
ABSTRACT
A key to longevity assurance is the nutrient-sensing mTOR pathway. Inhibition of mTOR extends lifespan in a variety of organisms. However, the downstream effectors of the mTOR pathway for lifespan regulation are elusive. In a recent report, we described the role of Maf1 as a critical lifespan regulator downstream of the mTOR pathway in fission yeast. Maf1 is the master negative regulator of RNA polymerase III-directed transcription (e.g. tRNAs and 5S rRNAs) and is regulated by mTOR-mediated phosphorylation. We demonstrated that Maf1 is required for lifespan extension under calorie restriction or when mTOR is inhibited. We also showed that Maf1 prevents DNA damage at tRNA genes, which appears to contribute to lifespan maintenance by Maf1. Here we highlight these observations and present additional results to discuss the role of the mTOR-Maf1-Pol III axis in promoting genomic integrity in the face of DNA replication-transcription conflicts in order to maintain normal lifespan.
Introduction: role of Maf1 as a major target of the mTOR pathway in lifespan regulation
The nutrient-sensing kinase, mTOR, functions as a lifespan modulator in response to nutrient availability [Citation1]. mTOR signaling consists of two separate protein complexes, TORC1 and TORC2, of which TORC1 is highly sensitive to rapamycin and well studied for its role in aging regulation [Citation2]. Inhibition of mTOR extends lifespan in various eukaryotic organisms ranging from yeast to mice. Inhibition of mTOR also ameliorates multiple age-related diseases in animal models including Alzheimer’s disease, Parkinson’s disease, and idiopathic cardiomyopathy [Citation1]. However, downstream regulators of mTOR responsible for these lifespan- and healthspan-promoting effects have not been fully defined.
In the course of identifying downstream mTOR effectors that regulate lifespan, we recently demonstrated that deficiency in Maf1, a TORC1 target, results in lifespan shortening in fission yeast [Citation3]. Maf1 is the master inhibitor of RNA polymerase III-directed transcription (tRNA and 5S rRNA) and is directly phosphorylated by TORC1 () [Citation4]. Intriguingly, inhibition of TORC1 largely failed to extend fission yeast lifespan in the absence of Maf1 [Citation3]. In addition, Maf1 is also necessary for lifespan extension by calorie restriction [Citation3]. Considering that TORC1 activity is reduced in calorie-restricted conditions [Citation1], our results indicate a critical role of Maf1 as a downstream effector of TORC1 in aging regulation [Citation3].
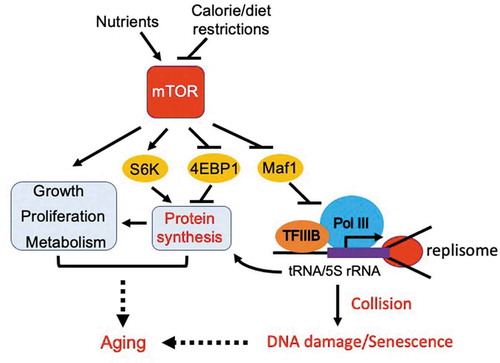
In response to nutrients, mTOR modulates metabolism to promote the biosynthesis of macromolecules required to support cell growth and proliferation () [Citation5]. This includes activation of protein synthesis via TORC1-mediated phosphorylation of 4E-BP1 and S6K1 [Citation6]. TORC1 also upregulates transcription by RNA polymerase I to supply rRNAs, which in turn supports ribosome biogenesis required for protein synthesis [Citation7]. In addition, TORC1-mediated inhibition of Maf1 results in elevated RNA polymerase III ac tivity to support tRNA synthesis [Citation4], which feeds into protein synthesis (). Energy consumption associated with TORC1-mediated protein synthesis may cause lifespan shortening when Maf1 is absent. Indeed, maf1-deleted fission yeast cells display elevated protein synthesis [Citation3]. However, our investigation revealed that elevated protein synthesis is not a cause of lifespan shortening in maf1-deleted cells. This conclusion is supported by evidence that inhibition of TORC1, which reduces protein synthesis, failed to rescue the short lifespan of maf1-deleted cells [Citation3]. Although these results may not exclude the possibility of protein synthesis as a driver of aging, TORC1-dependent Maf1 inactivation appears to cause aging through a different mechanism, which is discussed later in this article. Nevertheless, our recent study has demonstrated the role of Maf1 as a major lifespan regulator under mTOR control [Citation3].
Figure 1. Inhibition of TORC1 extends lifespan via activation of Maf1 and subsequent inhibition of Pol III. TORC1 phosphorylates and thus inactivates Maf1, leading to an elevated level of Pol III-directed transcription to promote tRNA and 5S rRNA synthesis. High levels of transcription lead to frequent collisions between the transcription machinery and the replisome, resulting in DNA damage and accelerated aging. mTOR also activates a variety of growth-promoting pathways, leading to aging. Calorie or dietary restrictions inhibit mTOR, minimizing aging processes via activation of Maf1 and subsequent inhibition of Pol III
Maf1 phosphorylation dictates lifespan
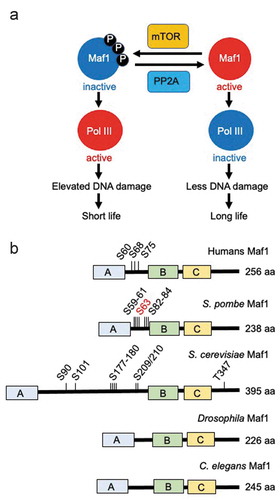
Maf1 is regulated by the TORC1 kinase via phosphorylation in a variety of organisms [Citation3,Citation4,Citation8]. This phosphorylation is reversed by PP2A phosphatases ()). Maf1 has three domains (A, B, and C) that are conserved among species, although the specific functions of these domains are not understood [Citation4,Citation9]. Multiple phosphorylation sites have been identified within the linker region between domains A and B in S. cerevisiae and human cells ()) [Citation4]. Phosphorylation of these sites negatively affects the role of Maf1 as an inhibitor of Pol III in human and yeast cells. When these sites are phosphorylated, Maf1 is inactivated; thus, Pol III-mediated tRNA and 5S rRNA synthesis are both elevated [Citation3,Citation4,Citation8].
In S. pombe, there are at least seven phosphorylatable serine residues within the same linker region ()) [Citation3]. When these sites are all changed to alanine residues, thus representing non-phosphorylatable Maf1, tRNA transcription is inhibited. In contrast, phosphomimetic substitution of these serine residues with glutamic acids resulted in elevated tRNA transcription. Strikingly, non-phosphorylatable maf1 mutants had a long lifespan, whereas phosphomimetic maf1 mutants showed a short lifespan under calorie restriction ()) [Citation3]. Consistently, torc1 mutant cells, which harbor hypophosphorylated Maf1, were long-lived, in contrast to PP2A mutants (ppa1∆ and ppa2∆ cells) that were short-lived with hyperphosphorylated Maf1 [Citation3]. In addition, Maf1 is hypophosphorylated under low glucose conditions (low TORC1 activity, long lifespan), while it undergoes hyperphosphorylation in response to high glucose (high TORC1 activity, short lifespan) [Citation3]. Thus, these results clearly indicate that Maf1 phosphorylation dictates lifespan under calorie restriction in fission yeast ()).
Our investigation also identified S63 as the primary phosphorylation site ()). When this site is mutated to alanine, Maf1 lost the detectable slow-migrating species associated with phosphorylation. However, the S63A single mutation was not enough to extend lifespan. Therefore, the other six phosphorylation sites also contribute to Maf1-mediated aging regulation [Citation3]. Future investigation is warranted to investigate how S63 phosphorylation influences Maf1 phosphorylation landscape in the linker region under calorie restriction.
Role of Maf1 in genomic integrity and transcription-replication conflicts
As is the case for other organisms including S. cerevisiae, C. elegans, and Drosophila, S. pombe lifespan is shortened under high-calorie conditions [Citation3]. In these conditions, TORC1 activity is elevated; thus, Maf1 is inactivated (phosphorylated), leading to increased Pol III activity. When TORC1 is inhibited by mutation or in response to calorie restriction, Maf1 is hypophosphorylated or dephosphorylated, resulting in reduced Pol III activity and lifespan extension ()). Intriguingly, a Pol III mutation, which reduces its transcriptional activity, was sufficient to rescue the short lifespan of maf1∆ cells [Citation3], suggesting that Maf1 controls lifespan by limiting Pol III activity. Consistently, inhibition of Pol III leads to lifespan extension in budding yeast, nematodes, and fruit flies [Citation10].
Figure 2. Maf1 phosphorylation dictates lifespan. (a) Maf1 is regulated by the TORC1 kinase via phosphorylation. This phosphorylation is reversed by PP2A phosphatases. Maf1 inactivation results in Pol III activation and elevated DNA damage, and thus lifespan shortening. Maf1 activation inactivates Pol III to limit DNA damage, leading to longer lifespan. (b) Schematic drawing of Maf1 orthologs from humans, S. pombe, S. cerevisiae, Drosophila, and C. elegans. Phosphorylation sites and total amino acid numbers are shown
Elevated Pol III activity has several consequences, including 1) increased tRNA/5S rRNA transcription, 2) increased protein synthesis supported by tRNA/5S rRNA transcription, and 3) DNA damage at Pol III-transcribed gene loci (tRNAs and 5S rRNA genes) due to conflicts between replication and transcriptional machineries. All of these factors might contribute to lifespan shortening (). However, we have excluded protein synthesis as a cause of lifespan shortening; as described earlier in this article, a reduction in protein translation failed to rescue the short lifespan of maf1∆ cells [Citation3].
A previous study suggested that the futile cycle of tRNA and 5S rRNA synthesis may result in high energy expenditure in mice [Citation11]. In support of this idea, we showed that maf1∆ cells have elevated levels of tRNA transcription. In addition, PP2A mutant (ppa1∆ and ppa2∆) cells, which harbor elevated Maf1 phosphorylation and Pol III activation, displayed increased tRNA synthesis and lifespan shortening [Citation3]. Therefore, we initially thought that increased energy expenditure associated with Pol III activity led to lifespan shortening in S. pombe. However, we obtained opposing results using another phosphatase mutant. It was previously shown that PP4 phosphatase also contributes to the dephosphorylation of Maf1 [Citation12]. When the PP4 phosphatase gene, pph3, was deleted, fission yeast cells failed to show lifespan shortening even though these cells had elevated Maf1 phosphorylation and tRNA synthesis [Citation3]. Rather, pph3∆ cells are slightly long-lived [Citation3]. Both PP2A and PP4 are known to dephosphorylate and thus activate Maf1 [Citation12]. However, PP4 has a negative role in the DNA repair process as PP4 dephosphorylates the checkpoint kinase Rad53 (Chk2 in humans) and histone H2AX to reduce checkpoint and DNA repair activity [Citation13–16]. Therefore, when PP4 is inactivated, cells have increased checkpoint activity, allowing additional time for DNA repair. In this situation, DNA repair capacity is elevated to reduce genomic instability. Indeed, pph3∆ cells have decreased levels of DNA damage [Citation3]. Furthermore, pph3 deletion resulted in alleviation of DNA damage and lifespan shortening of ppa1∆, ppa2∆, and maf1∆ cells [Citation3]. Considering that pph3∆ cells have elevated levels of tRNA transcription, our results suggest that energy expenditure associated with Pol III activity is unlikely to be a significant cause of lifespan shortening in maf1∆ cells.
Emerging evidence has suggested that high levels of transcription can cause DNA damage due to conflicts between DNA replication and transcription machinery, especially at highly transcribed genomic regions such as tRNA genes [Citation3,Citation17,Citation18]. It is also generally thought that DNA damage can induce aging processes, leading to a hypothesis that transcription-associated DNA damage limits lifespan [Citation19]. Consistent with this idea, our investigation revealed that tRNA genes break in the absence of Maf1 [Citation3]. Such DNA damage may shorten lifespan in maf1∆ cells. To address this question, it was important to create a situation where tRNA synthesis is elevated but DNA repair capacity is reduced. For this purpose, we have constructed maf1∆ rad52∆ mutant double cells. Rad52 is a protein required for DNA repair via homologous recombination. Therefore, rad52∆ cells have reduced DNA repair capacity. Importantly, the lifespan of maf1∆ rad52∆ double mutant cells was much shorter than that of either single mutant cells [Citation3]. Taken together with the fact that Pol III deficiency rescues the short lifespan of maf1∆ cells, our results are consistent with the notion that elevated DNA damage at tRNA and/or 5S rRNA genes contribute to lifespan shortening ().
tRNA genes break in the face of transcription-replication conflicts
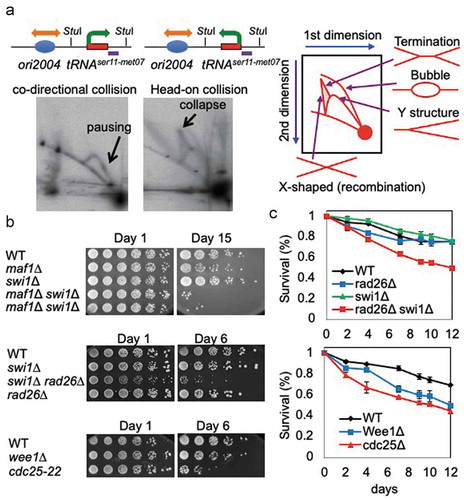
We further tested the idea that transcription-associated DNA damage is actually caused by replication-transcription conflicts at tRNA genes. For this purpose, we developed a system to monitor collisions between the replication and transcription machineries at a tRNA gene. One plasmid harbors the tRNASer11-Met07 gene in close proximity to ori2004, a well-characterized replication origin in S. pombe [Citation20]. In this construct, tRNA transcription is co-oriented with the replication fork that originates from ori2004, resulting in a co-directional collision between the replication and transcription machineries. In contrast, the other plasmid was designed to promote head-on collisions ()). These plasmids were introduced into wild-type S. pombe cells and analyzed by two-dimensional (2D) electrophoresis. Co-directional collisions led to an intense pausing signal on the Y-arc, indicative of replication fork pausing. In contrast, the head-on collisions between replication and transcription machineries resulted in the robust appearance of X-shaped DNA structures representative of replication fork breakage. We also observed a dramatic increase in replication termination structures ()). Such collisions associated with replication abnormalities may require a protein responsible for protecting replication fork structures. Indeed, loss of Swi1, involved in replication fork protection [Citation21,Citation22], further exacerbated the short lifespan of maf1∆ cells. As shown in ), maf1∆ swi1∆ double mutant cells had a much shorter lifespan than either single mutant cells. Therefore, these data suggest that transcription-replication conflicts induce DNA damage at tRNA genes, contributing to lifespan shortening.
It is important to note that, in the 2D-gel experiments shown in ), we used cells exponentially growing in rich medium. This medium allows robust cell growth, transcription, and DNA replication allowing us to efficiently determine replication fork structures. Although we haven’t tested how nutrient conditions affect fork integrity, cells that are slowly growing due to limited nutrient conditions may experience less fork collisions and subsequent DNA damage when compared to cells grown under rich conditions. This is consistent with the general observations that calorie or dietary restriction extends lifespan in many organisms.
Replication stress response prevents lifespan shortening
Interestingly, swi1∆ single-mutant cells are not short-lived ()). It is possible that replication-dependent DNA damage generated in swi1∆ cells is continuously repaired, leaving no lethal form of DNA damage. Indeed, DNA damage accumulated in swi1∆ cells activates the DNA damage checkpoint to facilitate DNA repair pathways [Citation22]. Therefore, to downregulate DNA repair activity, we deleted the rad26 gene. Rad26 is an S. pombe ortholog of the ATRIP protein. Rad26 is essential for the activation of the checkpoint kinase Rad3 (ATR homolog), which is involved in G2/M cell cycle arrest and DNA repair in response to DNA damage [Citation23]. Interestingly, swi1∆ rad26∆ double mutant cells showed a significant reduction in lifespan ()), suggesting that DNA damage repair and checkpoint-mediated G2/M regulation play an important role in the longevity of S. pombe cells. It is well understood that checkpoint’s final targets are Cdc25 and Wee1 to control CDK1 activity and arrest the cell cycle at the G2/M transition [Citation23]. Therefore, we tested the lifespan of cdc25 and wee1 mutants. Interestingly, we found that cdc25-22 mutants displayed a significantly shorter lifespan ()). We also observed a significant decrease of lifespan in wee1-50 mutant cells when we performed lifespan assays to obtain colony formation units of stationary liquid cell cultures ()). The effect of cdc25-22 and wee1-50 mutations on lifespan contrasts to that of rad26 deletion. Unlike Cdc25 and Wee1, which directly regulate the G2/M transition regardless of the status of DNA damage, Rad26 controls G2/M in response to DNA damage. Therefore, unless cells have DNA damage, rad26∆ cells are able to grow relatively normally without significant lifespan shortening ()). However, the function of Rad26 to arrest the cell cycle at G2/M becomes essential when the level of DNA damage is elevated in the absence of Swi1, thus accelerating aging processes ()). These results suggest that G2/M regulation minimizes lethal DNA damage, in order to promote lifespan extension.
Figure 3. Replication stress contributes to lifespan shortening. (a) 2D-gel analysis of replication forks in cells with pTKS-ori2004-tRNAser11met07Rear (left) or pTKS-ori2004-tRNAser11met07Head (right). Plasmids isolated from the cells were digested with StuI. The tRNAser11-met07 gene, within the StuI fragment, was analyzed via Southern blot. Schematic diagrams show the orientations of DNA replication and transcription. StuI restriction sites and the probed region are indicated. A diagram of the migration pattern of replication intermediates that can be detected in 2D-gel is shown. (b) Cells of the indicated genotypes were first grown under calorie restriction (0.1% glucose) for 1, 6, or 15 days in liquid medium. Fivefold serial dilutions of cells were then plated on solid agar medium containing 3% glucose to test cell viability. (c) The indicated cells were grown to stationary phase in liquid medium. The cultures were maintained at the indicated temperature for up to 12 days and cells were spread on agar medium to allow for colony formation. The number of colonies was counted to obtain colony formation unit per ml, and the survival rate of the 0-day culture was set to 1. Error bars correspond to standard deviations obtained from three independent experiments
mTOR-Maf1-Pol III axis as a therapeutic target to improve late life function
Preclinical models demonstrate that targeting mTOR provides late-life benefit, but there is a critical need to define additional targets of the mTOR pathway because the current inhibitors face difficulty transitioning into clinical use [Citation5]. As noted above, Maf1 is directly regulated via phosphorylation by TORC1 and dephosphorylation by PP2A and PP4 [Citation4]. When TORC1 is inhibited by calorie or dietary restriction, Maf1 becomes activated by dephosphorylation and then directly represses Pol III-directed transcription [Citation4] (). Calorie or dietary restriction extends lifespan [Citation1], and thus Maf1 activation and subsequent inhibition of Pol III activity by calorie or dietary restriction are expected to extend lifespan. Consistently, inhibition of Pol III extends lifespan in S. cerevisiae, C. elegans, and Drosophila [Citation10]. Conversely, elevated Pol III activity by Maf1 inactivation should lead to lifespan shortening. Indeed, S. cerevisiae maf1∆ cells have a shorter lifespan than wild-type cells under calorie restriction [Citation24,Citation25]. We have also demonstrated that S. pombe maf1∆ cells display elevated Pol III activity and lifespan shortening under calorie restriction [Citation3]. However, in C. elegans, inhibition of mafr-1 (Maf1 ortholog) extends lifespan, even though Pol III activity is elevated. This is attributed to elevated stress responses and autophagy, which seem to attenuate negative effects due to mafr-1 deficiency [Citation25]. Indeed, inhibition of Pol III still extends lifespan in this organism [Citation10], consistent with a negative role for Pol III activity in longevity, as also seen in S. cerevisiae and S. pombe [Citation3,Citation10]. In mice fed with standard chow diet, Maf1 knockout alters insulin signaling and prevents diet-induced obesity. This creates a calorie-restriction phenotype probably due to the increased turnover of tRNAs and lipids, leading to a small lifespan extension in females (a marginal effect in males) [Citation11]. However, this lifespan extension effect is slight, and this study did not investigate the effects of calorie or dietary restriction on lifespan regulation [Citation11]. Thus, only limited interpretation can be made from this single mouse study. In addition, in C. elegans and mammalian cells, Maf1 knockout results in lipogenic gene expression and lipid accumulation [Citation26,Citation27]. While in Drosophila, Maf1 deficiency stimulates insulin signaling, leading to increased growth and body mass [Citation28], which could shorten lifespan. These discrepancies are probably due to the differences in diets/nutrients, growth conditions, and organisms. Thus, the role of Maf1 in lifespan and growth control is critical, yet elusive.
In summary, we have discussed how the Maf1-Pol III axis of the mTOR pathway might regulate aging. Considering that mTOR regulates Maf1 and Pol III in a nutrient-dependent manner and Maf1-mediated Pol III regulation prevents DNA damage, we propose that the mTOR-Maf1-Pol III axis functions as a critical integration point for nutrient sensing and DNA damage response (). Further investigation is warranted to provide significant clinical implications as to how aging and nutritional conditions promote genetic instability and affect gene expression profiles in mammalian cells. In addition to the role of Maf1 in inhibiting Pol III, as noted above, Maf1 modulates the response to high-fat diet via regulation of lipid metabolism [Citation11,Citation26–Citation28]. This is potentially through Maf1’s role in regulating Pol II genes and/or an alteration in insulin signaling and mitochondrial function, which may also impact lifespan [Citation11]. Elucidating the role of Maf1 in cell homeostasis will provide insight into fundamental mechanisms linking nutrient sensing with DNA damage responses. We propose that, as a key mediator of the mTOR pathway, targeting Maf1/Pol III activity is a potential new approach for anti-aging therapies.
Materials and methods
Two-dimensional electrophoresis
Two-dimensional electrophoresis was performed as previously described [Citation29,Citation30]. Cells were transformed with the plasmid pTKS-ori2004-tRNAser11met07Rear or pTKS-ori2004-tRNAser11met07Head containing the origin 2004 and the tRNAser11met07 gene. Replication intermediates of the StuI fragments derived from these plasmids were detected.
Lifespan assays
Serial dilution growth assay for lifespan assessment was performed as previously described [Citation3]. The indicated strains were incubated for 1 to 15 days under calorie-restricted (0.1% glucose) conditions and tested for their viability on agar growth medium (3% glucose) at 25°C or 30°C. After incubation for 1 to 12 days as described above, cells were also spread on agar medium (3% glucose) to allow for colony formation. The number of colonies was counted to obtain colony formation unit per ml, and the survival rate of the 0-day culture was set to 1.
Acknowledgments
We thank members of the Noguchi laboratory for their support and encouragement. Leah Dobossy and Matthew Erlich are thanked for critical reading of this manuscript. This work was supported by Pennsylvania Department of Health Formula Grant (to EN) and the Aging Initiative at Drexel University College of Medicine (to EN).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345.
- Eltschinger S, Loewith R. TOR complexes and the maintenance of cellular homeostasis. Trends Cell Biol. 2016;26:148–159.
- Shetty M, Noguchi C, Wilson S, et al. Maf1-dependent transcriptional regulation of tRNAs prevents genomic instability and is associated with extended lifespan. Aging Cell. 2020;19:e13068.
- Zhang S, Li X, Wang HY, et al. Beyond regulation of pol III: role of MAF1 in growth, metabolism, aging and cancer. Biochim Biophys Acta. 2018;1861:338-343.
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183–203.
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35.
- Hannan KM, Brandenburger Y, Jenkins A, et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23:8862–8877.
- Michels AA, Robitaille AM, Buczynski-Ruchonnet D, et al. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol. 2010;30:3749–3757.
- Khanna A, Pradhan A, Curran SP. Emerging Roles for Maf1 beyond the regulation of RNA polymerase III activity. J Mol Biol. 2015;427:2577–2585.
- Filer D, Thompson MA, Takhaveev V, et al. RNA polymerase III limits longevity downstream of TORC1. Nature. 2017;552:263–267.
- Bonhoure N, Byrnes A, Moir RD, et al. Loss of the RNA polymerase III repressor MAF1 confers obesity resistance. Genes Dev. 2015;29:934–947.
- Oler AJ, Cairns BR. PP4 dephosphorylates Maf1 to couple multiple stress conditions to RNA polymerase III repression. Embo J. 2012;31:1440–1452.
- Keogh MC, Kim JA, Downey M, et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 2006;439:497–501.
- O’Neill BM, Szyjka SJ, Lis ET, et al. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc Natl Acad Sci U S A. 2007;104:9290–9295.
- Chowdhury D, Xu X, Zhong X, et al. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46.
- Heideker J, Lis ET, Romesberg FE. Phosphatases, DNA damage checkpoints and checkpoint deactivation. Cell Cycle. 2007;6:3058–3064.
- Duch A, de Nadal E, Posas F. Dealing with transcriptional outbursts during S phase to protect genomic integrity. J Mol Biol. 2013;425:4745–4755.
- Rudolph CJ, Upton AL, Stockum A, et al. Avoiding chromosome pathology when replication forks collide. Nature. 2013;500:608–611.
- Callegari AJ. Does transcription-associated DNA damage limit lifespan? DNA Repair (Amst). 2016;41:1–7.
- Ogawa Y, Takahashi T, Masukata H. Association of fission yeast Orp1 and Mcm6 proteins with chromosomal replication origins. Mol Cell Biol. 1999;19:7228–7236.
- Noguchi E, Noguchi C, Du LL, et al. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol. 2003;23:7861–7874.
- Noguchi E, Noguchi C, McDonald WH, et al. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol. 2004;24:8342–8355.
- Nyberg KA, Michelson RJ, Putnam CW, et al. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–656.
- Cai Y, Wei YH. Distinct regulation of Maf1 for lifespan extension by Protein kinase A and Sch9. Aging (Albany NY). 2015;7:133–143.
- Cai Y, Wei YH. Stress resistance and lifespan are increased in C. elegans but decreased in S. cerevisiae by mafr-1/maf1 deletion. Oncotarget. 2016;7:10812-10826.
- Khanna A, Johnson DL, Curran SP. Physiological roles for mafr-1 in reproduction and lipid homeostasis. Cell Rep. 2014;9:2180–2191.
- Palian BM, Rohira AD, Johnson SA, et al. Maf1 is a novel target of PTEN and PI3K signaling that negatively regulates oncogenesis and lipid metabolism. PLoS Genet. 2014;10:e1004789.
- Rideout EJ, Marshall L, Grewal SS. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci U S A. 2012;109:1139–1144.
- Tanaka A, Tanizawa H, Sriswasdi S, et al. Epigenetic regulation of condensin-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56 acetylation. Mol Cell. 2012;48:532–546.
- Zaratiegui M, Vaughn MW, Irvine DV, et al. CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature. 2011;469:112–115.