
ABSTRACT
Combining targeted therapeutic agents is an attractive cancer treatment strategy associated with high efficacy and low toxicity. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is an essential factor in DNA damage repair. Studies from us and others have revealed that DNA-PKcs also plays an important role in normal mitosis progression. Histone deacetylase (HDACs) inhibitors commonly lead to mitotic aberration and have been approved for treating various cancers in the clinic. We showed that DNA-PKcs depletion or kinase activity inhibition increases cancer cells’ sensitivity to HDACs inhibitors in vitro and in vivo. DNA-PKcs deficiency significantly enhances HDACs inhibitors (HDACi)-induced mitotic arrest and is followed by apoptotic cell death. Mechanistically, we found that DNA-PKcs binds to HDAC6 and facilitates its acetylase activity. HDACi is more likely to impair HDAC6-induced deacetylation of HSP90 and abrogate HSP90’s chaperone function on Aurora A, a critical mitotic kinase that regulates centrosome separation and mitotic spindle assembly in DNA-PKcs-deficient cells. Our current work indicates crosstalk between DNA-PKcs and HDACs signaling pathways, and highlights that the combined targeting of DNA-PKcs and HDACs can be used in cancer therapy.
Abbreviations: DNA-PKcs, DNA-dependent protein kinase catalytic subunit, HDACs, Histone deacetylases, DSBs, DNA double-strand breaks, ATM, ataxia telangiectasia mutated, ATR, ATM-Rad3-related
KEYWORDS:
Introduction
DNA-dependent protein kinase catalytic subunit (DNA-PKcs), a member of the PI3K-like kinase (PIKK) family, is widely known as a key component of non-homologous end joining (NHEJ), the primary pathway through which DNA double-strand breaks (DSBs) induced by endogenous (i.e. replication stress, metabolic reactions) or exogenous (i.e. ionizing radiation, IR) factors are repaired [Citation1]. In response to DNA DSBs, DNA-PKcs rapidly binds to lesions and is activated by the Ku70/Ku80 complex, which has high affinity for dsDNA ends. When recruited to damaged sites, DNA-PKcs is phosphorylated at Thr2609 and Ser2056 clusters by ataxia telangiectasia mutated (ATM) kinase and DNA-PKcs itself, respectively [Citation2,Citation3]. Phosphorylation of DNA-PKcs induces a conformational change and destabilizes the interaction between DNA-PKcs and DNA, which allows other NHEJ proteins to access DNA and, thus, facilitates processing and ligation [Citation4–7]. The Thr2609 cluster can also be phosphorylated by the ATM-Rad3-related (ATR) kinase in replicating cells when exposed to UV-induced replication stress [Citation8]. Recruiting DNA-PKcs to replication forks in response to replication stress depends on its binding with the apoptosis mediator p53-induced protein with a death domain (PIDD) protein. Disrupting the DNA-PKcs-PIDD complex not only leads to the dissociation of DNA-PKcs from ATR-DNA but also impairs ATR activation, DNA-PKcs phosphorylation, and intra-S-checkpoint maintenance [Citation9]. In addition, recent studies have revealed the novel functions of DNA-PKcs in regulating mitosis. DNA-PKcs is activated during normal mitosis progression in a Ku and DNA damage-independent manner [Citation10]. Interestingly, activated DNA-PKcs not only phosphorylates itself on both Thr2609 and Ser2056 sites [Citation11], but also stimulates Chk2-Brca1 [Citation12] and PLK1-Cdk1 [Citation13] signaling pathways. A mutual regulation between PLK1 and DNA-PKcs seems to occur. PLK1 also phosphorylates DNA-PKcs at its Ser3205 site in vitro. Phosphorylated DNA-PKcs are observed at all these sites (Thr2609, Ser2056, and Ser3205) to localize at the mitotic spindle apparatus [Citation10], suggesting that DNA-PKcs might play a critical role in regulating mitotic spindle assembly. Consistently, loss of DNA-PKcs expression or inhibition of DNA-PKcs kinase activity knowingly leads to uncontrolled microtubule organization around the centrosome, abnormal mitotic chromosome congression, and mitotic transition delay [Citation10–12]. The precise regulatory network of DNA-PKcs in mitosis is poorly understood and needs to be further explored.
Histone deacetylases (HDACs) remove acetyl groups from the lysine ε-amino acid on histone or non-histone proteins and are known to play important roles in different biological processes, including chromatin remodeling, gene transcription, DNA damage repair, apoptosis, and cell cycle progression [Citation14]. Because of their elevated levels in many cancer types, HDACs are attractive targets for clinical cancer treatment [Citation15,Citation16]. Histone deacetylase (HDAC) inhibitor (HDACi) showed anti-proliferative activity against cancer cells by perturbing cell cycle progression and inducing apoptosis [Citation14]. HDACi’s influence on mitosis has been observed in experiments in which impaired sister chromatid separation and chromosome segregation defects were induced in cells exposed to a pan-HDACi Trichostatin A (TSA) [Citation17]. These studies showed that HDACi treatment leads to defects in microtubule (MT)-kinetochore attachment and aberrant mitosis by releasing HP1 from the centromere region and destructing pericentric heterochromatin, which is critical in kinetochore organization [Citation18–20]. In addition to acting as microtubule anchors, kinetochores have shown to be important in generating spindle assembly checkpoint (SAC) signals. SAC is a cell cycle surveillance pathway that prevents premature chromosomal segregation before chromosomes firmly attach to microtubules from the bipolar spindle [Citation21]. Accordingly, HDACi treatment leads to aberrantly separated sister chromatids even in cells with activated SAC and, in turn, premature exit mitosis accompanied by failed cytokinesis [Citation22,Citation23]. HDAC-induced deacetylation of the H3 N-terminal tail is required for initiating Aurora B-mediated phosphorylation of H3 on Ser10, which is essential for mitotic chromosome condensation [Citation20]. HSP90, a molecular chaperone that prevents multi-protein degradation, is also an HDAC substrate. HDACi-induced high level acetylation of HSP90 attenuates its chaperone function. Both HDACi and HSP90 inhibitors, such as 17-AAG, have been reported to cause destabilization of HSP90 clients, including Aurora A and PLK1 [Citation24,Citation25]. Because of the interaction between HDACs and several important mitotic signal pathways, the combination of HDACi with other mitosis targeted agents has been recognized as a potentially effective therapeutic strategy for treating chemo-resistant cancers [Citation26–28]. Considering the important roles of DNA-PKcs in mitosis progression, we evaluated the combined cell killing effects of HDACi and DNA-PKcs inhibitor against cancer cells in vitro and in vivo, and further revealed the crosstalk between DNA-PKcs and HDAC6-HSP90 signaling pathway, as well as their role in spindle assembly during mitosis.
Materials and methods
Cell lines and treatment
Human cervical cancer HeLa cells were obtained from ATCC; human colorectal carcinoma HCT116 cells and derivative DNA-PKcs−/-, and Ligase 4−/ – cells were kindly provided by Pro. Hendrickson EA [Citation29]. All cells were maintained in a-minimum essential medium containing 10% fetal bovine serum and penicillin/streptomycin in a humidified incubator at 37°C with 5% CO2. Cells were treated with specified concentrations of Trichostatin A (TSA) or suberanilohydroxamic acid (SAHA) (Sigma, St Louis, MO, USA) for 2, 8, and 16 hours. In certain experiments, cells were treated together with DNA-PKcs or ATM kinase inhibitors (Nu7441 or Ku55933, respectively) (Sigma-Aldrich, St Louis, MO, USA). Cell transfection with small inhibitory RNA (siRNA) oligonucleotides or expression constructs of DNA-PKcs was performed using Lipofectamine 3000 (ThermoFisher Scientific, Carlsbad, CA, USA), according to the manufacturer’s instructions. SiRNA oligonucleotides against DNA-PKcs were used as previously described [Citation30].
Immunoblotting, immunofluorescent staining, and antibodies
Whole-cell lysate preparation and western blotting were performed as previously described [Citation11]. For immunofluorescent staining, cells were grown on poly-D-lysine-coated culture slides (BD Pharmingen, San Diego, CA, USA), washed in phosphate-buffered saline (PBS), fixed in PBS that contained 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, and blocked in PBS that contained 5% bovine serum albumin. Cells were incubated with primary antibodies for 2 hours, washed with PBS, and incubated with Alexa-568 – and Alexa-488-conjugated secondary antibodies (ThermoFisher Scientific, Carlsbad, CA, USA) for 1 hour. Cells were washed with PBS and mounted in Vectashield mounting medium with 4,6 – diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). Images were acquired using a Zeiss AxioImager M2 microscope system equipped with a Plan-Apochromat 63/NA 1.40 objective, an AxioCam MRm CCD camera and AxioVision software (Carl Zeiss, Oberkochen, Germany). Anti-Aurora A total, anti-PARP, anti-HSP90, anti-Acetyl lysine, anti-HDAC6 (Cell Signaling, Beverly, MA, USA), anti-α-tubulin, anti-acetylated-α-tubulin, anti-Flag (Sigma, St Louis, MO, USA), anti-phospho-histone H3 (EMD Millipore, Billerica, MA, USA), anti-Ku80 (Santa Cruz Biotechnology, TX, USA), anti-Crest (ImmunoVision, Springdale, AR, USA) antibodies were purchased from the indicated vendors. Antibodies against total DNA-PKcs were used as previously described [Citation11].
Clonogenic survival and MTT cell proliferation assays
Exponentially grown HCT116 cells were trypsinized, counted, and plated into 60-mm dishes in triplicate with indicated titration of TSA. Cells were fixed at 10–14 days and stained with 4% formaldehyde in PBS containing 0.05% crystal violet. Colonies containing more than 50 cells were scored under a microscope. For cell proliferation assays, 1 × 104 cells per well were seeded in a 96-well plate. Cells were cultured with an indicated titration of TSA, SAHA, Tubastatin A, or Nu7441 for 72 hours, and then analyzed by MTT assay [Citation12].
Mitotic index analysis
Cells were fixed in 70% ethanol, washed with PBS, and incubated with an anti-pH3 antibody for 3 hours followed by fluorescein isothiocyanate (FITC)-conjugated secondary antibody for 1 hour. Cells were then incubated in propidium iodide (PI) solution (0.1 mg/ml RNase A, 0.1% Triton X-100, and 20 mg/ml PI in PBS) for 30 minutes at 37°C. The mitotic cell population was analyzed by flow cytometry.
Apoptosis detection
Parental HCT116, DNA-PKcs−/-, and Ligase 4−/ – cells were treated with or without 20 ng/ml TSA for 16 hours and then harvested. Phycoerythrin (PE) Annexin V Apoptosis Detection Kit (BD, Pharmingen, San Diego, CA, USA) was used for detecting apoptosis according to the manufacturer’s protocol. After staining by PE-conjugated Annexin V and 7-Amino-Actinomycin (7-AAD), cells were analyzed by flow cytometry.
Immunoprecipitation assay
HeLa cells were lysed in lysis buffer [50 mM Tris HCl (pH 7.5), 150 mM NaCl, 1% Tween 20, 0.5% NP-40, and protease inhibitor cocktail] and incubated with 1 μg control IgG or target antibodies at 4 °C overnight; they were then incubated with protein A/G sepharose beads (Roche, Branford, CT) for 1 hour. The sepharose beads were washed with lysis buffer three times and resuspended in SDS-PAGE loading buffer for immunoblot analysis. For mapping the interaction domain between DNA-PKcs and HDAC6, Flag-tagged DNA-PKcs or HDAC6 truncated proteins were expressed in HeLa cells. Co-IP assay was performed using anti-Flag antibodies followed by immunoblot analysis.
Quantitative real-time PCR (qRT-PCR) analysis
Quantitative real-time PCR was performed as previously described [Citation31]. Briefly, extracted RNA was reverse transcribed to cDNA using a Prime ScriptTM RT reagent Kit (TAKARA, Otsu, Japan) according to the manufacturer’s protocol. The cDNA reactions were amplified with a SYBR Green Real-Time PCR assay kit (QIAGEN, Hilden, Germany). The expression levels of HDAC6 and GAPDH (internal standard) mRNAs were measured by real-time PCR in triplicates using a 7500 Real-Time PCR system (Applied Biosystems, Foster City, USA) with the following primers [Citation32]: HDAC6, 5ʹ-TCA GGT CTA CTG TGG TCG TT-3ʹ (3540–3559, forward), 5ʹ-TCT TCA CAT CTA GGA GAG CC-3ʹ (3691–3672, reverse); GAPDH, 5ʹ-AAA TCA AGT GGG GCG ATG CTG-3ʹ (forward), 5ʹ-GCA GAG ATG ATG ACC CTT TTG-3ʹ (reverse).
HDAC6 deacetylase activity detection
HDAC6 activity was measured using a HDAC6 Colorimetric Activity Assay kit (Biovision, Milpitas, CA, USA) as previously described [Citation33]. Briefly, HDAC6 was immunoprecipitated from HCT116 or HeLa cells treated with DMSO or Nu7441 for 16 hours, and mixed with an HDAC6 colorimetric substrate consisting of polypeptide chains with acetylated lysine side chains. The reaction was terminated by incubating with a developer for colorimetric detection at 405 nm. Relative HDAC6 activities were calculated.
Mouse xenograft tumors study
HCT116 cells (5x106 in 100 μl PBS) were injected subcutaneously into the right flank of female athymic nude mice. When tumors reached an optimal size (150–200 mm3), tumor-bearing mice were randomized to different treatment groups (N = 5): the control group received 0.9% PBS; the Nu7441 group received 25 mg/kg/day for 5 days by intraperitoneal (ip) injection; the SAHA group received 50 mg/kg/day for 5 days by ip injection; the combined Nu7441 and SAHA treatment received. The relative tumor volume was calculated by the ratio of tumor volume at the indicated day and tumor volume on the first day of treatment. The mice study was approved by the laboratory animal welfare ethics review at Soochow University. Data are presented as mean ± SEM.
Statistical analysis
Data is presented as the mean ± SD of at least three independent experiments. The results were tested for significance using the unpaired Student’s t test.
Results
Loss of DNA-PKcs enhances HDACi-induced inhibition of cancer cell proliferation
Both DNA-PKcs and HDACs signaling pathways can promote cancer cell proliferation by facilitating cell cycle progression. They play a role in various steps of mitosis, which also probably contributes to resistance against mitotic stress. We hypothesized that the sensitivity of cancer cells to HDACi might correlate with the status of DNA-PKcs. In order to test this possibility, human colon cancer cells (HCT116) and derivative DNA-PKcs knockout (DNA-PKcs−/-) cells were treated with increasing concentrations of TSA, and cell viability was determined using the MTT assay. Our findings indicated that DNA-PKcs−/ – cells were more sensitive to TSA treatment than parental HCT116 cells (). Because HDACi treatment can induce DNA damage and prevent DNA lesion repair [Citation34,Citation35], NHEJ-defective Ligase4−/ – cells were also tested but did not show increased sensitivity toward TSA (). Similar results were obtained using a clonogenic survival assay (), revealing that DNA-PKcs plays an NHEJ-independent role in regulating cancer cells’ sensitivity to HDACi. This notion was further investigated by using another pan-HDACi SAHA (or Vorinostat), which has been approved by the U.S. Food and Drug Administration (FDA) for treating cancer in the clinic. Exposure to SAHA also inhibited DNA-PKcs−/ – cell growth more significantly than parental HCT116 and Ligase 4−/ – cell growth (). In addition, MTT assay results indicated that DNA-PKcs-deficient M059J cells were more sensitive to SAHA and TSA (Fig. S1A and S1B) than DNA-PKcs-proficient M059K cells. To determine whether inhibiting DNA-PKcs’ kinase activity can sensitize cancer cells to HDACi, HeLa cells were treated with DNA-PKcs kinase inhibitor Nu7441 in combination with increasing doses of HDACi. As expected, Nu7441 exposure further augmented SAHA’s cell killing effect () or TSA (Fig. S1C). Finally, we analyzed tumor growth delay to further investigate the combined cell killing effect of Nu7441 and HDACi in vivo. HCT116 cells were implanted in athymic nude mice to induce subcutaneous tumor formation and then treated with Nu7441 and SAHA either alone or in combination. Tumor volumes (and animal body weights) were measured twice every week, and the relative tumor volume (RTV) was calculated. Our study revealed that combining Nu7441 and SAHA delayed tumor growth more significantly than either Nu7441 or SAHA alone (). Based on these results, targeting DNA-PKcs and HDACs signaling pathways shows promising outcomes as potential cancer treatment.
Figure 1. DNA-PKcs-deficiency or kinase activity inhibition sensitizes cancer cells to the HDAC inhibitor. (a and c) HCT116, DNA-PKcs−/-, and Ligase4−/ – cells were treated with TSA at the indicated concentration. (a) or SAHA (c) for 72 hours, and viability was determined, **P< 0.01. (b) HCT116, DNA-PKcs−/-, and Ligase 4−/ – cells were exposed to TSA at the indicated concentration for two weeks and were analyzed for clonal survival ability, **P< 0.01. (d) HeLa cells were exposed to increasing dosages of SAHA alone or in combination with 5 or 10 μM of Nu7441, and cell viability was determined by MTT assay, *P< 0.05, **P< 0.01. (e) Nu7441 in combination with SAHA treatment leads to significant tumor growth delay in HCT116 cells but not in Nu7441 or SAHA treatment alone, **P< 0.01
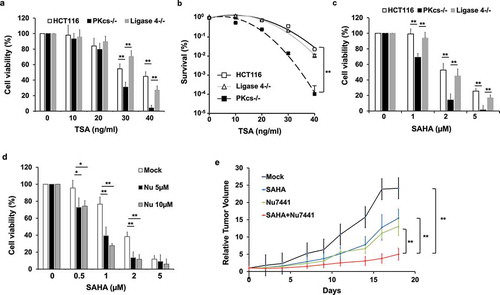
DNA-PKcs-deficiency accelerates HDACi treatment induced apoptosis
HDACi treatment commonly leads to apoptotic cell death. HCT116, DNA-PKcs−/-, and Ligase 4−/ – cells were treated with TSA for 16 hours to measure the induction of apoptosis by PE-labeled Annexin V in conjunction with DNA dye 7-Amino-Actinomycin (7-AAD). TSA induced cellular apoptosis more dramatically in DNA-PKcs-deficient cells at early (Annexin V positive and 7-AAD negative) and late (Annexin V positive and 7-AAD positive) stages than in parental HCT116 or Ligase 4−/ – cells (). An increase of TSA-induced apoptosis was also evidenced by cleavage of PARP-1 through immunoblot analysis. We noticed that both TSA and SAHA led to greater PARP-1 cleavage in DNA-PKcs−/ – cells than in HCT116 or Ligase 4−/ – cells (). Combining Nu7441 and TSA also significantly enhanced PARP-1 cleavage in HeLa cells, whereas ATM inhibitor Ku55933 only slightly increased TSA-mediated PARP-1 cleavage, which may be attributed to impaired DNA repair efficacy under ATM-deficient conditions (Fig. S2). These results suggest that combined inhibition of DNA-PKcs and HDAC dramatically induced apoptotic cell death.
Figure 2. The HDAC inhibitor leads to increased apoptosis in DNA-PKcs-deficient HCT116 cells. (a) TSA treatment (20 ng/ml, 16 hours)-induced apoptosis was determined by 7-AAD and Annexin V staining in flow cytometry. (b) Quantitative analysis of apoptotic cells (Annexin V-positive cells) was conducted in three independent experiments. **P< 0.01. (c and d) Apoptosis was determined by PARP-1 cleavage product (c-PARP) in cells exposed to TSA (c) or SAHA (d) for 16 hours
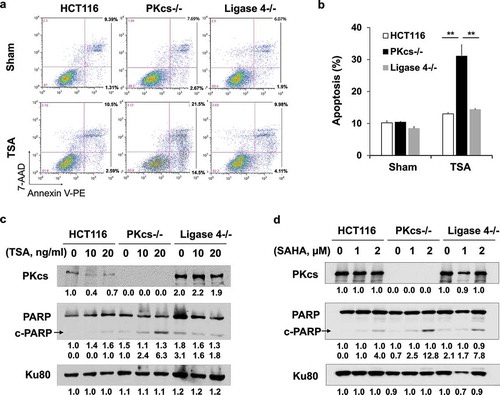
DNA-PKcs-deficiency augments HDACi-induced mitotic arrest and monopolar spindle formation
Aberrant mitosis and long-term stalling of the cell cycle in mitosis can be induced by HDACi and are associated with apoptotic cell death. To discriminate between mitotic arrest and cell death, HCT116, DNA-PKcs−/-, and Ligase 4−/ – cells were treated with TSA and analyzed for mitotic index by flow cytometry. Overnight (16 hours) incubation with TSA at 20 ng/ml induced mitotic arrest in DNA-PKcs−/ – cells, but not in HCT116 and Ligase 4−/ – cells (). Activation of the spindle assembly checkpoint by abnormal mitotic spindle is the most common reason for prolonged mitotic arrest. To further explore the involvement of DNA-PKcs in HDACi-mediated mitotic spindle disorganization, HCT116, DNA-PKcs−/-, and Ligase 4−/ – cells were exposed to TSA and treated for immunofluorescence staining with anti-α-tubulin and anti-Crest antibodies. During prometaphase to metaphase, bipolar spindles are formed and kinetochores of sister chromatid pairs attach to microtubules from opposite poles to align the chromosome at the central region of the spindle. However, increased amounts of the monopolar spindle were observed in all cell lines that responded to HDACi. Among them, DNA-PKcs−/ – cells displayed a higher proportion of the monopolar spindle than HCT116 and Ligase 4−/ – cells (), indicating a defect of centrosome duplication in DNA-PKcs−/ – cells.
Figure 3. The HDAC inhibitor induces mitotic arrest and monopolar spindle in DNA-PKcs-deficient HCT116 cells. (a) HCT116, DNA-PKcs−/-, and Ligase 4−/ – cells were exposed to sham or TSA (20 ng/ml) for 16 hours. The mitotic index was measured by flow cytometry with PI and anti-phospho-Histone H3 (pH3) antibody staining. Inset, % of mitosis cells. (b) Quantitative analysis of mitotic index was performed in three independent experiments. **P< 0.01. (c) The representative images show normal and monopolar spindle mitosis stained with anti-α-tubulin and anti-crest antibodies. (d) The percentage of monopolar spindle mitotic cells was determined with or without TSA treatment. Results were generated in three independent experiments. **P< 0.01
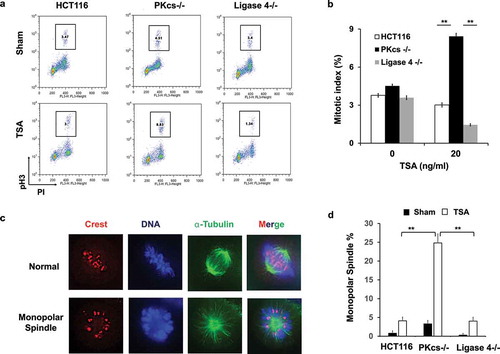
HDACi selectively downregulated Aurora A in DNA-PKcs-deficient cells
The Aurora A kinase localizes in mitotic centrosomes and plays a critical role in centrosome separation before assembling bipolar spindles [Citation36]. HDACi increases acetylation of the HSP90 chaperone and disrupts the interaction between Aurora A and HSP90, leading to subsequent destabilization of Aurora A [Citation24]. Therefore, the expression level of Aurora A was determined by immunofluorescence staining using an anti-Aurora A antibody. The fluorescence intensity of centrosomal Aurora A was analyzed, revealing that the average intensity of Aurora A was significantly lower in DNA-PKcs−/ – cells than in HCT116 or Ligase 4−/ – cells (). Consistently, immunoblot analysis indicated dramatic downregulation of Aurora A protein levels in DNA-PKcs−/ – cells upon TSA treatment (). To examine whether DNA-PKcs-deficiency contributes to HDACi-induced hyperacetylation of HSP90, the proteins were immunoprecipitated from cells that were either treated or not treated with TSA for 2 hours, which was followed by immunoblotting using anti-acetylated lysine and anti-HSP90 antibodies. This result proved that HDACi causes HSP90 acetylation in DNA-PKcs-deficient cells but not in HCT116 or Ligase 4−/ – cells (). We further isolated the Aurora A co-IP complex from cells that were either treated or not treated with TSA for only 2 hours, when Aurora A protein levels remained unaffected by TSA treatment (). The amount of HSP90 co-IP with Aurora A was significantly decreased in TSA-treated DNA-PKcs−/ – cells (). These data support our hypothesis that loss of DNA-PKcs attenuates protein association between Aurora A and HSP90, leading to Aurora A destabilization.
Figure 4. The HDAC inhibitor destabilizes Aurora A protein in DNA-PKcs-deficient HCT116 cells through HSP90 hyperacetylation. (a) HCT116, DNA-PKcs−/-, and Ligase 4−/ – cells treated with TSA (20 ng/ml, 8 hours) were immunostained with anti-α-tubulin (α-Tub) and anti-Aurora A (Aur A) antibodies. (b) The fluorescent intensities of Aurora A staining were determined from metaphase cells (N ≥ 100) and presented quantitatively. **P< 0.01. (c) TSA-treated HCT116 cells were immunoblotted for the expression of Aurora A and cleaved PARP. (d) HCT116 cells were collected at 2 hours after treating with TSA (20 ng/ml). Cells lysates were immunoprecipitated with an anti-HSP90 antibody followed by immunoblotting with anti-acetyl-lysine and anti-HSP90 antibodies. (e) Cells lysates were immunoprecipitated with an anti-Aurora A antibody or control IgG, and immunoblotted with anti-Aurora A and anti-HSP90 antibodies (left). The right panel shows “input” of Aurora A and HSP90 in cell lysates
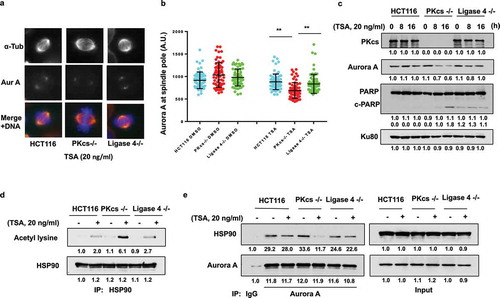
DNA-PKcs negatively regulates HDAC6 expression but facilitates its deacetylase activity
We further investigated the expression levels of HDAC6 in DNA-PKcs-deficient cells because HDAC6 is responsible for HSP90 deacetylation. We observed increased expression of HDAC6 in DNA-PKcs−/ – cells. Consistently, the acetylation of α-tubulin, a well-known target of HDAC6, was dramatically reduced in DNA-PKcs−/ – cells but not in HCT116 or Ligase 4−/ – cells (). DNA-PKcs-deficiency-induced upregulation of HDAC6 was further observed in DNA-PKcs-deficient M059J cells (Fig. S3). Similarly, siRNA-mediated depletion of DNA-PKcs in HeLa cells promoted HDAC6 expression while inhibiting α-tubulin acetylation (). To further examine the role of DNA-PKcs kinase activity in HDAC6 expression, HeLa cells were incubated for 16 hours with Nu7441, which effectively enhanced HDAC6 protein levels and decreased acetyl-α-tubulin in HeLa cells (). HDAC6 upregulation in DNA-PKcs-deficient cells is probably due to increased mRNA expression, as revealed by real time RT-PCR analysis in HCT116 and DNA-PKcs−/ – cells ().
Figure 5. DNA-PKcs negatively regulates HDAC6 expression but promotes its deacetylase activity in vitro. (a) HCT116 cell lysates were immunoblotted with anti-HDAC6, anti-acetylated-α-tubulin (Ac-Tub), anti-α-tubulin (α-Tub), anti-Ku80, and anti-DNA-PKcs antibodies. (b) HeLa cells were transfected with either control or specific siRNA against N – or C-terminus of DNA-PKcs mRNA for 48 hours, and were immunoblotted as shown. (c) HCT116 cells treated with Nu7441 or Ku55933 (10 μM each, 24 hours) were immunoblotted. (d) Expression of HDAC6 mRNA in HCT116 and DNA-PKcs−/ – cells was determined by RT-PCR analysis. **P< 0.01. (e, f) HDAC6 proteins immunoprecipitated from HCT116 and DNA-PKcs−/ – cells (e) or HeLa cells ± Nu7441 (10 µM, 24 hours) (f) were analyzed by colorimetric deacetylation assay. Upper panels show HDAC6 protein levels in the assay. **P< 0.01. (g) HCT116 cells treated with TubA for 72 hours were analyzed by MTT cell viability assay. **P< 0.01
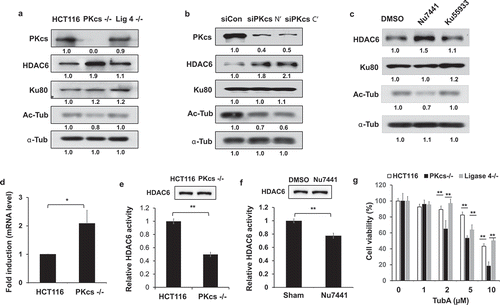
To validate whether DNA-PKcs regulates the deacetylase activity of HDAC6, HDAC6 proteins were immunoprecipitated from HCT116 and DNA-PKcs−/ – cell lysates. They were then processed through colorimetric deacetylase assay in vitro, as previously described [Citation33]. The relative deacetylase activity was calculated by detecting the fluorescent intensity of the released fluorophore. Unexpectedly, depletion or inhibition of DNA-PKcs impaired the deacetylases activity of HDAC6 (). Our data further showed that DNA-PKcs−/ – cells had higher sensitivity to an HDAC6-specific inhibitor Tubastatin A (TubA) than HCT116 or Ligase 4−/ – cells (), indicating dysregulation of HDAC6 without normal DNA-PKcs activity.
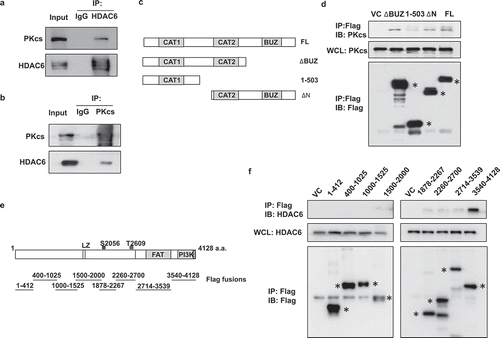
To investigate whether DNA-PKcs can physically interact with HDAC6, HeLa cell lysates were treated with Co-IP with anti-HDAC6 or anti-DNA-PKcs antibodies. Immunoblotting analysis proved that HDAC6 and DNA-PKcs could be mutually co-purified and are probably in the same protein complexes (). This notion was further supported by mapping the protein-protein association between HDAC6 and DNA-PKcs. Flag-tagged full length and truncated HDAC6 constructs were transfected into HeLa cells, followed by Co-IP treatment with an anti-Flag antibody and immunoblotting. Results indicated that the second catalytic domain of HDAC6 can interact with endogenous DNA-PKcs in HeLa cells ( and D). Conversely, anti-Flag antibody Co-IP treatment with various Flag-DNA-PKcs fusion fragments revealed that HeLa endogenous HDAC6 interacted with the C’-terminal PI3K domain (3540–4128) of DNA-PKcs. Taken together, our results indicate that DNA-PKcs negatively regulates HDAC6 expression. Meanwhile, DNA-PKcs can regulate HDAC6 deacetylases activity, probably through direct protein-protein interaction.
Figure 6. DNA-PKcs interacts with HDAC6. (a and b) HeLa cell lysates were immunoprecipitated with anti-HDAC6 (a) or anti-DNA-PKcs (b) followed by detecting DNA-PKcs and HDAC6 through immunoblotting. (c) Schema of Flag-tagged HDAC6 expression constructs. (d) Full length and truncated Flag-HDAC6 fusion proteins expressed in 293 T cells were immunoprecipitated and analyzed by immunoblotting, as indicated. (e) Schema of truncated Flag-DNA-PKcs expression constructs. (f) Various Flag-DNA-PKcs truncations were transfected into 293 T cells, immunoprecipitated, and analyzed by immunoblotting
Discussion
The HDAC6-HSP90 chaperone axis plays a critical role in safeguarding the structural maturity and stability of many client proteins, including nuclear receptors, transcription factors, and selected kinases [Citation37]. The HDAC6-HSP90 axis is known to be involved in multiple post-translation modifications across a diverse array of crucial cellular activities and has been exploited as a therapeutic target for treating different cancers [Citation38]. The central schema is that HSP90 deacetylation by HDAC6 affects the affinity of HSP90 with co-chaperones and client proteins. As an example, HSP90 deacetylation promotes protein homeostasis of mitotic kinase Aurora A, which is essential for centrosomal maturation and separation during mitosis. Therefore, inhibiting or depleting Aurora A leads to monopolar spindle formation [Citation39]. Our study showed that DNA-PKcs can help modulate the HDAC6-HSP90 axis because DNA-PKcs-deficiency enhances HDACi’s cell killing effects, Aurora A’s loss, and consequently, aberrant mitosis and programmed cell death. This is consistent with our previous reports on DNA-PKcs kinase activation in mitosis facilitating chromosome congression and mitotic progression, and DNA-PKcs affecting microtubule dynamic regulation via the Chk2-Brca1 signal pathway [Citation11,Citation12]. DNA-PKcs probably intersects with the HDAC6-HSP90 axis at multiple regulation layers. For example, HDAC6-mediated deacetylation of α-tubulin affects microtubule dynamics, yet it is unclear how DNA-PKcs and HDAC6 coordinate and regulate mitotic spindle organization. Furthermore, HSP90 is known to be phosphorylated by DNA-PKcs at threonine 5 and 7, and this phosphorylation influences its ability to carry out DNA damage response and programmed cell death [Citation40,Citation41]. The relationship between DNA-PKcs-mediated phosphorylation and HDAC6-regulated acetylation of HSP90 is unclear and warrants further investigation.
Our results showed DNA-PKcs’ negative effect on HDAC6 gene expression (). The nuclear factor-κB (NF-κB)-binding element exists within the HDAC6 promoter, and proinflammatory cytokine-dependent NF-κB activation has been shown to stimulate HDAC6 gene expression [Citation42]. Liu et al. reported that DNA-PKcs phosphorylates IκBα and increases its affinity to NF-κB, thus limiting the transcriptional properties of NF-κB [Citation43]. DNA-PKcs may negatively regulate NF-κB activation and restrict HDAC6 gene expression. We reported that DNA-PKcs-deficiency increased oxidative stress and hyperactivation of the ATM kinase [Citation44], both of which can potentiate NF-κB activity and increase HDAC6 expression. In addition, recent RNA immunoprecipitation (RIP)-coupled sequencing analysis revealed that DNA-PKcs may associate with mRNA expression [Citation45], though it is unclear whether DNA-PKcs can directly affect the stability of HDAC6 mRNA. Alternatively, increased expression of HDAC6 in DNA-PKcs-deficient cells can be due to a complementary effect against HDAC6’s reduced acetylase activity. On the other hand, HDAC6 can participate in various biological processes in an acetylase activity-independent manner. For example, the CAT1 domain acts as an E3 ligase that ubiquitinates and modulates the stability of MutS protein homolog 2 (MSH2), a crucial member of the mismatch repair pathway [Citation46]. DNA-PKcs may also influence HDAC6’s role other than its deacetylase activity, such as E3 ligase function.
Our work also reveals a direct association between DNA-PKcs and HDAC6 (), and a functional requirement for DNA-PKcs to promote HDAC6 deacetylase activity (). This is seemingly paradoxical for the upregulation of HDAC6, which is associated with DNA-PKcs-deficiency. The increased expression of HDAC6 may be induced by a compensatory feed-back loop due to attenuated HDAC6’s activity upon DNA-PKcs inhibition. HDAC6 has two catalytic domains (CAT1 and CAT2), both of which exhibit protein deacetylase activity. We found that the second catalytic domain is responsible for mediating protein-protein association between HDAC6 and DNA-PKcs. In fact, interacting with various protein partners is one of the important modifiers of HDAC6 deacetylase activity. Farnesyltransferase (FTase) can form a complex together with HDAC6 and microtubules. Treating with the FTase inhibitor induces FTase dissociation and attenuates HDAC6 deacetylase activity on α-tubulin [Citation47]. CYLD inhibits HDAC6 through direct protein binding in order to promote α-tubulin acetylation, microtubule stabilization, and mitotic progression [Citation48]. Similar to our finding, p62/SQSTM1 also binds to the CAT2 domain of HDAC6, but this interaction appears to suppress HDAC6 deacetylase activity [Citation33]. DNA-PKcs might competitively interact with the same region of HDAC6 to modulate binding and inhibition from p62/SQSTM1.
In addition to direct protein-protein interactions, several kinases have been reported to target HDAC6 phosphorylation and have affected its deacetylase activity. The extracellular signal-regulated kinase (ERK) phosphorylates HDAC6 at Serine 1035, which is beneficial to HDAC6 activation and HDAC6-mediated cell migration [Citation49]. GSK3β can also increase HDAC6 deacetylase activity via directly phosphorylation of Serine 22 [Citation50]. On the other hand, EGFR-mediated HDAC6 phosphorylation at Tyr570 within the CAT2 domain negatively regulates HDAC6 and, subsequently, hinders acetylated-α-tubulin-dependent EGFR endocytosis and degradation [Citation51]. We speculate that the relationship of DNA-PKcs with HDAC6 at the CAT2 region can help prevent the inhibitory phosphorylation of EGFR on HDAC6. Furthermore, it is possible that DNA-PKcs might directly phosphorylate HDAC6 because treating with the DNA-PKcs kinase inhibitor Nu7441 led to reduced HADC6 activity ().
Similar to HDACs, significant evidence has highlighted a role for DNA-PKcs in cancer development, thus various anti-DNA-PKcs strategies have been proposed as either monotherapy or in combination with chemo – and radiotherapy [Citation52]. We showed that the combined inhibition of both DNA-PKcs and HDAC6 leads to an increase in mitotic arrest and cellular apoptosis (), and also reduced cell viability and clonogenic survival ability (). Mechanistically, DNA-PKcs associates with HDAC6 and modulates its activity, with sublethal inhibition of DNA-PKcs and HDAC6 being sufficient to kill cancer cells efficiently. Our results shed new light on targeting both DNA-PKcs and HDACs as promising combination therapeutic strategies for treating cancer.
Author contributions statement
BPC, ZFS conceived and designed the study. BPC guided the project. LY, YL and JG coordinated and performed most experimental work. JG performed statistical analyses. BPC and ZFS wrote the manuscript, and JC provided review.
Supplemental Material
Download MS Word (251.2 KB)Acknowledgments
Plasmids expressing Flag tagged HDAC6 and truncated proteins were kindly provided by Pro. Geng Liu (School of Radiation Medicine and Protection, Medical College of Soochow University) (M New et al. Cell Death Differ. 2013 Oct; 20(10): 1306–1316.). We thank Dr. Damiana Chiavolini for scientific editing.
Disclosure statement
The authors declare no conflict of interest.
Supplementary Material:
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Chan DW, Chen BP, Chen DJ, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16(18):2333–2338.
- Chen BP, Chan DW, Chen DJ, et al. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280(15):14709–14715.
- Chen BPC, Uematsu N, Chen DJ, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282(9):6582–6587.
- Uematsu N, Weterings E, Chen DJ, et al. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177(2):219–229.
- Ding Q, Reddy YVR, Meek K, et al. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol. 2003;23(16):5836–5848.
- Merkle D, Douglas P, Lees-Miller SP, et al. The DNA-dependent protein kinase interacts with DNA to form a protein−DNA complex that is disrupted by phosphorylation. Biochemistry. 2002;41(42):12706–12714.
- Calsou P, Frit P, Salles B, et al. The DNA-dependent protein kinase catalytic activity regulates DNA end processing by means of Ku entry into DNA. J Biol Chem. 1999;274(12):7848–7856.
- Yajima H, Lee KJ, Chen BPC. ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol. 2006;26(7520–7528):7520–7528.
- Lin YF, Shih HY, Chen BPC, et al. PIDD mediates the association of DNA-PKcs and ATR at stalled replication forks to facilitate the ATR signaling pathway. Nucleic Acids Res. 2018;46(4):1847–1859.
- Douglas P, Ye RQ, Lees-Miller SP, et al. Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci Rep. 2014;34(3):e00113.
- Lee KJ, Lin YF, Chen BPC, et al. Involvement of DNA-dependent protein kinase in normal cell cycle progression through mitosis. J Biol Chem. 2011;286(14):12796–12802.
- Shang Z, Yu L, Chen BPC, et al. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis. 2014;3(2):e85.
- Lee KJ, Shang ZF, Chen BPC, et al. The catalytic subunit of DNA-dependent protein kinase coordinates with polo-like kinase 1 to facilitate mitotic entry. Neoplasia. 2015;17(4):329–338.
- Carey N, La Thangue NB. Histone deacetylase inhibitors: gathering pace. Curr Opin Pharmacol. 2006;6(4):369–375.
- Marks PA, Rifkind RA, Kelly WK, et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1(3):194–202.
- Dong L, Dong Q, Kung HF, et al. Novel HDAC5-interacting motifs of Tbx3 are essential for the suppression of E-cadherin expression and for the promotion of metastasis in hepatocellular carcinoma. Signal Transduct Target Ther. 2018;3:22.
- Cimini D, Mattiuzzo M, Degrassi F, et al. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol Biol Cell. 2003;14(9):3821–3833.
- Ma Y, Cai S, Zhang C, et al. Inhibition of protein deacetylation by trichostatin A impairs microtubule-kinetochore attachment. Cell Mol Life Sci. 2008;65(19):3100–3109.
- Taddei A, Roche D, Almouzni G, et al. The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy? EMBO Rep. 2005;6(6):520–524.
- Li Y, Kao GD, Lazar MA, et al. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006;20(18):2566–2579.
- Foley EA, Kapoor TM. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat Rev Mol Cell Bio. 2013;14(1):25–37.
- Stevens FE, Beamish H, Gabrielli B, et al. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008;27(10):1345–1354.
- Magnaghi-Jaulin L, Eot-Houllier G, Jaulin C, et al. Histone deacetylase inhibitors induce premature sister chromatid separation and override the mitotic spindle assembly checkpoint. Cancer Res. 2007;67(13):6360–6367.
- Park JH, Jong HS, Kim TY, et al. Inhibitors of histone deacetylases induce tumor-selective cytotoxicity through modulating Aurora-A kinase. J Mol Med. 2008;86(1):117–128.
- de Carcer G. Heat shock protein 90 regulates the metaphase-anaphase transition in a polo-like kinase-dependent manner. Cancer Res. 2004;64(15):5106–5112.
- Wissing MD, Mendonca J, Kachhap SK, et al. Targeting prostate cancer cell lines with polo-like kinase 1 innibitors as a single agent and in combination with histone deacetylase inhibitors. Faseb J. 2013;27:4279–4293.
- Kretzner L, Scuto A, Kirschbaum MH, et al. Combining histone deacetylase inhibitor vorinostat with Aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 2011;71(11):3912–3920.
- Dowling M, Voong KR, Kao GD, et al. Mitotic spindle checkpoint inactivation by trichostatin A defines a mechanism for increasing cancer cell killing by microtubule-disrupting agents. Cancer Biol Ther. 2005;4(2):197–206.
- Ruis BL, Fattah KR, Hendrickson EA. The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol Cell Biol. 2008;28(20):6182–6195.
- Peng YL, Zhang QM, Bedford JS, et al. Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer Res. 2002;62(22):6400–6404.
- Li M, Gu MM, Shang ZF, et al. The vanillin derivative VND3207 protects intestine against radiation injury by modulating p53/NOXA signaling pathway and restoring the balance of gut microbiota. Free Radical Bio Med. 2019;145:223–226.
- Zhang ZH, Yamashita H, Iwase H, et al. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10(20):6962–6968.
- Yan J, Seibenhener ML, Wooten MC, et al. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS One. 2013;8(9):e76016.
- Lee JH, Choy ML, Marks PA, et al. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010;107(33):14639–14644.
- Robert C, Rassool FV. HDAC inhibitors: roles of DNA damage and repair. Adv Cancer Res. 2012;116:87–129.
- Liu Q, Ruderman JV. Aurora A, mitotic entry, and spindle bipolarity. Proc Natl Acad Sci U S A. 2006;103(15):5811–5816.
- Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Bio. 2017;18(6):345–360.
- Kramer OH, Mahboobi S, Sellmer A. Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol Sci. 2014;35(10):501–509.
- Cowley DO, Rivera-Perez JA, Van Dyke T, et al. Aurora-A kinase is essential for bipolar spindle formation and early development. Mol Cell Biol. 2009;29(4):1059–1071.
- Solier S, Kohn KW, Pommier Y, et al. Heat shock protein 90 alpha (HSP90 alpha), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc Natl Acad Sci U S A. 2012;109(32):12866–12872.
- Quanz M, Herbette A, Dutreix M, et al. Heat shock protein 90 alpha (Hsp90 alpha) is phosphorylated in response to DNA damage and accumulates in repair foci. J Biol Chem. 2012;287(12):8803–8815.
- Ding G, Liu HD, Huang G, et al. HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett. 2013;587(7):880–886.
- Liu L, Kwak YT, Gaynor RB, et al. DNA-dependent protein kinase phosphorylation of I kappa B beta and I kappa B beta regulates NF-kappa B DNA binding properties. Mol Cell Biol. 1998;18(7):4221–4234.
- Li MX, Lin YF, Chen BPC, et al. The catalytic subunit of DNA-dependent protein kinase is required for cellular resistance to oxidative stress independent of DNA double-strand break repair. Free Radical Bio Med. 2014;76:278–285.
- Song ZQ, Xie Y, Zhou PK, et al. Genome-wide identification of DNA-PKcs-associated RNAs by RIP-Seq. Signal Transduct Target Ther. 2019;4(1):22.
- Zhang M, Xiang SY, Wang CG, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutS alpha. Mol Cell. 2014;55(1):31–46.
- Zhou J, Vos CC, Giannakakou P, et al. The protein farnesyltransferase regulates HDAC6 activity in a microtubule-dependent manner. J Biol Chem. 2009;284(15):9648–9655.
- Wickstrom SA, Masoumi KC, Massoumi R, et al. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. Embo J. 2010;29(1):131–144.
- Williams KA, Zhang M, Fang B, et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J Biol Chem. 2013;288(46):33156–33170.
- Chen SG, Owens GC, Edelman DB, et al. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5(5):e10848.
- Deribe YL, Wild P, Fetchko MJ, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal. 2009;2(102):ra84.
- Hsu FM, Zhang SC, Chen BPC. Role of DNA-dependent protein kinase catalytic subunit in cancer development and treatment. Transl Cancer Res. 2012;1(1):22–34.