
ABSTRACT
Breast cancer remains a general-threat event in the health of women. Currently, increasing records indicate that long non-coding RNA maternally expressed 3 (MEG3) plays a central role in breast cancer. The current research focused on the function of MEG3 in paclitaxel (PTX)-resistance and human breast cancer growth. Levels of MEG3, microRNA (miR)-4513, and phenazine biosynthesis-like domain-containing protein (PBLD) were evaluated using quantitative real-time polymerase chain reaction (qRT-PCR) or western blot assays. 3-(4.5-dimethylghiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) assay was performed to examine the IC50 of PTX and cell proliferation in breast cancer cells. In addition, cell apoptosis was determined utilizing flow cytometry. Transwell was conducted to assay cell migration and invasion in MCF-7 and MDA-MB-231 cells. The interaction between miR-4513 and MEG3 or PBLD was expounded via dual-luciferase reporter assay. Levels of MEG3 and PBLD were decreased, but miR-4513 level was triggered in breast cancer tissues and cell lines. Overexpression of MEG3 could reinforce cell apoptosis, impede proliferation, migration, invasion, and the IC50 of PTX in breast cancer cells. Moreover, the impact of miR-4513 inhibitor on cell progression and PTX-resistance was overturned by MEG3 deficiency. Interestingly, miR-4513 mimic could abolish the role of PBLD upregulation in cell behaviors and PTX-resistance in MCF-7 and MDA-MB-231 cells. Finally, the expression of PBLD was co-modulated by miR-4513 and MEG3 in vitro. MEG3/miR-4513/PBLD axis modulated PTX-resistance and the development of breast cancer cells, which might provide a promising therapeutic strategy for breast cancer.
Introduction
Breast cancer is one of the most frequent occurred diseases occurred diseases and represents the second leading cause of cancer-related deaths in women worldwide [Citation1,Citation2]. Because of the persistent development of comprehensive treatments, such as chemotherapy, radiotherapy, and surgical operation, the overall survival of patients with breast cancer is evidently improved in the past several decades. Nevertheless, the metastasis of cancer cells and drug-resistance are strictly related to the poor outcome of breast cancer patients in the clinic [Citation3,Citation4]. Accruing evidence has suggested that drug-resistance is occurred and developed in breast cancer cells against several chemotherapeutic drugs, including paclitaxel (PTX) [Citation5]. Understanding the precise molecular mechanism of breast cancer and PTX-resistance is utterly required.
Long non-coding RNAs (lncRNAs) belong to non-protein-coding RNAs and consist of more than 200 nucleotides (nts) [Citation6,Citation7]. Recently, the growing researchers pay attention to the interaction between lncRNAs and tumor biology, such as lncRNAs can affect the phenotypes of diverse cancers via dysregulation of interactive genes and biological progression [Citation8,Citation9]. In particular, numerous researches have analyzed the tumorigenic or tumor-suppressive role of lncRNA in human breast cancer. For example, maternally expressed 3 (MEG3) is dysregulated in multiple cancers [Citation10,Citation11]. More importantly, MEG3 has been demonstrated to be downregulated in breast cancer cells, and the low expression of MEG3 is deemed as an independent biomarker in the diagnosis and prognosis of breast cancer [Citation12]. However, the molecular mechanism of MEG3 in breast cancer is greatly uncharted.
Until now, microRNAs (miRNAs), composed of 18–24 nts, have been manifested to bind to the 3’-untranslated regions (3’-UTR) of targets [Citation13]. The key role of miRNAs in modifying cell growth and metastasis has been identified [Citation14], diverse miRNAs have been found to be dysregulated in various tumors [Citation15]. For example, miR-27a and miR-181 level are augmented in breast cancer tissues, thus playing an oncogenic role in the tumorigenesis and pathogenesis of breast cancer cells [Citation16,Citation17]. MiR-4513 is often associated with human diseases because of the polymorphisms in the seed sequence [Citation18,Citation19]. Currently, miR-4513 has been reported to be closely related to the progression of lung adenocarcinoma [Citation20], and the ectopic expression of miR-4513 can contribute to the progression of breast cancer by targeting tripartite motif-containing 3 (TRIM3) [Citation21]. Hence, the essential role of miR-4513 and the interrelation between miR-4513 and MEG3 in breast cancer were the targets of the present study. Besides, phenazine biosynthesis-like domain-containing protein (PBLD), also marked as MAWBP, is extensively distributed in human tissues, and the expression of PBLD is attenuated in gastric cancer [Citation22]. We speculated that PBLD could be involved in the tumorigenesis of breast cancer.
Herein, we investigated the expression patterns of MEG3, miR-4513, and PBLD in breast cancer cells. Meanwhile, the molecular mechanism among them was exposed to regulate cell behaviors and PTX-resistance in MCF-7 and MDA-MB-231 cells.
Materials and methods
Clinical samples and cell culture
Thirty-one patients with breast cancer were recruited in this study. All the donators were diagnosed and received surgical treatment at The First Affiliated Hospital of Zhengzhou University. We collected breast cancer tissues (n = 31) and adjacent non-tumor tissues (n = 31) from patients, and the specimens were maintained at −80°C until used. The written informed consents were received from all breast cancer patients, and the study was approved by the Ethics Committee of The First Affiliated Hospital of Zhengzhou University.
The human breast cancer cells (MCF-7 and MDA-MB-231) and the normal-like immortalized cells (MCF10A) were obtained from the Chinese Academy of Sciences (Shanghai, China). The tumor cells (MCF-7 and MDA-MB-231) were grown in Roswell Park Memorial Institute 1640 (RPMI-1640; Sigma, St. Louis, MO, USA; Catalog No. R8758) supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA), 100 U/mL penicillin (Gibco) and 100 mg/mL streptomycin (Gibco), and normal-like cell MCF10A was cultured in DMEM/F12 medium (Thermo Fisher Scientific, Waltham, MA, USA; Catalog No. 11,320,082) with 100 ng/ml cholera toxin, 0.01 mg/ml insulin, 20 ng/ml epidermal growth factor (EGF), 500 ng/ml hydrocortisone, and 5% chelex-treated horse serum. All these growth factors were obtained from Sigma. And then cells were incubated in a humidified atmosphere with 5% CO2. Besides, paclitaxel (PTX; Sigma) were added into cellular supernatant with different doses. Of which the 0 µM, 2 µM, 5 µM, 10 µM, or 20 µM of PTX was added to measure the effect of PTX on the level of MEG3, and 0 µM, 0.125 µM, 0.25 µM, 0.5 µM, 1 µM, 2 µM, 4 µM, 8 µM, 16 µM, or 32 µM was used to assess the IC50 of PTX in MCF-7 and MDA-MB-231 cells.
Transient transfection
Overexpression vectors of MEG3 (pcDNA-MEG3) and PBLD (pcDNA-PBLD), as well as their control (pcDNA3.1 empty vector), were designed and generated in Ribobio (Guangzhou, Guangdong, China). Small interference RNA (siRNA) targeting MEG3 (si-MEG3), siRNA negative control (si-NC), miR-4513 mimic (miR-4513) and inhibitor (anti-miR-4513), and their relative control (miR-NC for mimic, anti-miR-NC for inhibitor) were constituted and synthesized from KeyGEN Biotech (Jiangsu, China). Then, the corresponding sequences were transfected into MCF-7 and MDA-MB-231 cells, respectively, using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA; Catalog no. 11,668,030) in accordance with standard protocols.
Quantitative real-time polymerase chain reaction (qRT-PCR) assay
The Trizol reagent (Invitrogen) was used to extract the total RNA from breast cancer tissues and cell lines. With the administration of an Omniscript RT Kit (Qiagen, Dusseldorf, NRW, Germany; Catalog no. 205,113), cDNA was synthesized from the separated RNA according to the standard instructions. Then the mixture consisted of a template (cDNA), primers, and the reagents from SYBR Green Master Mix (Thermo Fisher Scientific; Catalog no. 4,364,344), and RNase/DNase-free water. After that, the reactive tubes were placed on a Bio-Rad system (Bio-Rad, Hercules, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were deemed as the internal control, and the gene levels were calculated via 2−ΔΔCt method. The primers were as follows: MEG3 (forward, 5’-CTCAGGCAGGATCTGGCATA-3’, reverse, 5’-CCTGGAGTGCTGTTGGAGAA-3’); miR-4513 (forward, 5’-ACACTCCAGCTGGGAGACTGACGGCTGGAG-3’, reverse, 5’-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGATGGGC-3’); PBLD (forward, 5’-GGGTCTGCACACGCTGTTC-3’, reverse, 5’-TAATGTCAACCCTTCCGTCT-3’); GAPDH (forward, 5’-GTGAAGGTCGGTGTGAACGG-3’, reverse, 5’-GATGCAGGGATGATGTTCTG-3’); U6 (forward, 5’-CTCGCTTCGGCAGCACA-3’, reverse, 5’-AACGCTTCACGAATTTGCGT-3’).
Cell proliferation detection
3-(4.5-Dimethylghiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT; Absin Bioscience, Shanghai, China; Catalog no. abs50010) was employed to determine cell proliferation and the IC50 of PTX in MCF-7 and MDA-MB-231 cells. Briefly, MCF-7 and MDA-MB-231 cells (5 × 103 cells/well) were loaded on a 96-well plate and cultivated for relative time (0 h, 24 h, 48 h or 72 h for cell proliferation assay, and 48 h for IC50 assay). Next, MTT reagent (20 µL) was added into each well, and then 100 µL dimethyl sulfoxide (DMSO; Sigma) was supplemented into cellular well to dissolve the formazan crystal. Lastly, the absorbance of lysates was recorded using a microplate reader (Bio-Rad).
Flow cytometry
MCF-7 and MDA-MB-231 cells (3 × 105 cells/well) were plated on the 6-well plate. Then, corresponding vectors or oligonucleotides were introduced into breast cancer cells. After transfection for 48 h, cells were trypsinized with trypsin (Gibco; Catalog no. 25,300,054) and washed using phosphate-buffered saline (PBS; Gibco; Catalog no. 10,010,049). Subsequently, the cells were stained with Annexin V-fluorescein isothiocyanate/propidium iodide reagent kit (Annexin V-FITC/PI; Sigma) according to the specifications. The apoptotic cells were stained by Annexin V-FITC and PI and distinguished using flow cytometry (Beckman Coulter, Kraemer Boulevard, CA, USA).
Transwell assay
Transwell chamber (Corning, Corning, NY, USA) was adopted to estimate cell mobility and invasiveness in breast cancer cells. Briefly, MCF-7 and MDA-MB-231 cells were treated accordingly, the transfected cells were re-suspended in serum-free media. Then, the cellular suspension was added into the upper chamber pre-coated with Matrigel (Corning; Catalog no. 354,671) (for cell invasion assay) or without (for cell migration assay), followed by addition with complete medium (200 µL) into the lower chamber. After incubation for 48 h, the moved cells were stained with 0.5% crystal violet (Sigma). The migrated and invaded cells were recognized and counted under a microscope (Olympus, Tokyo, Japan).
Western blot
We referenced the method described in a previous study [Citation23]. Briefly, transfected cells were lysed in RIPA lysis buffer (Thermo Fisher Scientific). Protein concentration was measured with the BCA Protein Assay Kit (Thermo Fisher Scientific). Then, protein (50 μg) samples were separated by 10% SDS-PAGE membranes and the isolated total protein was transfected onto PVDF membranes (Sigma). Next, 5% skim milk at 37°C for 2 h was used to block the membranes. Subsequently, the blots were incubated with primary antibodies overnight at 4°C after block with 5% milk. After that, membranes were incubated with corresponding secondary antibody to combine the primary antibody. The primer antibodies were purchased from Abcam (Cambridge, MA, USA), and their descriptions were as follows: PBLD (ab235947, 1:1000), GAPDH (ab181602, 1:10,000). The protein bands were scanned and visualized adopting an ECL System (Bio-Rad).
Dual-luciferase reporter assay
The complementary sequences between miR-4513 and MEG3 or PBLD were cloned into a luciferase reporter base (pmirGLO; Promega, Madison, WI, USA; Catalog no. E1330), thereby forming wildtype reporters of MEG3 (WT-MEG3) and PBLD (WT-PBLD). Simultaneously, the mutant vectors of MEG3 (MUT-MEG3) and PBLD (MUT-PBLD) were designed and generated. Then, dual-luciferase reporter assay was conducted by using the Dual-Luciferase Reporter Assay System (Promega; Catalog no. E1910) after breast cancer cells were treated accordingly. Finally, the relative firefly activity was normalized to renilla activity. All the assays were repeated thrice.
In vivo xenograft experiment
12 five-week-old BALB/c female nude mice (weight, 16–22 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). The housing environment was: 12-h light/dark cycle; room temperature (25°C); humidity (60%); easy access to food and water. The mice were randomly divided into two groups (n = 6 per group). MEG3 lentivirus plasmid was stably transfected into MDA-MB-231 cells using Lipofectamine 2000 (Invitrogen). Then, the transfected cells were subcutaneously injected into the left flank of nude mice. The tumor volumes were detected every 3 days and tumor weights were measured after the animals were sacrificed. The mice were killed by cervical dislocation after anesthesia with isoflurane (2%) after injection for 30 days. All the animal experiments were followed by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. In addition, the animal studies were approved by the Experimental Animal Ethics Committee of The First Affiliated Hospital of Zhengzhou University (Zhengzhou, China).
Statistical analysis
The data from three independent assays were presented as mean ± standard deviation (SD) after processing with SPSS 22.0 software. The Student’s t-test was administrated to compare the differences between two-tailed, whereas the differences in multiple groups were analyzed by Analysis of Variance (ANOVA) and Tukey post-hoc test. P-value less than 0.05 was regarded as statistically significant.
Results
PTX treatment could rescue the downregulation of MEG3 in MCF-7 and MDA-MB-231 cells
Firstly, the level of MEG3 was detected using qRT-PCR assay, the results disclosed that MEG3 was clearly downregulated in breast cancer tissues and cell lines ()). As shown in ), the overall survival of high MEG3 expression group was higher than the low expression group in breast cancer patients. Then, MCF-7 and MDA-MB-231 cells were added with PTX (0 µM, 2 µM, 5 µM, 10 µM, or 20 µM), qRT-PCR analysis suggested that MEG3 was significantly enhanced in breast cancer cells induced by PTX with different concentrates ()). And the expression of MEG3 had no obvious change in normal-like cells (MCF10A) treated by different amount of PTX (Fig. S1). All the data meant that MEG3 level was specially augmented, but the induction of PTX could trigger MEG3 level in MCF-7 and MDA-MB-231 cells.
Figure 1. PTX treatment could rescue the downregulation of MEG3 in MCF-7 and MDA-MB-231 cells. (a and b) The level of MEG3 in breast cancer tissues and cell lines. (c) The overall survival rate in breast cancer patients with high MEG3 expression compared to patients with low MEG3 expression was evaluated using Kaplan-Meier overall survival curve. (d and e) QRT-PCR analysis for the effect of PTX on MEG3 level in MCF-7 and MDA-MB-231 cells. *P < 0.05
Overexpression of MEG3 could promote cell apoptosis, suppress proliferation, migration and invasion, and the IC50 of PTX in MCF-7 and MDA-MB-231 cells
Given the ectopic expression of MEG3 in breast cancer cells, we further explored its role in cell growth and metastasis. Firstly, overexpression vector of MEG3 was employed to increase MEG3 level in vitro. As shown in ), MEG3 was strikingly upregulated after transfection with MEG3 in MCF-7 and MDA-MB-231 cells. As observed in the MTT assay, the IC50 was particularly reduced in MEG3 overexpression group (IC50 = 1.862 µM/L) compared with that of in control group (IC50 = 5.527 µM/L) ()). Simultaneously, the increase of MEG3 could weaken cell proliferation in breast cancer cells ()). However, the cell proliferation of MEG3 overexpression group was not significantly different from that of the control group (Fig. S2) in normal-like cells (MCF-10A). Besides, we found that cell apoptosis was obviously boosted after transfection with MEG3 in MCF-7 and MDA-MB-231 cells ()). Lastly, cell migration and invasion were assessed via transwell assay, the results expounded that upregulation of MEG3 could notably repress the mobility and invasiveness in vitro ()). In brief, the increase of MEG3 level could strengthen cell apoptosis, hinder cell proliferation, migration, invasion, and PTX-resistance in MCF-7 and MDA-MB-231 cells.
Figure 2. Overexpression of MEG3 could promote cell apoptosis, suppress proliferation, migration, invasion, and the IC50 of PTX in MCF-7 and MDA-MB-231 cells. (a-h) MEG3 or vector was transfected into MCF-7 and MDA-MB-231 cells, (a) and qRT-PCR analysis for the level of MEG3 in vitro. (b and c) The IC50 of PTX after MEG3 upregulation in breast cancer cells. (d and e) The impact of MEG3 overexpression on cell proliferation in MCF-7 and MDA-MB-231 cells. (f) Flow cytometric analysis for the change of cell proliferation after transfection with MEG3 or vector. (g and h) The ability of cell migration and invasion in breast cancer cells. *P < 0.05
MiR-4513 was directly targeted by MEG3
As mention above, MEG3 was an oncogenic gene in breast cancer, we guessed that MEG3 exerted its role via modulating the targets. Next, the analysis of LncBase Predicted v.2 [Citation24] showed that miR-4513 was a putative target of MEG3 ()). After co-transfection with WT-MEG3 or MUT-MEG3 and miR-4513 or miR-NC, the luciferase activity of WT-MEG3 was prominently decreased by miR-4513 overexpression, but miR-4513 mimic had no effect on the mutant reporter ()). Moreover, level of miR-4513 was evaluated by qRT-PCR in breast cancer tissues and cell lines, the results showed that miR-4513 level was apparently elevated in tumor tissues and cell lines (MCF-7 and MDA-MB-231) ()). To further probe the interaction between miR-4513 and MEG3, we firstly constructed knockdown vector of MEG3, qRT-PCR analysis indicated that level of MEG3 was clearly reduced in both MCF-7 and MDA-MB-231 cells ()). Expectedly, the level of miR-4513 was conspicuously hampered by MEG3 upregulation but was facilitated after transfection with si-MEG3 in MCF-7 and MDA-MB-231 cells ()). The evidence indicated that miR-4513 was targeted by MEG3.
Figure 3. MEG3 was a target of miR-4513. (a) The predictive binding sites between miR-4513 and MEG3. (b and c) Dual-luciferase reporter analysis for the interaction between miR-4513 and MEG3 in MCF-7 and MDA-MB-231 cells. (d and e) The level of miR-4513 in breast cancer cells and cell lines. (f) The knockdown efficiency of si-MEG3 in MCF-7 and MDA-MB-231 cells. (g) QRT-PCR analysis for the roles of si-MEG3 and MEG3 in miR-4513 level. *P < 0.05
The effect of miR-4513 inhibitor on cell behaviors and the IC50 of PTX was abolished by MEG3 detection in breast cancer cells
Based on the above evidence, the regulatory mechanism between miR-4513 and MEG3 needed to be further investigated. In the first place, anti-miR-NC, anti-miR-4513, anti-miR-4513+ si-NC, or anti-miR-4513+ si-MEG3 were introduced into MCF-7 and MDA-MB-231 cells, qRT-PCR analysis demonstrated that miR-4513 level was drastically repressed by transfection with miR-4513 inhibitor, while the suppressive role of miR-4513 inhibitor on miR-4513 level was rescued via simultaneous introduction of si-MEG3 in breast cancer cells ()). Meanwhile, the IC50 of PTX, which was inhibited by miR-4513 inhibitor, was strongly augmented via MEG3 silencing in MCF-7 and MDA-MB-231 cells ()). Functional assay indicated that reintroduction of si-MEG3 could relieve the restraining impact of miR-4513 inhibitor on cell proliferation in vitro ()). In addition, the roles of miR-4513 and MEG3 in cell apoptosis were explored, flow cytometric analysis determined that miR-4513 inhibitor could evidently expedite the apoptotic rate, but the acceleratory effect of miR-4513 inhibitor on cell apoptosis was reverted via MEG3 deficiency in breast cancer cells ()). Lastly, cell migration and invasion were identified by transwell assay, the results presented that the repressive impact of miR-4513 inhibitor on cell migration and invasion was overturned after co-transfection with si-MEG3 in vitro ()). These results strongly meant that MEG3 knockdown played a vital part in miR-4513 inhibitor-induced promotion of cell apoptosis, and repression of proliferation, migration, invasion, and PTX-resistance in breast cancer cells.
Figure 4. The effect of miR-4513 inhibitor on cell behaviors and the IC50 of PTX was abolished by MEG3 deletion in breast cancer cells
PBLD was a target of miR-4513
To search for the targets of miR-4513, Targetscan was performed to predict potential mRNAs that bound to miR-4513. As displayed in ), PBLD was predicted to contain the common fragments with miR-4513. Dual-luciferase reporter analysis confirmed that miR-4513 mimic specially constrained the luciferase activity of WT-PBLD, whereas had no inhibitory effect on MUT-PBLD reporter ()). Subsequently, we found that the mRNA and protein levels of PBLD were clearly hindered in breast cancer tissues and cell lines ()). To elucidate the mutual influence between miR-4513 and CCAT1, the efficiency of miR-4513 mimic was verified by qRT-PCR, the results manifested that miR-4513 mimic could drastically intensify miR-4513 level in MCF-7 and MDA-MB-231 cells ()). Then, MCF-7 and MDA-MB-231 cells were transfected with miR-NC, miR-4513, anti-miR-NC, or anti-miR-4513, respectively. As shown by qRT-PCR and western blot, miR-4513 downregulation led to an obvious increase of PBLD expression in the aspects of mRNA and protein, while miR-4513 mimic resulted in an efficient curb of mRNA and protein levels of PBLD ()). These data uncovered that miR-4513 could directly target PBLD in breast cancer cells.
Figure 5. PBLD was a target of miR-4513. (a) Partial complementary sequences between miR-4513 and PBLD. (b and c) The luciferase activity analysis for the luciferase activity after co-transfection with WT-PBLD or MUT-PBLD and miR-4513 or miR-NC in breast cancer cells. (d-g) The mRNA and protein levels of PBLD in breast cancer tissues and cell lines. (h) The acceleratory efficiency of miR-4513 mimic on miR-4513 level in MCF-7 and MDA-MB-231 cells. (i and j) QRT-PCR and western blot analyses for the mRNA and protein levels of PBLD in breast cancer cells after transfection with miR-4513 or anti-miR-4513. *P < 0.05
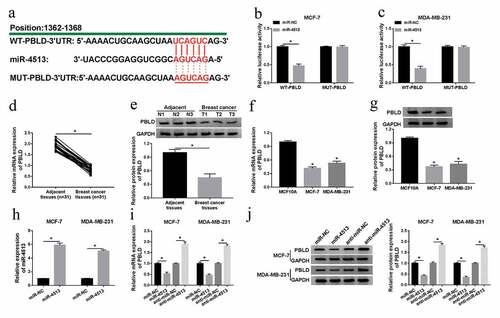
The impact of PBLD upregulation on cell behavior and the IC50 of PTX was abrogated by co-transfection with miR-4513 in vitro
According to the regulatory effect of MEG3 on miR4513 level, we paid attention to the functional mechanism between miR-4513 and MEG3 in breast cancer cells. QRT-PCR and western blot analyses clarified that introduction of PBLD could efficiently augment the mRNA and protein levels of PBLD, whereas the promoting effect of PBLD upregulation on PBLD level was attenuated via synchronous introduction of miR-4513 mimic in MCF-7 and MDA-MB-231 cells ()). Subsequently, the functional roles of PBLD and miR-4513 in the IC50 of PTX were exposed, MTT analysis displayed that PTX-resistance, which was weaken by PBLD overexpression, was regained after co-transfection with miR-4513 mimic ()). Besides, miR-4513 mimic relieved the suppressive impact of PBLD transfection on cell proliferation in vitro ()). At the same time, reintroduction of miR-4513 mimic segmentally abolished the acceleratory effect of PBLD increase on cell apoptotic rate in MCF-7 and MDA-MB-231 cells ()). What’s more, transwell analysis proved that PBLD overexpression could strikingly decreased the mobility and invasiveness of breast cancer cells, whereas the blocking influence of PBLD introduction in cell migration and invasion was ameliorated after co-transfection with miR-4513 mimic in vitro ()). All the data revealed that co-transfection with miR-4513 mimic could relieve the role of PBLD upregulation in cell behaviors and PTX-resistance in breast cancer cells.
Figure 6. The impact of PBLD upregulation on cell behavior and the IC50 of PTX was abrogated by co-transfection with miR-4513 in vitro. (a-j) MCF-7 and MDA-MB-231 cells were transfected with vector, PBLD, PBLD+miR-NC, or PBLD+miR-4513, respectively, (a-d) qRT-PCR and western blot analyses for the alteration of the mRNA and protein levels of PBLD in vitro. (e) The role of PBLD and miR-4513 in the IC50 of PTX in breast cancer cells. (f and g) MTT assay for measuring the change of cell proliferation in MCF-7 and MDA-MB-231 cells. (h) Flow cytometry estimation for the level of cell apoptotic rate in vitro. (i and j) The capacities of cell migration and invasion of MCF-7 and MDA-MB-231 cells treated accordingly. *P < 0.05
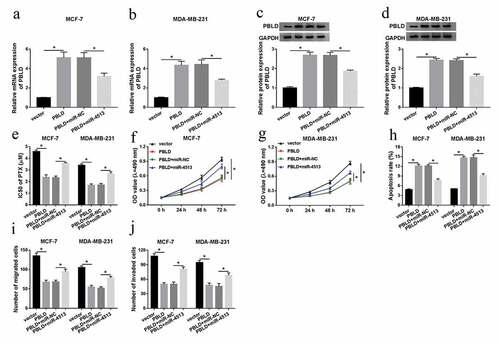
MEG3 regulated the effects of PBLD by miR-4513
As research above, we aimed to discover the molecular between PBLD and miR-4513 or MEG3. Firstly, miR-4513 alone or along with MEG3 was introduced into MCF-7 and MDA-MB-231 cells, qRT-PCR analysis suggested that the mRNA level of PBLD was particularly impeded by miR-4513 mimic, while the inhibiting effect of miR-4513 overexpression on PBLD mRNA level was restored after co-transfection with MEG3 in vitro ()). Furthermore, similar regulatory tendency in the protein level of PBLD was found and shown in ). Taken together, the evidence implied that PBLD was mediated by MEG3 and miR-4513 in MCF-7 and MDA-MB-231 cells. The detailed summary diagram is shown in ).
Figure 7. MEG3 regulated PBLD by miR-4513. (a-d) MiR-NC, miR-4513, miR-4513+ vector, or miR-4513+ MEG3 was introduced into MCF-7 and MDA-MB-231 cells, respectively. QRT-PCR and western blot analyses for the alteration of mRNA and protein levels of PBLD in breast cancer cells. (e) The summary diagram was shown. *P < 0.05
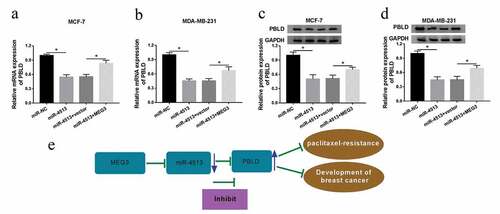
MEG3 overexpression restrained the tumor growth in xenograft model
To further study the function of MEG3 in breast cancer in vivo, we constructed the xenograft mouse model. After stably transfected MEG3 in MDA-MB-231 cells and then injected into nude mice for 8 days, we measured the tumor volume every 3 days and tumor weight after they were sacrificed. Our findings indicated that tumor volume and weight were significantly decreased after overexpression of MEG3 in vivo. Moreover, the expression of MEG3 in the transplanted tumors with MEG3 overexpression was detected by qRT-PCR, and the data showed that the expression levels of MEG3 were markedly increased after transfection with MEG3 ()).
Figure 8. Overexpression of MEG3 suppressed tumor growthin vivo. (a-c) MDA-MB-231 cells stable transfected with MEG3 were injected into nude mice for 8 days. (a) The tumor volume was measured every 3 days for 23 days after the injection. (b) The tumor weight was measused after the mices was sacrificed. (c) MEG3 expression in the transplanted tumors was examined. *P< 0.05
Discussion
During recent years, more and more researchers devote to unraveling the role of lncRNAs in tumorigenesis. Such as lncRNAs could modulate several cancer-associated pathways connected with the mediation of cell-cycle, differentiation, cell death, and cell metastasis [Citation25–27]. For instance, cancer susceptibility candidate 2 (CASC2) level was aberrantly decreased in breast cancer tissues and acted as a key factor in breast cancer tumorigenesis and malignancy [Citation28]. Accumulating records implicate that lncRNAs are involved in cellular behaviors and the development of pathology in human cancers, showing as the alteration of cell proliferation, metastasis, and angiogenesis [Citation29,Citation30]. In the present study, we explored that MEG3 level was unusually decreased in breast cancer tissues and cell lines. MEG3, regarded as a tumor-associated lncRNA, has been discovered to be downregulated in numerous tumors, such as lung cancer [Citation31] and glioma [Citation32]. Not only that, MEG3 is negatively associated with several tumor growths, such as the inhibition of cell proliferation, migration, and invasion in vitro [Citation33,Citation34]. PTX, as an efficient first-line chemotherapeutic drug, is used as single or combines with other drugs in the clinic [Citation35]. The impact of MEG3 on the PTX-resistance was another investigated object in this research. To further confirm the role of MEG3 in breast cancer progression, we introduced overexpression vector of MEG3 into MCF-7 and MDA-MB-231 cells, and subsequent analyses demonstrated that MEG3 upregulation could boost cell apoptosis, retard proliferation, migration, and invasion in breast cancer cells. Because of the PTX addition could clearly reinforce the MEG3 level, we also found that overexpression of MEG3 markedly reduced the IC50 of PTX in breast cancer cells. The findings were agreed with previous evidence [Citation36].
Our findings elucidated the critical role of MEG3 in modifying the homeostasis of breast cancer. Fundamentally, the interaction between lncRNAs and miRNAs go in for the regulatory mechanism of lncRNAs-regulated cellular behaviors and tumorigenesis [Citation37]. The prediction of lncBase Predicted v.2 showed that miR-4513 might be a target of MEG3, and the interrelation between miR-4513 and MEG3 was exposed in the subsequent assay. As for miR-4513, it was obviously elevated in breast cancer tissues and cell sublines. Then, we performed a series of in vitro assays to investigate the biological role of miR-4513 in MCF-7 and MDA-MB-231 cells. The results discovered that miR-4513 silencing could intensify cell apoptosis, weaken cell migration, invasion and PTX-resistance in breast cancer cells. Before this study, miR-4513 has been implied to be expedited in tumor tissues compared with noncancerous tissues [Citation38,Citation39]. What’s more, the upregulation of miR-4513 modulated cell proliferation and apoptosis via mediating CXC ligand 17 (CXCL17) [Citation39]. It was worth noting that miR-4513 has been proved to work as an oncogenic factor in the progression of breast cancer via regulating TRIM3 [Citation21]. Thereby, we further searched for the probable targets of miR-4513.
The essential function of miR-4513 in mediating the progression of breast cancer has been revealed previously. Online software analysis predicted that PBLD was a possible target gene of miR-4513, and the interrelation between them was confirmed subsequently. As the research of PBLD, the aberrant decrease of it is involved in the pathogenesis and process of hepatocellular carcinoma [Citation40,Citation41]. More importantly, PBLD, which was targeted by miR-548p, could constrain tumor progression and weaken drug-resistance in breast cancer [Citation42]. Based on the evidence, we clarified that PBLD functioned as a tumor suppressor in breast cancer tumorigenesis, exhibiting as the acceleration of cell apoptosis, and the curb of cell proliferation, migration, invasion, and PTX-resistance. Interestingly, the repressive role of PBLD overexpression on tumor growth was overturned by co-transfection with miR-4513 mimic in breast cancer cells. Above all, the expression of PBLD was co-modulated by MEG3 and miR-4513 in vitro.
In summary, the addition of PTX could induce the level of MEG3 in breast cancer cells, overexpression of MEG3 distinctly triggered cell apoptosis, impeded proliferation, migration, invasion, and the IC50 of PTX in MCF-7 and MDA-MB-231 cells. Mechanically, MEG3 exerted its tumor-suppressive role via miR-4513/PBLD axis in the pathogenesis of breast cancer. However, the evidence from this study is not enough, it is necessary to supplement xenograft tumor assay or more clinical samples for further investigation.
Highlights
PTX stimulation could relieve the downregulation of MEG3 in MCF-7 and MDA-MB-231 cells
MEG3 upregulation could repress cell proliferation, migration and invasion, induces apoptosis and PTX-resistance in breast cancer cells
MEG3 modified the PTX-resistance and progression of breast cancer cells via miR-4513/PBLD axis.
Disclosure of interest
The authors declare that they have no financial conflicts of interest.
Supplemental Material
Download Zip (97 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Related Research Data
References
- DeSantis C, Ma J, Bryan L, et al. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:1.
- O’Sullivan CC, Loprinzi CL, Haddad TC. Updates in the evaluation and management of breast cancer. Mayo Clin Proc. 2018;93:6.
- Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22.
- Hu G, Chong RA, Yang Q, et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell. 2009;15:1.
- Paridaens R, Biganzoli L, Bruning P, et al. Paclitaxel versus doxorubicin as first-line single-agent chemotherapy for metastatic breast cancer: a European organization for research and treatment of cancer randomized study with cross-over. J Clin Oncol. 2000;18:4.
- Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:6.
- Yang L, Froberg JE, Lee JT. Long noncoding RNAs: fresh perspectives into the RNA world. Trends Biochem Sci. 2014;39:1.
- Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21:11.
- Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:4.
- Kruer TL, Dougherty SM, Reynolds L, et al. Expression of the lncRNA maternally expressed gene 3 (MEG3) Contributes to the control of lung cancer cell proliferation by the Rb pathway. PLoS One. 2016;11:11.
- Zhang J, Yao T, Wang Y, et al. Long noncoding RNA MEG3 is downregulated in cervical cancer and affects cell proliferation and apoptosis by regulating miR-21. Cancer Biol Ther. 2016;17:1.
- Zhang JJ, Guo SH, Jia BQ. Down-regulation of long non-coding RNA MEG3 serves as an unfavorable risk factor for survival of patients with breast cancer. Eur Rev Med Pharmacol Sci. 2016;20:24.
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:9.
- Melo SA, Esteller M. Disruption of microRNA nuclear transport in human cancer. Semin Cancer Biol. 2014;27. DOI:10.1016/j.semcancer.2014.02.012
- Shin VY, Chu KM. MiRNA as potential biomarkers and therapeutic targets for gastric cancer. World J Gastroenterol. 2014;20:30.
- Jiang G, Shi W, Fang H, et al. miR27a promotes human breast cancer cell migration by inducing EMT in a FBXW7dependent manner. Mol Med Rep. 2018;18:6.
- Tian Y, Fu X, Li Q, et al. MicroRNA181 serves an oncogenic role in breast cancer via the inhibition of SPRY4. Mol Med Rep. 2018;18:6.
- Ghanbari M, de Vries PS, de Looper H, et al. A genetic variant in the seed region of miR-4513 shows pleiotropic effects on lipid and glucose homeostasis, blood pressure, and coronary artery disease. Hum Mutat. 2014;35:12.
- Ghanbari M, Erkeland SJ, Xu L, et al. Genetic variants in microRNAs and their binding sites within gene 3©UTRs associate with susceptibility to age-related macular degeneration. Hum Mutat. 2017;38:7.
- Zhang N, Li Y, Zheng Y, et al. miR-608 and miR-4513 significantly contribute to the prognosis of lung adenocarcinoma treated with EGFR-TKIs. Lab Invest. 2019;99:4.
- Li Y, Zhu H, Wang J, et al. miR-4513 promotes breast cancer progression through targeting TRIM3. Am J Transl Res. 2019;11:4.
- Li DM, Zhang J, Li WM, et al. MAWBP and MAWD inhibit proliferation and invasion in gastric cancer. World J Gastroenterol. 2013;19:18.
- Zhang Y, Wu J, Jing H, et al. Long noncoding RNA MEG3 inhibits breast cancer growth via upregulating endoplasmic reticulum stress and activating NF-kappaB and p53. Cell Biochem. 2019;120:4
- Paraskevopoulou MD, Vlachos IS, Karagkouni D, et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44(D1):D231–238. .
- Khaitan D, Dinger ME, Mazar J, et al. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 2011;71:11.
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:1.
- Yang X, Song JH, Cheng Y, et al. Long non-coding RNA HNF1A-AS1 regulates proliferation and migration in oesophageal adenocarcinoma cells. Gut. 2014;63:6.
- Zhang Y, Zhu M, Sun Y, et al. Upregulation of lncRNA CASC2 Suppresses Cell Proliferation and Metastasis of Breast Cancer via Inactivation of the TGF-beta Signaling Pathway. Oncol Res. 2019;27:3.
- Tripathi V, Shen Z, Chakraborty A, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9:3.
- Zhang A, Zhao JC, Kim J, et al. LncRNA HOTAIR enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate cancer. Cell Rep. 2015;13:1.
- Lu KH, Li W, Liu XH, et al. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13. DOI:10.1186/1471-2407-13-461.
- Wang P, Ren Z, Sun P. Overexpression of the long non-coding RNA MEG3 impairs in vitro glioma cell proliferation. J Cell Biochem. 2012;113:6.
- Zhang Y, Liu J, Lv Y, et al. LncRNA meg3 suppresses hepatocellular carcinoma in vitro and vivo studies. Am J Transl Res. 2019;11:7.
- Wang W, Xie Y, Chen F, et al. LncRNA MEG3 acts a biomarker and regulates cell functions by targeting ADAR1 in colorectal cancer. World J Gastroenterol. 2019;25:29.
- Akiyama M, Sowa Y, Taniguchi T, et al. Three combined treatments, a novel HDAC inhibitor OBP-801/YM753, 5-fluorouracil, and paclitaxel, induce G(2) phase arrest through the p38 pathway in human ovarian cancer cells. Oncol Res. 2017;25:8.
- Wang H, Li H, Zhang L, et al. Overexpression of MEG3 sensitizes colorectal cancer cells to oxaliplatin through regulation of miR-141/PDCD4 axis. Biomed Pharmacother. 2018;106. DOI:10.1016/j.biopha.2018.07.131.
- Zhang S, Leng T, Zhang Q, et al. Sanguinarine inhibits epithelial ovarian cancer development via regulating long non-coding RNA CASC2-EIF4A3 axis and/or inhibiting NF-kappaB signaling or PI3K/AKT/mTOR pathway. Biomed Pharmacother. 2018;102:302–308
- Ding H, Shi Y, Liu X, et al. miR-4513 promotes gastric cancer cell proliferation and epithelial-mesenchymal transition through targeting KAT6B. Hum Gene Ther Clin Dev. 2019. DOI:10.1089/humc.2019.094.
- Xu YX, Sun J, Xiao WL, et al. MiR-4513 mediates the proliferation and apoptosis of oral squamous cell carcinoma cells via targeting CXCL17. Eur Rev Med Pharmacol Sci. 2019;23:9.
- Neo SY, Leow CK, Vega VB, et al. Identification of discriminators of hepatoma by gene expression profiling using a minimal dataset approach. Hepatology. 2004;39:4.
- Long J, Lang ZW, Wang HG, et al. Glutamine synthetase as an early marker for hepatocellular carcinoma based on proteomic analysis of resected small hepatocellular carcinomas. Hepatobiliary Pancreat Dis Int. 2010;9:3.
- Liang Y, Song X, Li Y, et al. circKDM4C suppresses tumor progression and attenuates doxorubicin resistance by regulating miR-548p/PBLD axis in breast cancer. Oncogene. 2019. DOI:10.1038/s41388-019-0926-z.