
ABSTRACT
Triple-negative breast cancer (TNBC) is the most aggressive histological subtype of breast cancer and is characterized by poor outcomes and a lack of specific-targeted therapies. Transforming growth factor-β (TGF-β) acts as the key cytokine in the epithelial–mesenchymal transition (EMT) and the metastasis of TNBC. However, the regulatory mechanisms of the TGF-β signaling pathway remain largely unknown. In this study, we identified that the USP1/WDR48 complex could effectively enhance TGF-β-mediated EMT and migration of TNBC cells. Furthermore, lower phosphorylation of Smad2/3, Erk, Jnk, and p38 was noted on the suppression of the expression of endogenous USP1 or WDR48. Moreover, the USP1-WDR48 complex was found to downregulate the polyubiquitination of TAK1 and mediate its in vitro stability. Therefore, our findings have shed a light on the novel role of the USP1/WDR48 complex in promoting TGF-β-induced EMT and migration in TNBC via in vitro stabilization of TAK1.
Introduction
Triple-negative breast cancer (TNBC), which constitutes approximately 15%–20% of all breast cancer cases [Citation1], is characterized by the absence or minimal expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) [Citation2,Citation3]. Being the most aggressive histological subtype of breast cancer, TNBC typically affects young women and is characterized by poor prognosis [Citation4]. TNBC is associated with a high rate of metastasis, particularly to the visceral organs such as the lungs, liver, and brain. Owing to the lack of effective targets, chemotherapy remains the standard treatment for early stage as well as metastatic TNBC patients [Citation5]. However, only 20% of the TNBC patients respond to therapy. Therefore, there is a pressing need to search for effective molecular targets that are specific to TNBC.
Several previous studies have shown that transforming growth factor-β (TGF-β) plays a vital role in TNBC, such as in stemness regulation, epithelial–mesenchymal transition (EMT), apoptosis, and chemotherapy resistance [Citation6,Citation7]. TGF-β is a 25-kDa cytokine that regulates cell growth and differentiation, apoptosis, cell motility, angiogenesis, and cellular immune response [Citation8–10]. The TGF-β subfamily comprises three TGF-β ligands (namely, TGF-β1, TGF-β2, and TGF-β3), which have similar but not identical in vitro biological activities. Maris reported that TNBC cells, including MDA-MB-231, are the highest producers of TGF-β1 [Citation11].
TGF-β signal travels through a transmembrane receptor complex that comprises type I and type II receptor serine-threonine kinases. The activated receptor complexes initiate the so-called canonical TGF-β signaling through the C-terminal phosphorylation of the signaling transducers Smad2 and Smad3, which are associated with Smad4 and regulate the transcription [Citation12]. In addition to the stimulation of the Smad-dependent pathway, the activated receptor can also activate other intracellular pathways known as the non-Smad signaling pathways, including several branches such as MAPKs (i.e. Erk, Jnk, and p38) and AKT [Citation12]. Upon ligand binding, the TGF-β receptor complex interacts with TRAF6 and promotes its autoubiquitylation [Citation13]. TGF-β-activated kinase 1 (TAK1), also known as the mitogen-activated protein kinase 7 (MAP3K7), detected in the epithelial cells is stimulated in response to TGF-β via the phosphorylation of TGF-β-activated kinase 1 binding protein 1 (TAB1) [Citation14]. TRAF6 activates TAK1 through Lys63-linked polyubiquitylation [Citation15]. Consequently, the activated TAK1, in turn, activates target proteins such as IKKα/CHUK, IKKβ/IKBKB, and MKK3/6, which result in the phosphorylation and activation of MAPKs (i.e. p38, JNK, and ERK) in a Smad-independent manner [Citation16]. Subsequently, the phosphorylated Jnk and p38 phosphorylate the Smads (i.e. Samd2 and Smad3), thus regulating the Smad-dependent transcriptional responses [Citation17].
Increasing evidences indicate that both Smad-dependent and non-Smad signaling pathways can induce EMT [Citation18–21]. This process is characterized by alterations in the levels of three prominent biomarkers, namely E-cadherin, vimentin, and N-cadherin. This event leads to decreased adhesion of the cells, loss of polarity, and tight junctions, which results in the development of cellular malignant characteristics such as invasiveness and metastatic ability [Citation22]. Accumulating evidence has demonstrated that the activation of the TGF-β-activated kinase 1 (TAK1) plays a critical role in TGF-β-induced EMT [Citation13,Citation14,Citation20,Citation23,Citation24]. Thus, ascertaining the potential mechanisms regulating the stability of TAK1 protein is of utmost importance in cancer research.
In the present study, we identified that the USP1/WDR48 complex formed by ubiquitin-specific peptidase 1 (USP1) and WD repeat domain 48 (WDR48) is a novel enhancer of TGF-β signaling. More importantly, the inhibition of endogenous USP1 or WDR48 expression significantly decreased the TGF-β-induced EMT, migration, and invasion of the TNBC cells. Mechanistically, we also demonstrated that the USP1/WDR48 complex plays a key role in the transduction of TGF-β signaling by mediating TAK1 stability and therefore regulating the in vitro phosphorylation of Smad2/3, p38, Jnk, and Erk. Our results could be interpreted as the evidence of a new mechanism by which the USP1/WDR48 complex promotes TGF-β-induced EMT and migration of the TNBC cells.
Materials and methods
Cell lines and transfection
All the cells were purchased from the American Type Culture Collection (Manassas, VA, USA). MDA-MB-231, MDA-MB-468 cells were cultured in DMEM (Hyclone, South Logan, UT) with 10% fetal bovine serum (FBS) (GIBCO, Grand Island, NY), 100 µg/mL penicillin (Hyclone), and 100 µg/mL streptomycin (Hyclone). Cells in the medium were incubated in a humidified atmosphere containing 5% CO2 at 37°C. For the knockdown experiments, the small interfering RNA (siRNA) sequences for USP1 were as follows: 5′-GGCAAGUUAUGAGCUUAUA-3′ (siRNA 1), 5′-CGGCAAGGUUGAAGAACAA-3′(siRNA 2), and 5′-GGAGAGCUCUGAAAUUUCU-3′(siRNA 3); The sequences for WDR48 siRNA were as follows: 5′-GGUCGAGACUCCAUCAUAA-3′(siRNA 1); 5′-GGAACAAAGACUCCAUUUA-3′(siRNA 2) and 5′-GCCCGA CCAAGUUAUUAAA-3′(siRNA 3). Transfection was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol.
RNA isolation and qPCR assay
Total RNA was extracted from cultured cells with Trizol reagents (Invitrogen) according to the manufacturer’s instructions. Total RNA was reverse-transcribed to cDNA using PrimeScript RT reagent Kit (Takara, Tokyo, Japan), Real-time PCR was performed with the SYBR Premix Ex Taq (Tli RNaseH Plus) (Takara) with Light Cycler 480 II Real-Time PCR System (Roche, Basel, Switzerland). GAPDH was used as the endogenous control for the detection of mRNA expression level.
Co-immunoprecipitation assay
For immunoprecipitation (IP), whole-cell extracts were collected 36 h after transfection and were lysed in Western and IP lysis buffer (Beyotime, Shanghai, China) with protease inhibitors (Sigma, St. Louis, MO, US). After centrifugation at 12,000 g for 10 min at 4°C, supernatants were collected and incubated with 1 µg monoclonal anti-Flag, 1 µg anti-Myc, or 1 µg anti-TAK1 for 1 h. Then, protein G Plus-Agarose Immunoprecipitation reagent (Santa Cruz) was used to incubate together with monoclonal antibody. After 8 h of incubation, protein G Plus-Agarose beads were washed five times with IP buffer. Immunoprecipitates were eluted by boiling with 1% (wt/vol) SDS sample buffer for 5 mins.
Western blot analysis and regents
After indicated treatment, cells were collected and lysed with Western and IP lysis buffer (Beyotime, Shanghai, China) with protease inhibitors (Sigma, St. Louis, MO, US), then centrifuged at 12000g for 15mins at 4°C, supernatant was transferred into a 1.5ml EP and 5×SDS was added, boiling for 5mins. Equal amounts of proteins were loaded on SDS-PAGE gels and then transferred to a PVDF membrane (Millipore, Carlsbad, CA, US). After blocking with 5% non-fat milk, the membrane was incubated overnight at 4°C with the primary antibody and then with horseradish peroxydase-coupled secondary antibody (CST, Danvers, MA, US). Signal was detected with enhanced chemiluminescence (Millipore, Bedford, MA, US). USP1(#14346-1-AP, 1:1000), WDR48(#16503-1-AP, 1:1000), E-cadherin (#20874-1-AP, 1:1000) and N-cadherin (#16503-1-AP, 1:1,000) antibodies were purchased from Proteintech (Rosemount, IL, USA). β-actin (#3700, 1:1000), Jnk (#9252, 1:1000), p-Jnk (#9251, 1:1000), Erk (#4695, 1:1000), p-Erk (#4370, 1:1000), p38 (#8690, 1:1000), p-p38 (#4511, 1:1000), Smad2 (#5339, 1:1000), p-Smad2 (#18338, 1:1000), Smad3 (#9523, 1:1000), p-Smad3 (#9520, 1:1000), Flag (#14793, 1:1000), Tak1 (#4505S, 1:1000), Myc (#2276, 1:1000), HA (#5017, 1:1000) and Vimentin (#5741, 1:1000) antibody were purchased from CST. ML323 was from Selleck Chemicals (Houston, TX, US). TGF-β (98726-64-8) was purchased from PeproTech (Rocky Hill, NJ, US).
Transwell migration and invasion assays
The cell migration and invasion assays were performed using Transwell chambers, which were coated with (invasion assay) or without (migration assay) Matrigel, briefly, for invasion assay, the polycarbonate membranes were coated with 40μL matrigel at 37°C for 2h to form a reconstituted basement membrane, 700μL of the medium with 20% FBS was added to the lower well of each chamber, and 1×105 cells suspended in serum-free medium with or without TGF-β and ML323 were added to the upper inserts. After 24-48h, the cells located on the lower surfaces were fixed with methanol for 5mins and stained with 0.1% crystal violet for 15mins. The stained cells were photographed in six representative fields, and relative cell number was calculated.
Statistical analysis
The GraphPad Prism 8 was used for statistical analysis. All data from at least three independent experiments were presented as means ± SD unless stated otherwise. A Student’s t-test and one-way analysis of variance were performed to determine significance. Confidence level of 0.95% was used for all statistical analyses (α-value = 0.05).
Results
Expression pattern of USP1 and WDR48 during TGF-β stimulation
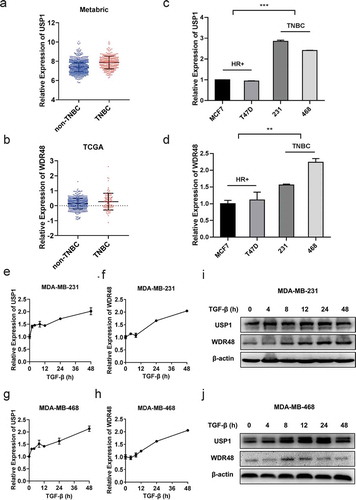
By segregating the patient samples in the published Metabric and TCGA datasets, we found that the expressions of USP1 and WDR48 were higher in TNBC than in non-TNBC (). Furthermore, the USP1 and WDR48 expressions were observed to be higher in the TNBC cell lines than in the non-TNBC cell lines (–d). Owing to the important regulatory role of TGF-β in TNBC [Citation6,Citation7], we examined the expression status of USP1 and WDR48 during TGF-β stimulation. The results revealed that the USP1 and WDR48 expression levels were markedly elevated at a later stage of TGF-β stimulation in MDA-MB-231 and MDA-MB-468, which are both TNBC cells ( e–j). These data illustrate the dynamic changes in USP1 and the WDR48 expression during TGF-β stimulation, suggesting the potential involvement of the USP1/WDR48 complex in the TGF-β signaling pathway.
Figure 1. Expression pattern of USP1 and WDR48 during TGF-β stimulation. (a–b) USP1 and WDR48 expression in TNBC and non-TNBC breast cancer patients according to the data in METABRIC (a) and TCGA (b). (c–d) The relative expression of USP1 and WDR48 in different cell lines, including hormone receptor (HR) positive cell lines, M7 and T47D, and triple-negative cell lines, MDA-MB-231 and MDA-MB-468 (c–d). (e–h) RT-PCR analysis of the expression pattern of USP1 (e, g) and WDR48 (f, h) in MDA-MB-231 and MDA-MB-468 cells during TGF-β stimulation. (i–j) Western blot analysis of USP1 and WDR48 expression in MDA-MB-231 (i) and MDA-MB-468 (j) cells stimulated with TGF-β for the indicated time periods
USP1/WDR48 complex enhanced TGF-β-induced migration and invasion in the TNBC cells
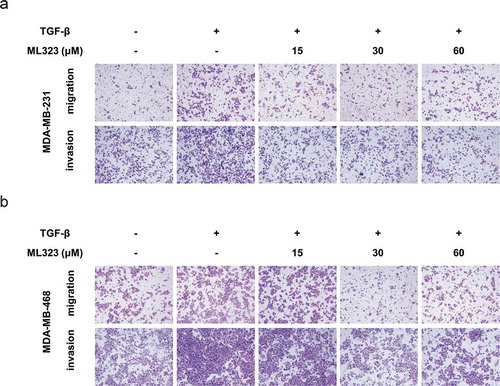
TGF-β has been proven to be a crucial activator of breast cancer metastasis [Citation25]. To further investigate the effect of the USP1/WDR48 complex in TGF-β-mediated TNBC cell metastasis, we performed the Transwell assay. We noted that 10 ng/mL of TGF-β significantly increased the migration and invasion abilities of the MDA-MB-231 and MDA-MB-468 cells. Nevertheless, ML323, a specific USP1/WDR48 inhibitor, significantly reversed the abovementioned process in a dose-dependent manner (–b). These results suggest that the USP1/WDR48 complex is essential for the TGF-β-induced TNBC cell migration and invasion.
Figure 2. USP1/WDR48 complex enhanced TGF-β-induced migration and invasion in the TNBC cells. (a–b) Transwell migration and invasion assays for MDA-MB-231 (a) and MDA-MB-468 cells (b) treated with or without TGF-β and ML323. All representative images were obtained from at least three independent experiments
USP1/WDR48 complex promoted TGF-β-induced EMT
EMT plays a key role in the TGF-β-induced breast cancer cell migration and invasion [Citation26]. To further investigate whether the USP1/WDR48 complex promoted TNBC migration and invasion through the regulation of TGF-β-induced EMT, we used ML323 to inhibit the function of the endogenous USP1/WDR48 complex. Subsequently, we applied reverse transcription-polymerase chain reaction (RT-PCR) and western blotting to detect the expression pattern of the EMT markers. Our results showed that ML323 could significantly increase the E-cadherin expression in the TNBC cells, which was diminished by TGF-β. Simultaneously, TGF-β-induced N-cadherin and vimentin expression were decreased (). We did detect the E-cadherin expression in MDA-MB-231 cells via western blotting, but we also found that the endogenous E-cadherin expression was too low to be detected because of their mesenchymal characteristics.
Figure 3. USP1/WDR48 complex promoted TGF-β-induced EMT. (a–b) MDA-MB-231 and MDA-MB-468 cells treated with or without TGF-β and ML323 for 12 h, the protein levels of EMT markers were analyzed by western blot. (c) The mRNA expression levels were analyzed by RT-PCR. (d–e) Western blot and RT-PCR analysis of USP1 (d) or WDR48 (e) expression in MDA-MB-231 and MDA-MB-468 cells transfected with the indicated siRNA for 48 h. (f–g) MDA-MB-231 and MDA-MB-468 cells were transfected with control siRNA, USP1 siRNA 2, or WDR48 siRNA 1 for 48 h and then stimulated with PBS or TGF-β for 24 h. Western blot analysis of the EMT markers expression pattern. All representative images were obtained from at least three independent experiments. *P < 0.05, **P < 0.01, *** P < 0.001, **** P < 0.0001 by Student’s t-test. Error bars are defined as s.d
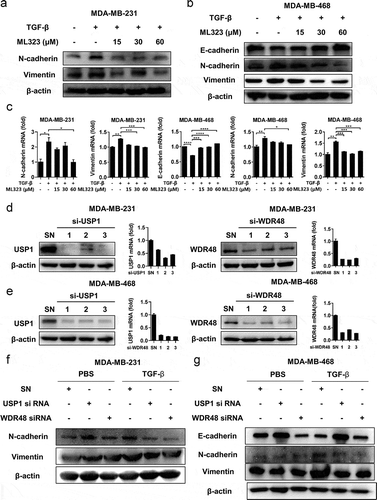
To confirm the effect of the USP1/WDR48 complex in TGF-β-induced EMT, we used synthesized interfering RNAs targeting human USP1 or WDR48 and suppressed endogenous USP1 or WDR48 expression. USP1 siRNA 2 and WDR48 siRNA 1, which showed high efficiencies in inhibiting the expression of the target proteins (), were used in the subsequent experiments. Similar to ML323, both USP1 and WDR48 knockdown markedly inhibited TGF-β-induced EMT in MDA-MB-231 () and MDA-MB-468 cells ().
USP1/WDR48 knockdown inhibited TGF-β-induced-Smad2/3 and MAPK activation
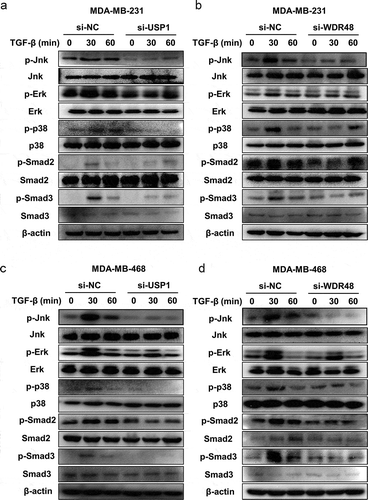
Considering the fact that the USP1/WDR48 complex could enhance the in vitro migration and invasion of the TGF-β-induced TNBC cells, we next investigated the effect of the USP1/WDR48 complex on the key components of the TGF-β signaling pathway. Accordingly, MDA-MB-231 and MDA-MB-468 cells were transfected with USP1 or WDR48 siRNA and were stimulated with TGF-β at specific time periods. Consistently, USP1 and WDR48 knockdown significantly decreased the TGF-β-induced Smad2/3, Jnk, Erk, and p38 phosphorylation (). In addition, these changes were not caused by protein degradation because the Smad2/3, Jnk, Erk, and p38 protein levels were not affected by USP1 or WDR48 siRNA transfection (). Collectively, these data indicate that the USP1/WDR48 complex is a positive regulator of the TGF-β signaling pathway.
Figure 4. USP1/WDR48 knockdown inhibited TGF-β-induced-Smad2/3 and MAPK activation. (a–d) MDA-MB-231 (a–b) and MDA-MB-468 (c–d) cells were transfected with control siRNA, USP1 siRNA 2, or WDR48 siRNA 1 for 48 h and stimulated with TGF-β for the indicated time periods. Western blot analysis of Jnk/p-Jnk, Erk/p-Erk, p38/p-p38, Smad2/p-Smad2, and Smad3/p-Smad3 expression
USP1/WDR48 complex directly binds to TAK1 and mediates its stability by downregulating its polyubiquitination
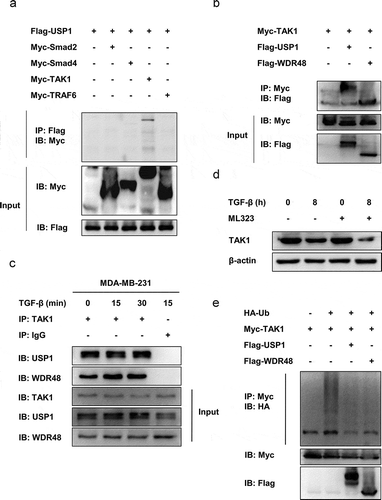
To determine the molecular targets of the USP1-WDR48 deubiquitinase complex in the TGF-β signaling pathway, Myc-tagged Smad2, Smad4, TAK1, TRAF6, and Flag-tagged USP1 were cotransfected into the HEK293T cells. As shown in , USP1 could interact with TAK1, but not with Smad2, Smad4, or TRAF6. Moreover, Myc-tagged TAK1 and Flag-tagged USP1 and WDR48 were used to further verify the interaction between the USP1-WDR48 complex and the TAK1 in HEK293T cells. As shown in , TAK1 was co-precipitated with both USP1 and WDR48. Furthermore, endogenous interaction was observed in the TGF-β-stimulated MDA-MB-231 cells (). Collectively, these results imply that USP1 and WDR48 specifically bind to TAK1. To further illustrate the roles of the USP1-WDR48 complex after TGF-β stimulation, ML323 was used to inhibit the function of the endogenous USP1-WDR48 complex. As seen in , the administration of ML323 greatly decreased the TAK1 expression in the MDA-MB-231 cells. As previously described, the USP1-WDR48 complex possessed potent deubiquitinase activity. We thus investigated the effects of USP1 and WDR48 on TAK1 ubiquitination. We found that USP1 and WDR48 significantly inhibited TAK1 ubiquitination (). Together, these results allude that the USP1-WDR48 complex promotes TGF-β signaling by directly binding to TAK1 and downregulating its polyubiquitination.
Figure 5. USP1/WDR48 complex directly binds to TAK1 and mediates its stability by downregulating its polyubiquitination. (a) Lysates from HEK293T cells transiently cotransfected with Flag-USP1 and Myc-Smad2, Myc-Smad4, Myc-TAK1, or Myc-TRAF6 were subjected to immunoprecipitation with anti-Flag antibody followed by Western blot analysis with anti-Myc antibody. (b) Lysates from HEK293T cells transiently cotransfected with Myc-TAK1 and Flag-WDR48 or Flag-USP1 were subjected to immunoprecipitation with anti-Myc antibody followed by Western blot analysis with anti-Flag antibody. (c) Lysates from MDA-MB-231 cells stimulated with TGF-β for indicated periods were subjected to immunoprecipitation with the TAK1 antibody or control IgG followed by Western blot analysis with the indicated antibodies. Protein in whole-cell lysate was used as positive control (Input). (d) Western blot analysis of extracts from MDA-MB-231 cells treated with 30 µM ML323 for 4 h and then stimulated with TGF-β for the indicated time periods. (e) Western blot analysis of lysates from HEK293T cells transfected with HA-tagged ubiquitin (HA-Ub), Myc-TAK1 and Flag-USP1, or Flag-WDR48, followed by immunoprecipitation with anti-Myc, probed with anti-HA
Discussion
TNBC is a long-lasting orphan disease with only little therapeutic progress made in the past several decades, and it is associated with a high rate of metastasis. Once metastasis occurs, TNBC has a high predisposition to involve the critical visceral organs, eventually leading to a significantly shorter median overall survival than the other subtypes [Citation1]. However, the molecular mechanism of TNBC progression and metastasis remains largely unknown.
TGF-β signaling plays a vital role in tumor progression. Tumor cells secrete abundant TGF-β, which is the most potent inducer of EMT in epithelial cancers and is a promoter of cell invasion and metastasis [Citation27,Citation28]. Anti-TGF-β therapies have proved to be efficacious in inhibiting cancer invasion and metastasis in animal models [Citation29]. A previous study reported that the levels of TGF-β1 and TGF-β2 are higher in TNBC cells than in non-TNBC cells [Citation30], the aberrant expression of TGF-β1 is associated with poor prognosis in breast cancer patients [Citation31]. TAK1, which is one of the best characterized non-Smad signal transducers, plays a critical role in TGF-β-induced EMT and metastasis by activating the Jnk and p38 MAPK cascade [Citation13,Citation14,Citation32]. In breast cancer, past studies have established that TAK1 plays a critical role in mediating the effects of TRAF4 [Citation33], UBC13 [Citation34], CCN6 [Citation35], and mir-892b [Citation36] on tumor progression and metastasis. Veenu et al. found that TGF-β-induced alternative splicing of TAK1 promotes EMT and drug resistance in breast cancer cells [Citation23]. Wang et al. reported that NG25, a synthesized TAK1 inhibitor, greatly enhances the Dox treatment efficacy in a panel of breast cancer cell lines [Citation37]. Data from TCGA demonstrate that the TAK1 levels exhibit a higher expression in TNBC than in the other subtypes [Citation38]. Oihana et al. demonstrated that TAK1 mediates microenvironment-triggered autocrine signals and promotes in vitro and in vivo lung metastasis of TNBC [Citation38].
Accumulating data have confirmed that several USP family members play essential roles in TGF-β-induced cancer progression as well as in a diverse set of other human diseases [Citation39]. For example, USP4 directly deubiquitylates TβRI, maintaining sustained TβRI levels, and reinforcing pro-tumorigenic functions induced by the TGF-β receptor in breast cancer cells [Citation40]. USP18 inhibits cardiac hypertrophy and postpones cardiac dysfunctioning by modulating the TGF-β-activated p38/Jnk signaling [Citation41]. In addition, the USP15 overexpression in systemic sclerosis fibroblasts decreases ubiquitin-mediated degradation of the proteins involved in TGF-β signaling [Citation42]. As a member of the USP family, USP1 can deubiquitinate a wide range of substrates, and its deubiquitinase activity can be enhanced by its binding partner WDR48 [Citation43,Citation44]. Yu demonstrated that the USP1/WDR48 complex is a physiological enhancer of TLR3/4-, RIG-I-, and cGAS-induced antiviral signaling by targeting TBK1 [Citation45]. Sonego demonstrated the USP1-linked platinum resistance of cancer cell dissemination by regulating Snail stability [Citation46]. Ma established that USP1 promotes breast cancer metastasis by stabilizing KPNA2 [Citation47]. However, the potential roles of USP1 in TGF-β-induced EMT have not been reported yet.
In the present study, we identified the USP1/WDR48 complex as the new regulator of TGF-β-mediated TNBC migration and invasion. Data from both Metabric and TCGA databases demonstrated a high expression of USP1 and WDR48 in TNBC than in the non-TNBC tissues. Moreover, the expression pattern of the USP1-WDR48 complex in several cell lines was confirmed by RT-PCR. Meanwhile, TGF-β markedly increased the USP1 and WDR48 expressions in the TNBC cells. ML323 or knocking-down of the USP1/WDR48 complex could potentially decrease TGF-β-induced TNBC cell migration and EMT. Moreover, depleting the USP1/WDR48 complex in TNBC decreased the phosphorylation of TGF-β-induced Smad2/3, Erk, Jnk, and p38. Moreover, USP1 and WDR48 were found to directly bind with TAK1 and downregulate its polyubiquitination. Cumulatively, these results suggest that the USP1/WDR48 complex could potentially enhance the TGF-β-induced EMT and migration of the TNBC cells by targeting TAK1.
In conclusion, we identified that USP1 and WDR48 are TGF-β-inducible genes. Furthermore, we inferred that the USP1/WDR48 complex is a positive regulator of TGF-β-induced EMT and migration of the TNBC cells. Our study also defined a very important mechanism for the regulation of the USP1/WDR48 complex according to TGF-β-induced activation of Smad2/3, Erk, Jnk, and p38 by directly interacting with TAK1 and mediating its stability. Thus, we propose that targeting the USP1/WDR48 complex may be a promising therapy for TGF-β-related TNBC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81672613; No. 81874119; No. 82072912), Special Foundation for Taishan Scholars (No. ts20190971), Special Support Plan for National High-Level Talents (Ten Thousand Talents Program W01020103), National Key Research and Development Program (No. 2018YFC0114705), Funded by Clinical Research Center of Shandong University (No.2020SDUCRCA015), Qilu Hospital Clinical New Technology Developing Foundation (No. 2018-7; No. 2019-3).
Disclosure statement
All authors read and approved the final version of the manuscript, and the authors have no conflict interest to declare.
Additional information
Funding
References
- Denkert C, Liedtke C, Tutt A, et al. Molecular alterations in triple-negative breast cancer-the road to new treatment strategies. Lancet. 2017;389(10087):2430–2442.
- Hammond ME, Hayes DF, Dowsett M, et al. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28(16):2784–2795.
- Wolff AC, Hammond MEH, Allison KH, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of american pathologists clinical practice guideline focused update. J Clin Oncol. 2018;36(20):2105–2122.
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948.
- Zeichner SB, Terawaki H, Gogineni K. A review of systemic treatment in metastatic triple-negative breast cancer. Breast Cancer: Basic and Clinical Research. 2016;10:25–36.
- Xu X, Zhang L, He X, et al. TGF-beta plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem Biophys Res Commun. 2018;502(1):160–165.
- Bhola NE, Balko JM, Dugger TC, et al. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123(3):1348–1358.
- Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129.
- Dumont N, Arteaga CL. Targeting the TGF beta signaling network in human neoplasia. Cancer Cell. 2003;3(6):531–536.
- Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67(1):753–791.
- Maris P, Blomme A, Palacios AP, et al. Asporin is a fibroblast-derived TGF-beta1 inhibitor and a tumor suppressor associated with good prognosis in breast cancer. PLoS Med. 2015;12(9):e1001871.
- Derynck R, Budi EH. Specificity, versatility, and control of TGF-beta family signaling. Sci Signal. 2019;12(570):eaav5183.
- Sorrentino A, Thakur N, Grimsby S, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10(10):1199–1207.
- Yamaguchi K, Shirakabe K, Shibuya H, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270(5244):2008–2011.
- Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78(1):769–796.
- Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33(10):522–530.
- Yumoto K, Thomas PS, Lane J, et al. TGF-β-activated kinase 1 (Tak1) mediates agonist-induced Smad activation and linker region phosphorylation in embryonic craniofacial neural crest-derived cells. J Biol Chem. 2013;288(19):13467–13480.
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196.
- Zavadil J, Bitzer M, Liang D, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci U S A. 2001;98(12):6686–6691.
- Landström M. The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol. 2010;42(5):585–589.
- Davies M, Robinson M, Smith E, et al. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem. 2005;95(5):918–931.
- Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem. 2003;278(23):21113–21123.
- Tripathi V, Shin JH, Stuelten CH, et al. TGF-β-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene. 2019;38(17):3185–3200.
- Dong N, Tang X, Yuan XY, et al. [TAK1 promotes epithelial-mesenchymal transition of lens epithelial cells]. Zhonghua Yan Ke Za Zhi. 2016;52:278–284.
- Suriyamurthy S, Baker D, Ten Dijke P, et al. Epigenetic reprogramming of TGF-β signaling in breast cancer. Cancers (Basel). 2008;10(5):726.
- Pang MF, Georgoudaki AM, Lambut L, et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene. 2016;35(6):748–760.
- Han G. Distinct mechanisms of TGF- 1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest. 2012;33(7):1714–1723.
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2010;42(6):1420–1428.
- Korpal M, Yan J, Lu X, et al. Imaging transforming growth factor-β signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med. 2003;278(8):960–966.
- Kim S, Lee J, Jeon M, et al. Elevated TGF-beta1 and -beta2 expression accelerates the epithelial to mesenchymal transition in triple-negative breast cancer cells. Cytokine. 2015;75(1):151–158.
- Kim S, Lee J, You D, et al. Berberine suppresses cell motility through downregulation of TGF-β1 in triple negative breast cancer cells. Cellular Physiology and Biochemistry. 2019;38(2):795–807.
- Yamashita M, Fatyol K, Jin C, et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31(6):918–924.
- Zhang L, Zhou F, García de Vinuesa A, et al. TRAF4 promotes TGF-β receptor signaling and drives breast cancer metastasis. Mol Cell. 2013;51(5):559–572.
- Wu X, Zhang W, Font-Burgada J, et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1-p38 MAP kinase cascade. Proc Natl Acad Sci U S A. 2014;111(38):13870–13875.
- Pal A, Huang W, Li X, et al. CCN6 modulates BMP signaling via the Smad-independent TAK1/p38 pathway, acting to suppress metastasis of breast cancer. Cancer Res. 2012;72(18):4818–4828.
- Jiang L, Yu L, Zhang X, et al. miR-892b silencing activates NF-κB and promotes aggressiveness in breast cancer. Cancer Res. 2009;119(5):1101–1111.
- Wang Z, Zhang H, Shi M, et al. TAK1 inhibitor NG25 enhances doxorubicin-mediated apoptosis in breast cancer cells. Sci Rep. 2016;6(1):32737.
- Iriondo O, Liu Y, Lee G, et al. TAK1 mediates microenvironment-triggered autocrine signals and promotes triple-negative breast cancer lung metastasis. Nat Commun. 2018;9(1):1994.
- Nolte M, Margadant C. Controlling Immunity and Inflammation through integrin-dependent regulation of TGF-beta. Trends Cell Biol. 2020;30(1):49–59.
- Zhang L, Zhou F, Drabsch Y, et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-beta type I receptor. Nat Cell Biol. 2012;14(7):717–726.
- Ying X, Zhao Y, Yao T, et al. Novel protective role for ubiquitin-specific protease 18 in pathological cardiac remodeling. Hypertension. 2016;68(5):1160–1170.
- Galant C, Marchandise J, Stoenoiu MS, et al. Overexpression of ubiquitin-specific peptidase 15 in systemic sclerosis fibroblasts increases response to transforming growth factor beta. Rheumatology (Oxford). 2019;58(4):708–718.
- Cohn MA, Kee Y, Haas W, et al. UAF1 is a subunit of multiple deubiquitinating enzyme complexes. J Biol Chem. 2009;284(8):5343–5351.
- Sowa ME, Bennett EJ, Gygi SP, et al. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138(2):389–403.
- Yu Z, Song H, Jia M, et al. USP1-UAF1 deubiquitinase complex stabilizes TBK1 and enhances antiviral responses. J Exp Med. 2017;214(12):3553–3563.
- Sonego M, Pellarin I, Costa A, et al. USP1 links platinum resistance to cancer cell dissemination by regulating snail stability. Sci Adv. 2019;5(5):eaav3235.
- Ma A, Tang M, Zhang L, et al. USP1 inhibition destabilizes KPNA2 and suppresses breast cancer metastasis. Oncogene. 2019;38(13):2405–2419.