
ABSTRACT
High-mobility group AT-hook2 (HMGA2), serving as an architectural transcription factor, participates in plenty of biological processes. Our study is aimed at illustrating the effect of HMGA2 on hypoxia-induced HUVEC injury and the underlying mechanism. To induce hypoxia-related cell injury, HUVECs were exposed to hypoxic condition for 12–24 h. Molecular expression was determined by Western blot analysis, real-time PCR and immunofluorescence staining. Cell migration was monitored by wound healing assay and Transwell chamber assay. Cell proliferation and apoptosis were measured by MTT assay kits and TUNEL staining. In this study, we discovered that HMGA2 was upregulated in hypoxia-induced HUVECs. Overexpression of HMGA2 promoted cell migration, decreased the apoptosis ratio in response to hypoxia stimulation, while HMGA2 knockdown inhibited cell migration and accelerated apoptosis in HUVECs under hypoxic condition. Mechanistically, we found that HMGA2 induced increased expression of HIF-1α,VEGF, eNOS and AKT. eNOS knockdown significantly reduced HMGA2-mediated pro-migration effects, and AKT knockdown strikingly counteracted HMGA2-mediated anti-apoptotic effect. Hence, our data indicated that HMGA2 promoted cell migration by regulating HIF-1α/VGEF/eNOS signaling and prevented cell apoptosis by activating HIF-1α/VGEF/AKT signaling in HUVECs.
1. Introduction
Myocardial dysfunction can be caused by apoptosis of endothelial cells induced by hypoxia in the heart [Citation1]. Hypoxia-induced vascular endothelial cell damage is a common pathogenesis of cardiovascular disease (CVD) such as coronary atherosclerotic heart disease [Citation2]. Myocardial microvascular endothelial cells play an important role in participating in substance exchange, preventing platelet aggregation and thrombosis, and directly affecting myocardial cells by secreting active substances. Endothelial cells respond to stress in the absence of oxygen, including cell proliferation, migration, inflammation, and apoptosis [Citation1,Citation3]. Imbalance of vascular structure and functional homeostasis caused by hypoxia are important pathophysiological mechanisms leading to a variety of functional disorders of organs and systems. Therefore, studying the molecular mechanisms of hypoxia-induced injury of vascular endothelial cells (VECs) is of great significance for understanding the pathogenesis of cardiovascular disease and discovery of new treatment.
The high mobility group protein A2(HMGA2) is a small non-histone chromosomal protein with no intrinsic transcriptional activity, but can regulate transcription by altering chromatin structure [Citation4]. HMGA2 is highly expressed during embryogenesis and in many human malignancies [Citation5].The research field of HMGA2 is mainly focused on tumors. Accumulated studies have indicated that HMGA2 could promote epithelial-to-mesenchymal transition (EMT) in cancer and contribute to proliferation, migration and invasion of carcinoma cells [Citation6,Citation7]. However, its role in cardiovascular disease has gradually been revealed in recent years. Previous research found that it has a crucial impact on the normal development of the heart [Citation8]. Inhibition the expression of HMGA2 protects sepsis mice from cardiac dysfunction [Citation9]. Impeding HMGA2 expression can inhibit the apoptosis of myoblast H9c2 cells and primary neonatal rat ventricular cardiomyocytes(NRVM) and protect these cells against hypoxia injury [Citation10]. HMGA2 can improve the remodeling response of heart due to pressure overload by activating PPARγ [Citation11]. Nevertheless, its mechanism of action on vascular endothelial cell injury induced by hypoxia has not been clearly elucidated.
Therefore, the objective of this study was to determine whether HMGA2 could affect hypoxia-induced endothelial cell injury and meanwhile to elucidate the potential mechanisms of HMGA2 in hypoxia-induced endothelial cell injury.
2. Materials and methods
2.1. Materials
Adenovirus expressing HMGA2 (Ad-HMGA2) (NM_003484) or green fluorescent protein (Ad-GFP) were constructed and generated by Vigene Bioscience (Shangdong, China).
HMGA2 siRNA (si-HMGA2), VEGFR2 siRNA (si-VEGFR2), eNOS siRNA (si-eNOS), AKT siRNA (si-AKT) and siRNA negative control (si-NC) were purchased from Thermo Fisher. Lipo6000 ™ transfection reagent, Caspase 3 Activity Assay Kit and Total Nitric Oxide Assay Kit were bought from Beyotime Biotechnology (Shanghai, China). Primary antibodies against HMGA2(ab97276), HIF-1α(ab2185), HIF-2α(ab109616) and VEGF(ab46154) were acquired from Abcam (Cambridge, UK). Primary antibodies against P-Erk1/2 (4370P), AKT (4691), Bcl-2(2870), Bax(2722) and GAPDH(2118) were purchased from Cell Signaling Technology(Boston, USA). eNOS (sc-654) was obtain from Santa Cruz Biotechnology(USA). The secondary antibodies for western blotting and for immunofluorescence staining were purchased from Abbkine and Thermo Fisher Scientific, respectively. The BCA protein assay kit was from Thermo Fisher Scientific. ApopTag® Plus In Situ Apoptosis Fluorescein Detection Kit (S7111) was bought from Millipore Corporation. Human vascular endothelial growth factor (VEGF) ELISA Kit(HM10057) was obtained from Bio-Swamp (Wuhan, China).
2.2. Cell culture and treatments
Human umbilical vein endothelial cell line was purchased from YrGene (Changsha, China). HUVECs were cultured with RPMI1640 medium containing 10% FBS and incubated in a 37°C, 5% CO2 incubator. When the cells had reached the appropriate density, they were digested with 0.25% trypsin. The cell suspension seeded in 6-well plates were collected for Western blots and RNA analysis, in 24-well plates for TUNEL staining and immunofluorescence staining analysis, and in 96-well plates for MTT assay. HUVECs were transfected with Ad-HMGA2 to overexpress HMGA2 (MOI = 50) for 8 h. To knock down certain target genes, cells were incubated with si-RNA by Lipofectamine 6000 following the manufacturer’s instructions. To establish a model of hypoxia-induced cell injury, cells were exposed in hypoxic conditions with 1%O2, 5% CO2 and 94% N2 for 12–24 h. Cells in the control group were cultured in a 5% CO2 and 95% air at 37°C. Observed under an optical microscope, the cells adhered to the culture plate and suspended dead cells increased in hypoxic condition compared with the normoxic environment.
2.3. Western blotting
Western blotting was performed referring to our previous studies [Citation12]. Briefly, HUVECs in each group were dissolved in RIPA lysis buffer, and the supernatant was taken after sonication and high-speed centrifugation. BCA Protein Assay Kit was used to measure the protein concentrations. Subsequently, protein was separated by 10% SDS-PAGE and then transferred to PVDF membranes. After incubation with the corresponding primary antibodies at 4°C overnight and secondary antibodies at 37°C for 1 h, the membranes were detected using ECL chemiluminescent system. Image J software analysis system analyzed gray value of bands(n = 6) and relative expressions of target proteins were normalized to GAPDH.
2.4. Quantitative real-time PCR
Total RNA was extracted from HUVECs by TRIzol reagent (Invitrogen,), and the total RNA was reverse transcribed to cDNA using a Transcriptor First Strand cDNA Synthesis Kit(Roche). LightCycler 480 SYBR Green 1 Master Mix(Roche) was used for the amplification of the target genes, and the PCR results were analyzed with the 2-ΔΔCq method and quantitatively analyzed. The mRNA expression levels of target genes were uniformly normalized to GAPDH gene expression.
2.5. Immunofluorescence staining
HUVECs were plated in coverslips and subjected to different treatments. Cells were fixed with 4% formaldehyde and perforated with 0.2% Triton X-100. Then, cells were blocked with 10% goat serum for 1 h, followed by incubation with anti-HMGA2 overnight at 4°C. The Alexa Fluor 488-goat anti-rabbit secondary antibody was employed to label target proteins and 4ʹ,6-diamidino-2-phenylindole was used to stain the cell nucleus. Finally, we used Olympus DX51 fluorescence microscope (Tokyo, Japan) to detect the expression of the protein of interest and capture image.
2.6. MTT
The digested cells were seeded into 96-well plates. After the cells received different treatments, the old medium was discarded and 100 μL of medium containing 10 μL of MTT solution was added to each well. Then, the plates were incubated for 4 h. After that, the cells were treated with100μL of Formazan lysate and incubated for 3–4 h. The absorbance value (A) of each well at a wavelength of 560 nm was measured using a microplate reader to calculate the relative survival rate of the cells.
2.7. Cell migration assay
The influence of HMGA2 on the migration of hypoxia-induced HUVECs was detected through the wound healing assay and Transwell chamber assay. For the wound healing assay, cells were seeded in six-well plates and subjected to different treatments according to the experimental design. Next, the cells were scraped with a 200 μl sterile micropipette tip. Wound recovering was observed and photographed by an inverted microscope at different time point. For Transwell chamber assay, a total of 1 × 105 pretreated cells in 200 µl of serum-free growth medium were seeded into the upper chamber and 500 µl of complete media with 20% fetal bovine serum was added to the lower compartment. After incubation for 24 h, cells in the bottom of the well were fixed with 70% ethanol for 10 min and stained using 0.5% crystal violet for 20 min. Cells in five randomly selected fields were counted at a 200× magnification.
2.8. Detection of caspase-3 activity, nitric oxide production and VEGF
Caspase3 activity in HUVECs was measured according to the manufacturer’s instructions (Beyotime Biotechnology, Shanghai, China). Nitric oxide(NO) production was measured as NADPH dependent nitrate reductase method using the Total Nitric Oxide Assay Kit according to the manufacturer’s instructions(Beyotime Biotechnology, Shanghai, China). The levels of VEGF in HUVECs were determined by the corresponding ELISA kits in accordance with the manufacturer’s instructions (Bioswamp, Wuhan, China).
2.9. TdT-mediated dUTP nick end-labeling (TUNEL) staining
Cells were given different treatments as required by the experiment and stained according to the manufacturer’s instructions of Apoptosis Fluorescein Detection Kit. Briefly, the cells were fixed with 4% paraformaldehyde and incubated with a mixture of ethanol and acetic acid. Subsequently, the cells were incubated with the equilibration buffer, then, given with TdT Enzyme working solution and Stop/WashBuffer working solution. After supplemented with Anti-Digoxigenin Fluorescein working solution for 30 min in the dark, the nuclei were stained with DAPI (Invitrogen, USA). The TUNEL-positive cells were observed using an Olympus fluorescence microscope. The cell slide was magnified 200 times in the microscope. We randomly selected 10 fields and calculated the ratio of the number of TUNEL positive cells to the total number of cells in each field.
2.10. Statistical analysis
All quantitative data were presented as the mean ± SD and were analyzed with SPSS 23.0 software. Student’s unpaired t-test was used for comparisons between two groups. Comparisons among three or more groups were analyzed by two-way ANOVA followed by a post hoc Tukey test. A value of P < 0.05 (two-tailed) was considered statistically significant.
3. Results
3.1. The expression level of HMGA2 in HUVEC under hypoxic condition
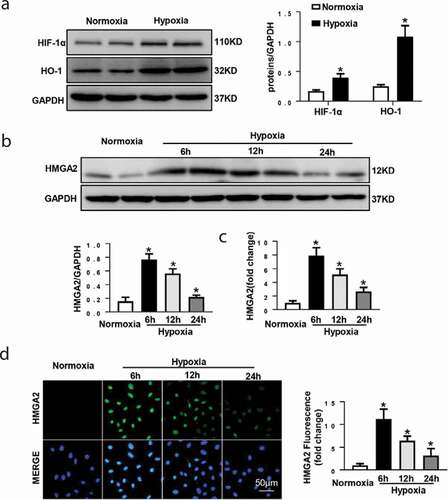
To determine the expression level of HMGA2 in HUVEC under hypoxic condition, the HUVECs received hypoxia induction for 6 h, 12 h, and 24 h. Hypoxia stimulation for 12 h induces an increase in the expression of hypoxia markers [Citation13], including HIF-1α, HO-1 ()).The expression level of HMGA2 increased from 6 h until 12 h after induction of hypoxia then decreased at 24 h after induction of hypoxia (). The above results suggest that HMGA2 may participate in hypoxia-induced myocardial injury during pathological insult.
Figure 1. The expression level of HMGA2 in HUVEC under hypoxic condition
3.2. HMGA2 overexpression accelerates hypoxia-induced cell migration and attenuates hypoxia-induced apoptosis in HUVECs
To further probe the effects of HMGA2 in HUVECs undergoing hypoxia insult, HUVECs were transfected with Ad-HMGA2 to overexpress HMGA2 ()). After hypoxia stimulation for 24 h, the HUVECs revealed a significantly decreased cell viability. In contrast, HMGA2 overexpression improved the hypoxia-induced decrease in cell viability ()).
Figure 2. HMGA2 overexpression accelerated hypoxia-induced cell migration and attenuated hypoxia-induced apoptosis in HUVECs
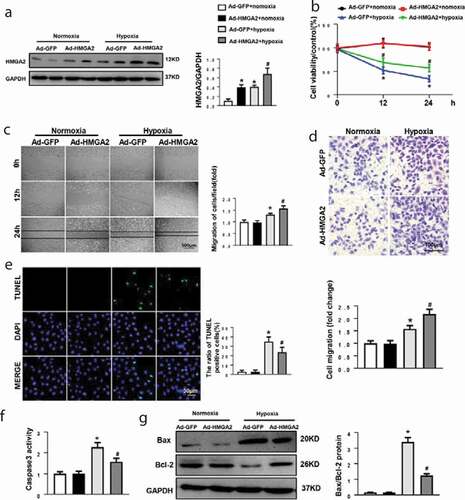
We also evaluate the influence of HMGA2 on migration in HUVECs with exposure to hypoxia. Data indicated that 24 h after hypoxia stimulation in wound healing assay and in Transwell chamber assay, HUVECs showed a large increase in cell migration (). Additionally, in HUVECs overexpressing HMGA2, the number of migrating cells is more significant (). The increase in caspase3 activity triggered by 12 h of hypoxia is attenuated in HMGA2 overexpressing cells ()). TUNEL staining displayed that hypoxia insult for 12 h triggered a striking increase in apoptosis in HUVECs and the overexpression of HMGA2 significantly inhibited the apoptosis ()). Furthermore, western blotting demonstrated that HUVECs with exposure of hypoxia for 12 h resulted in increasing the expression of proapoptotic protein Bax and reducing the expression of antiapoptotic protein Bcl-2. However, overexpression of HMGA2 in HUVECs could ameliorate these alterations ()).
3.3. HMGA2 knockdown suppresses hypoxia-induced cell migration and aggravate hypoxia-induced apoptosis in HUVECs
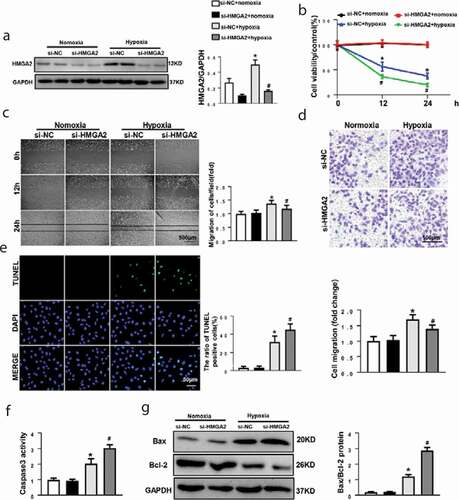
Nest, HUVECs received transfection of small interfering RNA of HMGA2 (si-HMGA2) to downregulate HMGA2 ()). After that, we found that HMGA2 knockdown exacerbated the hypoxia-induced reduction of cell viability ()), HUVECs with HMGA2-knockdown illustrated a weaker migration activity when exposed to hypoxic condition (). Additionally, caspase3 activity ()), the TUNEL positive cells ()) and the ratio of Bax/Bcl-2 ()) increased in the HMGA2-knockdown group.
Figure 3. HMGA2 knockdown suppressed hypoxia-induced cell migration and aggravated hypoxia-induced apoptosis in HUVECs
3.4. HMGA2 regulates HIF-1α/VEGF pathway
To investigate the underlying mechanism of HMGA2 in HUVECs under hypoxia conditions, HIF(including HIF-1α and HIF-2α) and its downstream signaling molecules P-Erk1/2, eNOS and AKT were detected. As a result, the expression of HIF-1α, HIF-2α, P-Erk1/2, VEGF and eNOS proteins increased, while the AKT protein decreased after 12 h of hypoxia induction. Overexpression HMGA2 increased the levels of HIF-1α,VEGF, eNOS and AKT proteins after hypoxia, but had no effect on the expression of HIF-2α ()). Consistent with Western Blotting result, ELISA analysis also showed that HMGA2 increased VEGF levels ()). Additionally, NO production was increased by the Overexpression of HMGA2 ()).
Figure 4. HMGA2 regulated HIF-1α/VEGF pathway
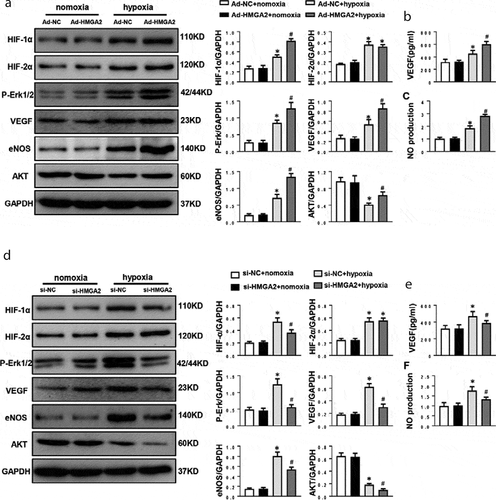
Contrary to the function of HMGA2 overexpression in endothelial cells stimulated by hypoxia, we found that knockdown of HMGA2 downregulated proteins expression of HIF-1α,VEGF, eNOS and AKT, reduced VEGF release, decreased NO production, and also had no effect on the expression of HIF-2α ().
3.5. HMGA2 promotes migration by activating eNOS and inhibits apoptosis by regulating AKT
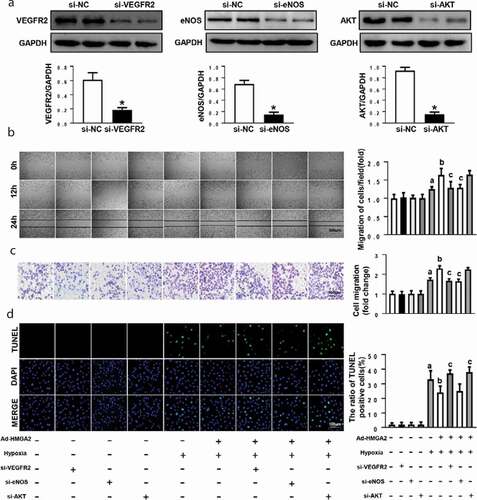
Subsequently, the rescue experiments by knockdown of VEGFR2, eNOS and AKT via transfection of small interfering RNA were performed. Si-VEGFR2, si-eNOS and si-AKT could significantly inhibit the expression of VEGFR2, eNOS and AKT, respectively ()). As illustrated in (b-c), hypoxia-induced increases in cell migration were markedly aggravated in HUVECs overexpressing HMGA2, and the pro-migration effect of HMGA2 was significantly inhibited after VEGFR2 knockdown and eNOS knockdown but not AKT knockdown. Moreover, hypoxia-induced apoptosis was ameliorated in HUVECs overexpressing HMGA2, and the anti-apoptosis effect of HMGA2 was markedly blocked by VEGFR2 knockdown and AKT knockdown but not by eNOS knockdown ()).
Figure 5. HMGA2 promoted migration by activating eNOS and inhibited apoptosis by regulating AKT
4. Discussion
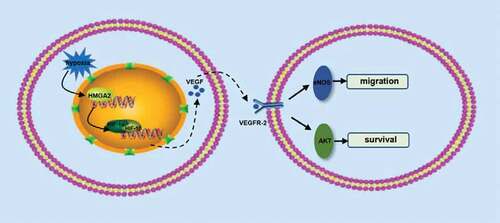
Here, we disclosed that HMGA2 exerted a previously unrecognized role in hypoxia-induced endothelial cell injury. Our findings demonstrated that HMGA2 has a protective effect on endothelial cell injury induced by hypoxia, which can inhibit hypoxia-induced apoptosis and promote cell migration. The protective effect of HMGA2 on endothelial cells may be mediated by activating HIF-1α/VEGF. Inhibition of eNOS abolished HMGA2-mediated pro-migration effect and knockdown of AKT can counteract HMGA2-mediated anti-apoptotic effects, which suggest that HMGA2 exerts pro-migration effect may depend on the activation of eNOS and anti-apoptotic effect may rely on the regulation of AKT .
Figure 6. A putative scheme illustrating the mechanism by which HMGA2 contributes to pro-migration and anti-apoptosis of HUVECs in hypoxia-induced injury
NO is mainly secreted by endothelial cells. Under physiological conditions, eNOS is the main catalytic enzyme for NO production, which allows vascular endothelial cells to continuously release NO and maintain vascular homeostasis [Citation14]. In a hypoxic environment, blood oxygen partial pressure and oxygen saturation decrease, and vascular endothelial cells first sense hypoxia and release NO, causing vasodilation to increase local tissue blood supply [Citation15]. Therefore, NO is a hypoxic messenger molecule between vascular endothelial cells and smooth muscle cells, and mediates the compensation regulation of vascular homeostasis after hypoxia. The mechanism of NO induced by hypoxia in vascular endothelial cells is the result of HIF transcription up-regulating the expression of eNOS gene, and HIF-1 is the core initiating molecule for cells to sense hypoxia [Citation16]. Under normoxic conditions, the HIF-1β subunit is stably expressed in the cytoplasm, while the HIF-1α subunit is degraded by the ubiquitin-protease hydrolysis complex. In a hypoxic state, degradation of the HIF-1α subunit is inhibited, and HIF-1α and HIF-1β subunits form an active HIF-1 transcription complex, which transfers into the nucleus and interacts with the hypoxic response elements in the promoter region of their target genes to regulate transcription and expression of many downstream genes [Citation17]. VEGF is an important downstream target of HIF-1 and HIF-1 can induce the transcription of VEGF in the bone marrow [Citation18]. It has been found that hypoxia activated the HIF-1α/VEGF signaling pathway [Citation19], which was involved in the angiogenesis of myocardial infarction [Citation20]. VEGF could stimulate cell migration and promote the formation of new capillaries [Citation21,Citation22]. Research shows VEGF interacts with VEGFR-2 on the surface of endothelial cells could stimulate the formation of new blood vessels in hypoxic tumor tissues to fulfill the oxygen supply and nutrient requirements [Citation23–25]. Furthermore, VEGF promotes eNOS protein expression and NO production in EPCs (endothelial progenitor cells) and ECs [Citation26,Citation27], which may be mediated by the activation of ERK1/2 signaling pathway [Citation28]. eNOS has been demonstrated to promoted angiogenesis and contribute to enhance capillary density [Citation29] and eNOS/NO signaling is a significant regulator of angiogenesis [Citation30]. Previous studies have shown that HMGA2 can promote the metastasis of pancreatic cancer cells [Citation31] and the migration and invasion of breast cancer cells [Citation32], which suggests that HMGA2 is closely related to the ability of cells to migrate and invade. Our study found that HMGA2 can up-regulate the expression of HIF-1α, thereby activating the VEGF/eNOS signaling pathway, which promote migration of endothelial cell in response to hypoxia insult.
Protein Kinase B (AKT) is an important serine-threonine protein kinase, which is a crucial molecule involved in various biological behaviors such as cell proliferation, survival and life cycle [Citation33]. Once activated, AKT phosphorylated certain downstream molecules to repress the intrinsic pathway of hypoxia-induced apoptosis [Citation34]. It can inactivate pro-apoptotic factors caspase-9 by phosphorylating Ser196 site, thereby inhibiting the cascade activation of Caspase-3/8/9 and exerting anti-apoptosis effect [Citation35,Citation36]. Inhibition of VEGF/AKT signaling pathway can hinder cell proliferation and survival [Citation37]. Results emerging from study of Li indicate that down-regulating HMGA2 expression can induce apoptosis and inhibits cell proliferation [Citation38].In line with that, we also found that the HMGA2 can promote cell survival and decrease cell apoptosis after hypoxia treatment. Moreover, our data showed that knockdown of AKT abolished the anti-apoptotic effects but not the effects of promoting migration, indicating the involvement of the AKT pathway in anti-apoptotic effect induced by HMGA2.
From the above discussion, the conclusion can be reached that HMGA2 promotes cell migration by activating the HIF-1α/VGEF/eNOS signaling pathway and inhibits cell apoptosis through the HIF-1α/VGEF/AKT signaling pathway in response to hypoxia. However, information concerning the genes affected and other potential targets of HMGA2 has not been fully revealed. In the near future, we can perform the comparative transcriptomic analysis of HUVECs to further determine which molecular targets are specifically regulated.
Supplemental Material
Download PDF (294.1 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Cassavaugh J, Lounsbury KM. Hypoxia-mediated biological control. J Cell Biochem. 2011;112(3):735–744.
- Cheng F, Lan J, Xia W, et al. Folic acid attenuates vascular endothelial cell injury caused by hypoxia via the inhibition of ERK1/2/NOX4/ROS pathway. Cell Biochem Biophys. 2016;74(2):205–211.
- Tabata Y, Yoshino D, Funamoto K, et al. Migration of vascular endothelial cells in monolayers under hypoxic exposure. Integr Biol (Camb). 2019;11(1):26–35.
- Hammond SM. Sharpless NE HMGA2, microRNAs, and stem cell aging. Cell. 2008;135(6):1013–1016.
- Oliveira-Mateos C, Sanchez-Castillo A, Soler M, et al. The transcribed pseudogene RPSAP52 enhances the oncofetal HMGA2-IGF2BP2-RAS axis through LIN28B-dependent and independent let-7 inhibition. Nat Commun. 2019;10(1):3979.
- Huang W, Li J, Guo X, et al. miR-663a inhibits hepatocellular carcinoma cell proliferation and invasion by targeting HMGA2. Biomed Pharmacother. 2016;81:431–438.
- Li Y, Zhao Z, Xu C, et al. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett. 2014;355(1):130–140.
- Monzen K, Ito Y, Naito AT, et al. A crucial role of a high mobility group protein HMGA2 in cardiogenesis. Nat Cell Biol. 2008;10(5):567–574.
- Zhu J, Lin X, Yan C, et al. microRNA-98 protects sepsis mice from cardiac dysfunction, liver and lung injury by negatively regulating HMGA2 through inhibiting NF-kappaB signaling pathway. Cell Cycle. 2019;18(16):1948–1964.
- Wong LL, Saw EL, Lim JY, et al. MicroRNA Let-7d-3p contributes to cardiac protection via targeting HMGA2. Int J Mol Sci. 2019;20(7). DOI:10.3390/ijms20071522
- Wu QQ, Xiao Y, Liu C, et al. The protective effect of high mobility group protein HMGA2 in pressure overload-induced cardiac remodeling. J Mol Cell Cardiol. 2019;128:160–178.
- Wu QQ, Xiao Y, Duan MX, et al. Aucubin protects against pressure overload-induced cardiac remodelling via the beta3 -adrenoceptor-neuronal NOS cascades. Br J Pharmacol. 2018;175(9):1548–1566.
- Halder A, Yadav K, Aggarwal A, et al. Activation of TNFR1 and TLR4 following oxygen glucose deprivation promotes mitochondrial fission in C6 astroglial cells. Cell Signal. 2020;75:109714.
- Katusic ZS. Austin SA Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2014;35(14):888–894.
- Jaitovich A, Jourd’heuil D. A brief overview of nitric oxide and reactive oxygen species signaling in hypoxia-induced pulmonary hypertension. Adv Exp Med Biol. 2017;967:71–81.
- Balamurugan K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer. 2016;138(5):1058–1066.
- Luo W, Wang Y. Epigenetic regulators: multifunctional proteins modulating hypoxia-inducible factor-alpha protein stability and activity. Cell Mol Life Sci. 2018;75(6):1043–1056.
- Ahn GO, Seita J, Hong BJ, et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc Natl Acad Sci U S A. 2014;111(7):2698–2703.
- Zhang PC, Liu X, Li MM, et al. AT-533, a novel Hsp90 inhibitor, inhibits breast cancer growth and HIF-1alpha/VEGF/VEGFR-2-mediated angiogenesis in vitro and in vivo. Biochem Pharmacol. 2020;172:113771.
- Du Y, Ge Y, Xu Z, et al. Hypoxia-Inducible Factor 1 alpha (HIF-1alpha)/Vascular Endothelial Growth Factor (VEGF) pathway participates in angiogenesis of myocardial infarction in muscone-treated mice: preliminary study. Med Sci Monit. 2018;24:8870–8877.
- Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2(10):795–803.
- Amini A, Masoumi Moghaddam S, Morris DL, et al. The critical role of vascular endothelial growth factor in tumor angiogenesis. Curr Cancer Drug Targets. 2012;12(1):23–43.
- Izzedine H, Ederhy S, Goldwasser F, et al. Management of hypertension in angiogenesis inhibitor-treated patients. Ann Oncol. 2009;20(5):807–815.
- Cook KM, Figg WD. Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin. 2010;60(4):222–243.
- Bos R, Zhong H, Hanrahan CF, et al. Levels of hypoxia-inducible factor-1 alpha during breast carcinogenesis. J Natl Cancer Inst. 2001;93(4):309–314.
- Yang L, Guan H, He J, et al. VEGF increases the proliferative capacity and eNOS/NO levels of endothelial progenitor cells through the calcineurin/NFAT signalling pathway. Cell Biol Int. 2012;36(1):21–27.
- Kim JH, Lee KS, Lee DK, et al. Hypoxia-responsive microRNA-101 promotes angiogenesis via heme oxygenase-1/vascular endothelial growth factor axis by targeting cullin 3. Antioxid Redox Signal. 2014;21(18):2469–2482.
- Park JS, Hong GR, Baek SW, et al. Expression and regulation of endothelial nitric oxide synthase by vascular endothelial growth factor in ECV 304 cells. J Korean Med Sci. 2002;17(2):161–167.
- Jujo K, Ii M, Sekiguchi H, et al. CXC-chemokine receptor 4 antagonist AMD3100 promotes cardiac functional recovery after ischemia/reperfusion injury via endothelial nitric oxide synthase-dependent mechanism. Circulation. 2013;127(1):63–73.
- Kolluru GK, Sinha S, Majumder S, et al. Shear stress promotes nitric oxide production in endothelial cells by sub-cellular delocalization of eNOS: A basis for shear stress mediated angiogenesis. Nitric Oxide. 2010;22(4):304–315.
- Xiao G, Wang X, Yu Y. CXCR4/Let-7a axis regulates metastasis and chemoresistance of pancreatic cancer cells through targeting HMGA2. Cell Physiol Biochem. 2017;43(2):840–851.
- Wu J, Zhang S, Shan J, et al. Elevated HMGA2 expression is associated with cancer aggressiveness and predicts poor outcome in breast cancer. Cancer Lett. 2016;376(2):284–292.
- Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868.
- Matsui T, Li L, Del Monte F, et al. Adenoviral gene transfer of activated phosphatidylinositol 3ʹ-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100(23):2373–2379.
- Nakano N, Matsuda S, Ichimura M, et al. PI3K/AKT signaling mediated by G proteincoupled receptors is involved in neurodegenerative Parkinson’s disease (Review). Int J Mol Med. 2017;39(2):253–260.
- Sangawa A, Shintani M, Yamao N, et al. Phosphorylation status of Akt and caspase-9 in gastric and colorectal carcinomas. Int J Clin Exp Pathol. 2014;7(6):3312–3317.
- Wan X, Zhu Y, Zhang L, et al. Gefitinib inhibits malignant melanoma cells through the VEGF/AKT signaling pathway. Mol Med Rep. 2018;17(5):7351–7355.
- Li Y, Wang D, Ren H, et al. Metformin alleviates breast cancer through targeting high-mobility group AT-hook 2. Thorac Cancer. 2020;11(3):686–692.