turnover and membrane protrusion at the front of the cells [Citation42–46]. Moreover, previous in vivo work has demonstrated that targeted deletion of FAK in myogenic cells impairs skeletal muscle regeneration [Citation47] and that paxillin is hyper-phosphorylared in dystrophin-deficient mdx muscle [Citation48]. Therefore, differences in abundance of FAK and paxillin could explain the observed variation in ERK contribution to cell migration among myogenic cells.
Multiple lines of evidence support a pro-migratory role of CDK2 though stathmin, a phosphorylation-regulated tubulin-binding protein. Stathmin has been shown to be phosphorylated by CDK2 at Ser25, which is located within the consensus CDK/MAPK phosphorylation motif PXS*P [Citation49]. In agreement with our model, several papers have demonstrated that p27, the cyclin-dependent kinase inhibitor, inhibits cell migration by interacting with CDK2 and stathmin [Citation50–53]. Furthermore, p27 knockout mice showed increased body weight along with muscle weight [Citation54], whereas stathmin knockout mice developed age-dependent myopathy [Citation55]. Interestingly, the expression of stathmin has been suggested to increase as myoblasts undergo differentiation [Citation56–58]. Further study is needed to elucidate the precise mechanisms by which CDK2 and stathmin interact to promote cell migration.
We speculate that the cell cycle-dependent migration in myogenic cells may contribute to efficient regeneration and differentiation, mediated by CDK2, p21, and the myogenic master transcription factor MyoD. Of note, crosstalk between cell-cycle regulators and myogenic regulatory factors has been well characterized in vitro. Expression of MyoD peaks in mid-G1, and is reduced to its minimum level at G1/S transition [Citation59]. In late G1, MyoD is degraded by the ubiquitin proteasome system, which is triggered by cyclin E/CDK2-dependent phosphorylation of MyoD at Ser200 [Citation60–62]. In turn, MyoD inhibits CDK2 activity by inducing expression of the cyclin-dependent kinase inhibitor p21 [Citation63,Citation64]. Although satellite cell-derived myogenic cells need to proliferate and migrate into the site of injury, they also need to stop migrating and differentiate into myotubes, by fusing to each other or to the remaining myofibers. We speculated that such migration control is important especially where cells migrate stochastically along the long axis of myofibers ()). Thus, higher motility of myogenic cells in S/G2 would help to supply myogenic cells at the site of injury, whereas lower motility of myogenic cells in G1 would be beneficial to induce efficient differentiation into myofibers.
We note that the percentage of proliferative myogenic cells at 5 dpi in our study ()) is markedly lower compare to those in the other studies (Table S1) [Citation65–68]. This difference could be attributed to the methods used to identify proliferative cells. We detect the cells in S/G2/M phase by mVenus-tagged Geminin, while the other studies detect the cells in the S phase by EdU or BrdU incorporation. The cells labeled with EdU or BrdU at 4 dpi and measured at 5 dpi will include cells that have already entered into G1 phase. Moreover, the timing of Pax7-Cre activation will also affect the percentage of proliferative cells. We labeled Pax7 lineage cells before injury, whereas Evano et al. labeled them at 4 dpi. Thus, we speculate that the total cell population in our study includes more differentiated cells such as myoblasts, myotubes, and myofibers, which decreased the apparent percentage of mVenus-hGeminin positive cells. Likewise, the total population in our study includes more differentiated cells than studies by Joe et al. and Rocheteau et al., where flow cytometry was used to isolate satellite cells at 5 dpi. The fact that different methods return varying percentages of proliferative myogenic cells may be informative for future research into the control of myogenic cell fate decisions.
In summary, we demonstrated that myogenic cells migrate in a cell cycle-dependent manner and that both ERK and CDK2 contribute to migration during muscle regeneration in vivo, indicating that the ERK-CDK2 axis is crucial to efficient muscle regeneration. These findings highlight the importance of studying fundamental processes such as cell migration and cell-cycle progression—previously characterized in vitro—in living tissue through intravital imaging.
Materials and methods
Reagents and antibodies
PD0325901 (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan), roscovitine (Sigma-Aldrich, St. Louis, MO), RO-3306 (Tokyo Chemical Industry, Tokyo, Japan), and palbociclib (Chemietek, Indianapolis, IN) were applied as inhibitors for MEK, CDK1/2, CDK1, and CDK4/6, respectively. Anti-p44/42 MAPK (Erk1/2) antibody (L34F12) (#4696; Cell Signaling Technology, Danvers, MA) was applied at 1:100. Anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody (D13.14.4E) (#4370; Cell Signaling Technology) was applied at 1:200.
Cell culture
MCF10A cells (CRL-10,317; ATCC, Manassas, VA) were cultured in phenol red-free DMEM/F12 (Thermo Fisher Scientific, Waltham, MA) supplemented with 5% horse serum (ATCC), 20 ng/mL EGF (PeproTech, Rocky Hill, NJ), 10 µg/mL insulin (Sigma-Aldrich), 0.5 µg/mL hydrocortisone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich), 50 U/mL penicillin and 50 µg/mL streptomycin (Thermo Fisher Scientific). Cells were co-transfected with pPBbsr2-EKAREV-NLS and a piggyBac transposase as previously described [Citation69].
Live-cell imaging and immunofluorescence
MCF10A cells were plated onto glass-bottom 96-well plates (Cellvis, Mountain View, CA), which were coated with 30 µg/mL bovine collagen I (Advanced BioMatrix, San Diego, CA) and imaged on an Eclipse Ti2 microscope (Nikon, Tokyo, Japan) in humidified 37°C chambers with 5% CO2. For immunofluorescence, cells were fixed in 4% PFA for 15 minutes at room temperature, washed three times in PBS, and blocked with blocking buffer (10% fetal bovine serum, 1% bovine serum albumin, 0.1% Triton X-100, and 0.01% NaN3 in PBS) for 1 hour. Cells were stained overnight at 4°C with primary antibodies, washed three times in PBS, and stained with Alexa Fluor secondary goat antibodies (Thermo Fisher Scientific, 1:2000) for 1 hour.
Transgenic mice
Gt(ROSA)26Sortm1(CAG-loxP-tdKeima-loxP-EKAREV-NLS) (hereafter called R26R-EKAREV-NLS) mice have been developed previously [Citation35]. This mouse line is designed to express the tdKeima fluorescent protein before Cre-mediated recombination and EKAREV after recombination, under the CAG promoter in the ROSA26 locus. Gt(ROSA)26Sortm1(Fucci2aR)Jkn (hereafter called R26Fucci2aR) mice have been developed previously [Citation70]. B6;129-Pax7tm2.1(cre/ERT2)Fan/J (hereafter called Pax7-CreERT2) mice have been developed previously [Citation36] and were provided by Atsuko Sehara-Fujisawa (Kyoto University, Kyoto, Japan). This mouse line is designed to express a tamoxifen-inducible Cre recombinase–estrogen receptor fusion protein, CreERT2 under the endogenous promoter in the Pax7 locus.
To develop transgenic mice expressing EKAREV-NLS or Fucci in Pax7 lineage cells specifically, R26R-EKAREV-NLS or R26Fucci2aR mice were crossed with Pax7-CreERT2 mice. To induce Cre mediated recombination, tamoxifen (Sigma-Aldrich) dissolved in corn oil (Sigma-Aldrich) were injected into intraperitoneally (75 mg/kg) once a day consecutively for 5 days. Mice were housed in a specific-pathogen-free facility and received a routine chow diet and water ad libitum. Adult female and male mice of 2 to 6 months of age were used for the in vivo imaging. The animal protocols were reviewed and approved by the Animal Care and Use Committee of Kyoto University Graduate School of Medicine (No.14079, 15064, 16038, 17539, and 18086).
Muscle injury with cardiotoxin
To investigate muscle regeneration, muscle damage was induced by cardiotoxin. The skin over the skeletal muscle was shaved and cleaned with 70% ethanol. The skeletal muscle was injected with 10 µL of cardiotoxin (Sigma-Aldrich or Latoxan, Portes lès Valence, France) in DDW (1 mg/mL).
In vivo imaging of skeletal muscle
For repeated observations, custom-made imaging windows were implanted in the femoral region as described previously [Citation37] before cardiotoxin injection. For a single observation, the skin over the tibialis anterior (TA) was shaved and incised to expose approximately 1 cm2 of the TA muscle as described previously [Citation71] after cardiotoxin injection. Mice were anesthetized with 1 to 1.5% isoflurane (FUJIFILM Wako Pure Chemical Corporation) mixed with oxygen delivered at 1 L/min. Drugs were injected intravenously during imaging.
Tissue clearing
For tissue clearing, the TA muscle was collected from mice and fixed in 4% PFA overnight in 4°C. The fixed organs were immersed in CUBIC-1 reagent for 5 days and then further immersed in CUBIC-2 reagent. ScaleCUBIC-1 (reagent-1A) was prepared as a mixture of 10 wt% urea (Nacalai Tesque, Kyoto, Japan), 5 wt% N, N, N’, N’-tetrakis (2-hydroxypropyl) ethylenediamine (Tokyo Chemical Industry), 10 wt% Triton X-100 (Nacalai Tesque), and 25 mM NaCl (Nacalai Tesque). ScaleCUBIC-2 (reagent 2) was prepared as a mixture of 50 wt% sucrose (Nacalai Tesque), 25 wt% urea, 10 wt% 2, 2’, 2”-nitrilotriethanol (FUJIFILM Wako Pure Chemical Corporation), and 0.1% (v/v) Triton X-100 [Citation72].
Muscle fixation and histological analysis
Tibialis anterior muscles were isolated and frozen in liquid nitrogen-cooled isopentane (Nacalai Tesque). Transverse cryosections (16 µm) were fixed with 4% PFA for 15 min at room temperature. TUNEL assays were performed using ApopTag Fluorescein In Situ Apoptosis Detection Kit (Sigma-Aldrich) according to manufacturer’s instructions. Histological sections were counter-stained with 1 µg/mL Hoechst (Thermo Fisher Scientific) for 5 min at room temperature. Images were collected using a Fluoview FV1000/IX83 confocal microscope (Olympus, Tokyo, Japan) equipped with a UPlanSApo 30x/1.05NA silicon oil-immersion objective lens (Olympus).
Two-photon excitation microscopy
For repeated observations, living mice were observed with an FV1200MPE-BX61WI upright microscope (Olympus) equipped with an XLPLN25XWMP water-immersion objective lens (Olympus), where the pixel size was 1.59 um/pixel. For a single observation, living mice were observed with an FV1200MPE-IX83 inverted microscope (Olympus) equipped with a UPlanSApo 30x/1.05NA silicon oil-immersion objective lens (Olympus), where the pixel size was 1.325 um/pixel. The microscopes were equipped with an InSight DeepSee Ultrafast laser (0.95 W at 900 nm) (Spectra Physics, Mountain View, CA). The scan speed was set at 2 to 10 µs/pixel. The excitation wavelength for CFP, GFP, and RFP was 840, 960, and 1040 nm, respectively. Fluorescent images were acquired with the following filters and mirrors: (1) an infrared-cut filter BA685RIF-3 (Olympus), (2) two dichroic mirrors DM505 (Olympus) and DM570 (Olympus), and (3) four emission filters FF01-425/30 for second harmonic generation (SHG) (Semrock, Rochester, NY), BA460-500 for CFP/SHG (Olympus), BA520-560 for FRET/GFP (Olympus), and 645/60 for RFP (Chroma Technology, Bellows Falls, VT). The microscopes were equipped with a two-channel GaAsP detector unit and two multialkali detectors. FLUOVIEW software (Olympus) was used to control the microscope and to acquire images, which were saved in the multilayer 12-bit tagged image file format.
Lightsheet microscopy
Images of cleared tissues were acquired with a Lightsheet Z.1 microscope (Zeiss, Oberkochen, Germany) equipped with a single sided light sheet and two lenses: an EC Plan-Neofluar 5x/0.16 detection objective lens and LSFM clearing 5x/0.1 illumination objective lens. The excitation wavelength for mVenus and mCherry was 488 and 561 nm, respectively. The light sheet thickness was 12.67 µm. A laser blocking filter, LBF 405/488/561/640, secondary beam splitters, SBS LP490 and SBS LP560, and emission filters, BP505-545 and BP575-615, were used. Images were saved in the multilayer 16-bit tagged image file format. ZEN software (Zeiss) was used to control the microscope and to acquire images. Samples were immersed in a 1:1 mixture of silicon oil TSF4300 (Momentive Performance Materials Japan, Tokyo) and mineral oil (Sigma-Aldrich) during image acquisition.
Image processing
Acquired images were processed with ImageJ (National Institutes of Health, Bethesda, MD) and MATLAB software (MathWorks, Natick, MA).
ImageJ software was used to obtain x- and y-coordinates of the nuclei centroid. First, z-stack images were aligned using an ImageJ plug-in “Correct 3D drift” [Citation73]. The CFP or SHG images were used as landmarks for the correction. Corrected z-stack images were processed with a median filter (5x5x5 pixels) and subtracted background noise with a top-hat filter (11 × 11 pixels). Filtered images were maximum intensity projected along the z-axis. The nuclei were tracked with an ImageJ plug-in “Trackmate” [Citation74]. For efficient tracking, CFP images were contrast adjusted using an ImageJ plug-in “Stack Contrast Adjustment” [Citation75]. The parameters in Trackmate were set as follows:
Detector: LoG detector
Estimated blob diameter: 5 pixels
Intensity threshold: 0
Median filter: false
Sub-pixel localization: true
Local maxima: 3 (for SECFP in EKAREV-NLS), 5 (for mVenus in Fucci), and 1 (for mCherry in Fucci)
Tracker: Simple LAP tracker
Linking max distance: 10 pixels
Gap-closing max distance: 10 pixels
Gap-closing max frame: 2
MATLAB standard and custom-written scripts were used to obtain the FRET/CFP ratio and the speed. The FRET/CFP ratio was calculated by dividing the averaged FRET intensity by the averaged CFP in the radial distance of 1-pixel from the centroid. The speed was calculated by dividing the displacement of the centroid by the time.
Statistical analysis
Graphing and statistical analysis was performed with MATLAB software. Statistical differences between two experimental groups were assessed by Student’s two-sample two-sided t-test. Statistical differences among experimental groups more than two were assessed by Scheffe’s F-test. Statistical significances were indicated by asterisks (*p < 0.05; **p < 0.01; ***p < 0.001).
Supplemental Material
Download PDF (4.4 MB)Supplemental Material
Download MS Word (12.4 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Ceafalan LC, Popescu BO, Hinescu ME. Cellular players in skeletal muscle regeneration. Biomed Res Int. 2014;2014(957014):1–21.
- Tedesco FS, Dellavalle A, Diaz-Manera J, et al. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J Clin Invest. 2010;120:11–19.
- Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93:23–67.
- Almada AE, Wagers AJ. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat Rev Mol Cell Biol. 2016;17:267–279.
- Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nat Med. 2015;21:854–862.
- Sousa-Victor P, Garcia-Prat L, Serrano AL, et al. Muscle stem cell aging: regulation and rejuvenation. Trends Endocrinol Metab. 2015;26;287–296.
- Kuang S, Charge SB, Seale P, et al. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol. 2006;172:103–113.
- Oustanina S, Hause G, Braun T. Pax7 directs postnatal renewal and propagation of myogenic satellite cells but not their specification. EMBO J. 2004;23:3430–3439.
- Seale P, Sabourin LA, Girgis-Gabardo A, et al. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102(6):777–786.
- Corcoran A, Del Maestro RF. Testing the “go or grow” hypothesis in human medulloblastoma cell lines in two and three dimensions. Neurosurgery. 2003;53:174–184; discussion 184–175.
- Garay T, Juhász É, Molnár E, et al. Cell migration or cytokinesis and proliferation?–revisiting the “go or grow” hypothesis in cancer cells in vitro. Exp Cell Res. 2013;319:3094–3103.
- Giese A, Loo MA, Tran N, et al. Dichotomy of astrocytoma migration and proliferation. Int J Cancer. 1996;67:275–282.
- Bouchard G, Bouvette G, Therriault H, et al. Pre-irradiation of mouse mammary gland stimulates cancer cell migration and development of lung metastases. Br J Cancer. 2013;109:1829–1838.
- Yano S, Miwa S, Mii S, et al. Invading cancer cells are predominantly in G0/G1 resulting in chemoresistance demonstrated by real-time FUCCI imaging. Cell Cycle. 2014;13:953–960.
- Haass NK, Beaumont KA, Hill DS, et al. Real-time cell cycle imaging during melanoma growth, invasion, and drug response. Pigment Cell Melanoma Res. 2014;27:764–776.
- Kagawa Y, Matsumoto S, Kamioka Y, et al. Cell cycle-dependent Rho GTPase activity dynamically regulates cancer cell motility and invasion in vivo. PLoS One. 2013;8(12):e83629.
- Burstyn-Cohen T, Kalcheim C. Association between the cell cycle and neural crest delamination through specific regulation of G1/S transition. Dev Cell. 2002;3:383–395.
- Rajan SG, Gallik KL, Monaghan JR, et al. Tracking neural crest cell cycle progression in vivo. Genesis. 2018;56(6–7):e23214.
- Sakaue-Sawano A, Kurokawa H, Morimura T, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132(3):487–498.
- Jones NC, Fedorov YV, Rosenthal RS, et al. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J Cell Physiol. 2001;186:104–115.
- Suzuki J, Yamazaki Y, Li G, et al. Involvement of Ras and Ral in chemotactic migration of skeletal myoblasts. Mol Cell Biol. 2000;20:4658–4665.
- Koyama T, Nakaoka Y, Fujio Y, et al. Interaction of scaffolding adaptor protein Gab1 with tyrosine phosphatase SHP2 negatively regulates IGF-I-dependent myogenic differentiation via the ERK1/2 signaling pathway. J Biol Chem. 2008;283(35):24234–24244.
- Rommel C, Clarke BA, Zimmermann S, et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286(5445):1738–1741.
- Yokoyama T, Takano K, Yoshida A, et al. DA-Raf1, a competent intrinsic dominant-negative antagonist of the Ras-ERK pathway, is required for myogenic differentiation. J Cell Biol. 2007;177:781–793.
- Michailovici I, Harrington HA, Azogui HH, et al. Nuclear to cytoplasmic shuttling of ERK promotes differentiation of muscle stem/progenitor cells. Development. 2014;141(13):2611–2620.
- Le Grand F, Grifone R, Mourikis P, et al. Six1 regulates stem cell repair potential and self-renewal during skeletal muscle regeneration. J Cell Biol. 2012;198:815–832.
- Floss T, Arnold HH, Braun T. A role for FGF-6 in skeletal muscle regeneration. Genes Dev. 1997;11:2040–2051.
- Neuhaus P, Oustanina S, Loch T, et al. Reduced mobility of fibroblast growth factor (FGF)-deficient myoblasts might contribute to dystrophic changes in the musculature of FGF2/FGF6/mdx triple-mutant mice. Mol Cell Biol. 2003;23:6037–6048.
- Doukas J, Blease K, Craig D, et al. Delivery of FGF genes to wound repair cells enhances arteriogenesis and myogenesis in skeletal muscle. Mol Ther. 2002;5(5):517–527.
- Nobis M, Warren SC, Lucas MC, et al. Molecular mobility and activity in an intravital imaging setting - implications for cancer progression and targeting. J Cell Sci. 2018;131:jcs206995–jcs206995.
- Pittet MJ, Weissleder R. Intravital imaging. Cell. 2011;147:983–991.
- Webster MT, Manor U, Lippincott-Schwartz J, et al. Intravital imaging reveals ghost fibers as architectural units guiding myogenic progenitors during regeneration. Cell Stem Cell. 2016;18:243–252.
- Mercier L, Böhm J, Fekonja N, et al. In vivo imaging of skeletal muscle in mice highlights muscle defects in a model of myotubular myopathy. Intravital. 2016;5(1):e1168553.
- Hotta K, Behnke BJ, Masamoto K, et al. Microvascular permeability of skeletal muscle after eccentric contraction-induced muscle injury: in vivo imaging using two-photon laser scanning microscopy. J Appl Physiol. 2018;125(2):369–380.
- Konishi Y, Terai K, Furuta Y, et al. Live-cell FRET imaging reveals a role of extracellular signal-regulated kinase activity dynamics in thymocyte motility. Food Sci Hum Wellness. 2018;10:98–113.
- Lepper C, Conway SJ, Fan CM. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–631.
- Takaoka S, Kamioka Y, Takakura K, et al. Live imaging of transforming growth factor-β activated kinase 1 activation in Lewis lung carcinoma 3 LL cells implanted into syngeneic mice and treated with polyinosinic:polycytidylic acid. Cancer Sci. 2016;107(5):644–652.
- Tomura M, Sakaue-Sawano A, Mori Y, et al. Contrasting quiescent G0 phase with mitotic cell cycling in the mouse immune system. PLoS One. 2013;8(9):e73801.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166.
- Ogawa R, Ma Y, Yamaguchi M, et al. Doublecortin marks a new population of transiently amplifying muscle progenitor cells and is required for myofiber maturation during skeletal muscle regeneration. Development. 2015;142:51–61.
- Ruiz-Bonilla V, Perdiguero E, Gresh L, et al. Efficient adult skeletal muscle regeneration in mice deficient in p38beta, p38gamma and p38delta MAP kinases. Cell Cycle. 2008;7(14):2208–2214.
- Hauck CR, Hsia DA, Schlaepfer DD. Focal adhesion kinase facilitates platelet-derived growth factor-BB-stimulated ERK2 activation required for chemotaxis migration of vascular smooth muscle cells. J Biol Chem. 2000;275:41092–41099.
- Liu ZX, Yu CF, Nickel C, et al. Hepatocyte growth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin-focal adhesion kinase association. J Biol Chem. 2002;277:10452–10458.
- Singh J, Sharma K, Frost EE, et al. Role of PDGF-A-activated ERK signaling mediated FAK-paxillin interaction in oligodendrocyte progenitor cell migration. J Mol Neurosci. 2019;67:564–573.
- Subauste MC, Pertz O, Adamson ED, et al. Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. J Cell Biol. 2004;165:371–381.
- Teranishi S, Kimura K, Nishida T. Role of formation of an ERK-FAK-paxillin complex in migration of human corneal epithelial cells during wound closure in vitro. Invest Ophthalmol Vis Sci. 2009;50:5646–5652.
- Quach NL, Biressi S, Reichardt LF, et al. Focal adhesion kinase signaling regulates the expression of caveolin 3 and beta1 integrin, genes essential for normal myoblast fusion. Mol Biol Cell. 2009;20:3422–3435.
- Sen S, Tewari M, Zajac A, et al. Upregulation of paxillin and focal adhesion signaling follows dystroglycan complex deletions and promotes a hypertensive state of differentiation. Eur J Cell Biol. 2011;90:249–260.
- Chi Y, Welcker M, Hizli AA, et al. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008;9(10):R149.
- Baldassarre G, Belletti B, Nicoloso MS, et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7:51–63.
- Nadeem L, Brkic J, Chen YF, et al. Cytoplasmic mislocalization of p27 and CDK2 mediates the anti-migratory and anti-proliferative effects of nodal in human trophoblast cells. J Cell Sci. 2013;126:445–453.
- Schiappacassi M, Lovat F, Canzonieri V, et al. p27Kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol Cancer Ther. 2008;7(5):1164–1175.
- Schiappacassi M, Lovisa S, Lovat F, et al. Role of T198 modification in the regulation of p27(Kip1) protein stability and function. PLoS One. 2011;6(3):e17673.
- Kiyokawa H, Kineman RD, Manova-Todorova KO, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of P27Kip1. Cell. 1996;85(5):721–732.
- Liedtke W, Leman EE, Fyffe RE, et al. Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am J Pathol. 2002;160:469–480.
- Balogh A, Mege RM, Sobel A. Growth and cell density-dependent expression of stathmin in C2 myoblasts in culture. Exp Cell Res. 1996;224:8–15.
- Casadei L, Vallorani L, Gioacchini AM, et al. Proteomics-based investigation in C2C12 myoblast differentiation. Eur J Histochem. 2009;53(4):e31.
- Gonnet F, Bouazza B, Millot GA, et al. Proteome analysis of differentiating human myoblasts by dialysis-assisted two-dimensional gel electrophoresis (DAGE). Proteomics. 2008;8(2):264–278.
- Kitzmann M, Carnac G, Vandromme M, et al. The muscle regulatory factors MyoD and Myf-5 undergo distinct cell cycle-specific expression in muscle cells. J Cell Biol. 1998;142:1447–1459.
- Kitzmann M, Vandromme M, Schaeffer V, et al. cdk1- and cdk2-mediated phosphorylation of MyoD Ser200 in growing C2 myoblasts: role in modulating MyoD half-life and myogenic activity. Mol Cell Biol. 1999;19(4):3167–3176.
- Song A, Wang Q, Goebl MG, et al. Phosphorylation of nuclear MyoD is required for its rapid degradation. Mol Cell Biol. 1998;18:4994–4999.
- Tintignac LAJ, Sirri V, Leibovitch MP, et al. Mutant MyoD lacking Cdc2 phosphorylation sites delays M-phase entry. Mol Cell Biol. 2004;24(4):1809–1821.
- Guo K, Wang J, Andres V, et al. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol Cell Biol. 1995;15:3823–3829.
- Halevy O, Novitch B, Spicer D, et al. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267(5200):1018–1021.
- Evano B, Khalilian S, Le Carrou G, et al. Dynamics of asymmetric and symmetric divisions of muscle stem cells in vivo and on artificial niches. Cell Rep. 2020;30:3195–3206 e3197.
- Joe AWB, Yi L, Natarajan A, et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12(2):153–163.
- Rocheteau P, Gayraud-Morel B, Siegl-Cachedenier I, et al. A subpopulation of adult skeletal muscle stem cells retains all template DNA strands after cell division. Cell. 2012;148:112.
- Zhang C, Li Y, Wu Y, et al. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J Biol Chem. 2013;288:1489–1499.
- Komatsu N, Aoki K, Yamada M, et al. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol Biol Cell. 2011;22(23):4647–4656.
- Mort RL, Ford MJ, Sakaue-Sawano A, et al. Fucci2a: a bicistronic cell cycle reporter that allows cre mediated tissue specific expression in mice. Cell Cycle. 2014;13(17):2681–2696.
- Konagaya Y, Terai K, Hirao Y, et al. A highly sensitive FRET Biosensor for AMPK exhibits heterogeneous AMPK responses among cells and organs. Cell Rep. 2017;21(9):2628–2638.
- Susaki EA, Tainaka K, Perrin D, et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell. 2014;157(3):726–739.
- Parslow A, Cardona A, Bryson-Richardson RJ. Sample drift correction following 4D confocal time-lapse imaging. J Vis Exp. 2014;1–4. DOI:10.3791/51086
- Tinevez J-Y, Perry N, Schindelin J, et al. TrackMate: an open and extensible platform for single-particle tracking. Methods. 2017;115(80):80–90.
- Čapek M, Janáček J, Kubínová L. Methods for compensation of the light attenuation with depth of images captured by a confocal microscope. Microsc Res Tech. 2006;69:624–635.