
ABSTRACT
Amyotrophic Lateral Sclerosis (ALS) is a deadly neuromuscular disorder caused by progressive motor neuron loss in the brain and spinal cord. Over the past decades, a number of genetic mutations have been identified that cause or are associated with ALS disease progression. Numerous genes harbor ALS mutations, and they encode proteins displaying a wide range of physiological functions, with limited overlap. Despite the divergent functions, mutations in these genes typically trigger protein aggregation, which can confer gain- and/or loss-of-function to a number of essential cellular processes. Nuclear processes such as mRNA splicing and the response to DNA damage are significantly affected in ALS patients. Cytoplasmic organelles such as mitochondria are damaged by ALS mutant proteins. Processes that maintain cellular homeostasis such as autophagy, nonsense-mediated mRNA decay and nucleocytoplasmic transport, are also impaired by ALS mutations. Here, we review the multiple mechanisms by which mutations in major ALS-associated genes, such as TARDBP, C9ORF72 and FUS, lead to impairment of essential cellular processes.
KEYWORDS:
Introduction
Amyotrophic Lateral Sclerosis (ALS) is one of the deadliest neurodegenerative diseases with no cures currently available. Onset typically begins with weakness in limbs followed by whole-body paralysis due to progressive motor neuron (MN) degeneration in brain and spinal cord [Citation1–4]. Respiratory failure is a common cause of death at the terminal stage of the disease [Citation5,Citation6]. Although tremendous effort has been expended in previous decades to develop treatments for ALS, therapeutic options remain few because of insufficient understanding to the mechanisms underlying MN death. Only 10% of ALS cases are caused by inherited mutations, while the other 90% are sporadic without clear familial history [Citation7]. Although the fraction of ALS cases that result from a known mutation is small, the affected genes and loci, which number as many as 26, spread across a wide spectrum [Citation8], suggesting that MN death can be initiated through diverse mechanisms.
A prominent feature of many ALS mutations is that they lead to the appearance of protein aggregates. Such aggregates frequently arise from aberrant phase-separation of cellular proteins, often involving nuclear proteins aberrantly localized to the cytoplasm [Citation9–12]. The pathological role of ALS-associated protein aggregates has been elusive though, as protein aggregation can serve a protective role in the cellular response to stress [Citation13]. For example, it was reported that accumulation of aggregates containing mutant superoxide dismutase 1 (SOD1) in the CNS of ALS SOD1 mutant mice was inversely correlated with disease progression, while high levels of misfolded but soluble SOD1 were more toxic. This apparent neuroprotective role of the SOD1 aggregates was proposed to be to sequester the toxic soluble species [Citation14]. It was also reported that SOD1 oligomers are cytotoxic while large fibril aggregates may play a protective role against cell death [Citation15]. Nonetheless, evidence has suggested that unresolved ALS-associated protein aggregates, including SOD1, can disrupt critical cellular processes such as mRNA metabolism, mitochondrial functions and nucleocytoplasmic transport [Citation16–20]. Genes harboring ALS mutations have also been reported to acquire novel functions, resulting in unwanted activation or suppression of certain pathways, which may contribute to MN death [Citation21–23]. Finally, ALS mutations can cause protein mis-localization and/or sequestration, and thus lead to loss-of-function at their normal functioning sites [Citation24–26].
In this review, we discuss how multiple essential cellular processes are disrupted by ALS mutations, how these mutations affect the encoded proteins, and how these diverse pathways lead to MN cell death. The interested reader is referred to several recent related reviews [Citation27–31].
ALS mutations profoundly alter biochemical properties and molecular functions of the encoded proteins and in one case RNA
Aggregation of RNA binding proteins (RBPs) in neural tissue is a pathological hallmark of ALS. Many of these RBPs contain low complexity domain(s) (LCD) involved in physiological granule formation through liquid-liquid phase separation (LLPS) [Citation28,Citation32]. ALS mutations in LCD-containing RBPs, FUS for instance, have been shown to undergo excessive LLPS and aggregate formation through LCD interactions [Citation12,Citation33]. Under normal circumstances, FUS is imported by the nuclear import receptor TNPO-1 through interactions with its nuclear localization signal (NLS) [Citation34]. Binding of TNPO-1 to the NLS and other FUS regions not only shuttles FUS from the cytoplasm to nucleus, but also inhibits FUS LLPS by preventing FUS–FUS interactions [Citation35,Citation36]. The NLS, located at the protein’s C-terminus, is a hotspot for ALS mutations [Citation37]. Mutations in this region disrupt FUS-TNPO-1 interactions and lead to FUS cytoplasmic accumulation [Citation34]. It has been shown that RNA serves as a buffering system to prevent excessive FUS LLPS. Since RNA concentrations are lower in the cytoplasm than in the nucleus, ALS mutations that lead to cytoplasmic localization appear to also promote FUS LLPS by this mechanism [Citation33].
Phase separation is an important step in formation of RBP condensates under diverse cellular contexts. Physiological functions of RBP condensates have been suggested in organismal development, stress responses and synaptic transmission [Citation38–40]. Although the detailed mechanisms remain unclear, RBP condensates are thought to achieve selective gene regulation under certain conditions by altering cellular protein and RNA stoichiometry via specific sequestration. For example, stress granules specifically recruit the autophagy inhibitor complex TORC1 during heat stress to protect cells from DNA damage [Citation41]. Paraspeckles are nuclear condensates that sequester multiple nuclear-encoded mitochondrial mRNAs during mitochondrial stress [Citation42]. Therefore, extensive RBP condensate formation in ALS patient cells can be part of stress response pathways. Nonetheless, physiological RBP condensates such as stress granules are dynamic structures that constantly exchange proteins and RNAs with the external environment [Citation43,Citation44]. Physiological stress granules also undergo assembly-disassembly cycles depending on the existence or absence of stress [Citation45]. Persistent stress granules can further transit into solid-state protein aggregates [Citation12,Citation46]. As proteins and RNAs sequestered by the overly stable aggregates are not available for cellular processes, aggregate-mediated sequestration may contribute to diseases such as neurodegeneration and cancer [Citation47,Citation48]. This will be discussed further below in the context of specific ALS mutations.
In addition to aberrant phase separation/aggregation of mutant proteins, ALS mutations can also lead to expression of novel toxic proteins or RNAs. For example, expansion of a GGGGCC hexanucleotide in an intron of the C9ORF72 gene results in toxic gain-of-function through protein sequestration and production of dipeptide repeat (DPR) proteins via repeat-associated non-AUG (RAN) translation [Citation49–53]. Both processes are found in ALS, and in frontotemporal dementia (FTD), a neurological disorder that shares overlapping clinical manifestations and pathological features with ALS, and are suggested to induce neurotoxicity [Citation54,Citation55]. Cytoplasmic mis-localization of ALS-associated RBPs may impart novel functions due to altered protein or RNA interactions. Indirect evidence based on protein and RNA interactome studies have shown that cytoplasmic FUS and TDP-43 display distinct protein and RNA interacting partners [Citation17,Citation56,Citation57]. These aberrant interactions between mis-localized RBPs and cytoplasmic components likely impair a range of pathways underlying ALS pathogenesis.
mRNA processing
Pre-mRNA alternative splicing (AS) greatly expands the diversity of mRNAs and their encoded proteins. By regulating expression of mRNA isoforms, gene activities can be modulated under different cellular contexts [Citation58]. Transcriptomic analyses of familial ALS patient brains and cells expressing ALS mutant genes show wide-spread RNA mis-splicing [Citation59,Citation60]. Several lines of evidence suggest that mis-splicing may be associated with sequestration of splicing factors or regulatory RBPs by toxic proteins or RNA repeats. The GGGGCC expansion in the C9ORF72 gene, the most common known mutation in ALS/FTD, is found in 40–50% of familial ALS patients and in 5–10% of sporadic patients [Citation61]. The GC-rich repeats are transcribed as part of an intronic region in C9ORF72, and bind RBPs, notably hnRNP H. Sequestration of hnRNP H by the RNA repeats depletes functional, soluble hnRNP H from the nucleoplasm, resulting in mis-splicing of hnRNP H-target transcripts [Citation49,Citation51]. Notably, several of these mis-spliced transcripts are either involved in pathways implicated in excitotoxicity and protein degradation, or directly associated with ALS/FTD and other neurological disorders [Citation49]. Consistent with this notion, a recent study has shown that C9 ALS/FTD brains with high levels of insoluble hnRNP H display significantly elevated intron retention events, especially in transcripts encoding proteasome machinery subunits [Citation62]. Interestingly, formation of insoluble hnRNP H-containing aggregates and mis-splicing of target transcripts occurs not only in C9 ALS/FTD, but also in a significant fraction of sporadic patients without C9ORF72 expansions [Citation63]. In some cases, aberrant splicing may be caused by sequestration of spliceosomal components. Aggregates of ALS mutant FUS have been reported to retain U11 and U12 snRNPs and thereby deregulate splicing of minor introns [Citation64]. Additionally, spliceosomal U1 and U2 snRNAs have also been suggested to be sequestered in mutant FUS aggregates, contributing to ALS-associated splicing dysregulation [Citation65]. TDP-43 cytoplasmic aggregates also affect mRNA processing, in one example by compromising the protein’s ability to repress cryptic exon splicing [Citation66]. Loss of nuclear TDP-43 due to cytoplasmic mis-localization was shown to result in increased cryptic exon inclusion, leading to frameshifts and mRNA substrates for nonsense-mediated decay (NMD). However, the consequences of splicing factor mislocalization and how this contributes to ALS pathogenesis require further investigation.
Alternative polyadenylation (APA) is also an important mechanism of gene control [Citation65]. Citation67Similar to aberrant AS, wide-spread differential APA has been observed in C9-ALS patient brains [Citation59]. Since splicing factors including hnRNP H can also function in APA regulation [Citation68,Citation69], it is not surprising that APA is affected along with AS by RBP sequestration. Other ALS-associated RBPs, such as FUS and TDP-43, have been shown to control APA [Citation70,Citation71], and mutations that cause RBP aggregation and mis-localization may thus also dysregulate APA. Moreover, mRNA 3’UTRs contain binding sites for microRNAs and a number of RBPs that control mRNA stability, localization and translation [Citation72]. Therefore, APA events that modulate 3’UTRs potentially alter mRNA functions in disease. Indeed, fibroblasts or iPSCs derived from C9-ALS patients show RNA destabilization and differential protein expression [Citation73]. Interestingly, the destabilized transcripts and downregulated proteins are enriched in mitochondrial gene products, which may contribute to mitochondrial dysfunction in ALS patient MNs (see also below). In spite of these findings, however, direct links between APA dysregulation and abnormal mRNA stability in ALS patient brains remain to be established.
Many RBPs, including FUS and TDP-43, possess the ability to autoregulate their expression through alternative mRNA processing. When levels of nuclear FUS are high, the protein binds to exon 7 in its own pre-mRNA and thereby promotes exon 7 skipping. The resultant spliced variant contains a premature stop codon, which leads to NMD [Citation74]. Elevated TDP-43 expression promotes formation of an alternatively spliced TDP-43 mRNA isoform that tends to undergo nuclear retention via an unknown mechanism [Citation75]. Since a number of splicing factors are sequestered in ALS-associated protein aggregates or by toxic RNA repeats, it is conceivable that autoregulation of LCD-containing RBPs may be compromised and lead to excessive RBP production, which in turn could accelerate LLPS and aggregate formation. It has been shown that overexpressing wild-type FUS or TDP-43 alone is sufficient to induce protein aggregation and MN degeneration in mice [Citation76,Citation77]. Therefore, loss of LCD-containing RBP autoregulation could provide a link between RNA processing dysregulation and ALS pathogenesis.
Nonsense-mediated decay
NMD is one of the major pathways that regulates mRNA stability. NMD is coupled with both splicing and translation and is activated when a premature stop codon occurs within an open reading frame within a certain distance upstream of an exon-exon junction [Citation78]. Differential NMD activity has been widely observed in ALS patients and cell models [Citation17,Citation79–81]. Mutant FUS derivatives were shown to aberrantly increase NMD activity in transfected cells and ALS patient fibroblasts [Citation17]. Proteomic analysis has shown that ALS mutant FUS aggregates are enriched in translation and NMD factors, which promotes premature translation termination and enhanced interaction of NMD factors [Citation17]. Immunoprecipitation of the NMD factor UPF1 from mutant FUS-transfected N2a cells shows increased co-precipitation of NMD drivers including UPF3B, phosphorylated UPF1 and SMG6 endonuclease relative to cells expressing wild-type FUS. In addition, mutant FUS-ALS patient fibroblasts display increased levels of NMD positive regulators UPF1 and UPF3B, and downregulation of the NMD repressor UPF3A, compared to wild-type FUS patients [Citation17].
Increased NMD activity was also observed in C9-ALS patient cells. A recent study reports that eRF1, an essential translation termination and NMD factor, accumulates at the nuclear envelope along with other NMD factors to prevent export of C9 repeat-containing RNA and facilitate its degradation in C9-ALS patient-derived MNs [Citation79]. Overexpression of eRF1 in transgenic flies with C9 expansions reduces neurotoxicity and DPR expression [Citation79]. While NMD hyperactivation was found in mutant FUS and C9ORF72 expansion expressing cells, other studies suggest that DPRs inhibit NMD in C9 ALS patient brains and cultured neurons [Citation80,Citation81]. The reasons for the differential observations remain unclear. Nonetheless, it has been consistently observed that overexpression of Upf1 ameliorates neurotoxicity caused by mutant C9ORF72 C9-ALS[Citation79–81]. Furthermore, NMD activation not only reduces neuronal death caused by mutant C9ORF72, but also improves the survival rate of mutant TDP-43- and FUS-expressing neurons [Citation82]. The improved cell survival is possibly achieved by enhancing degradation of mRNAs encoding mis-folded proteins following NMD activation.
Overall, these findings suggest that enhanced NMD may play a protective role in ALS patient MNs. However, hyperactivation of NMD may not be beneficial since phosphorylated UPF1 can repress translation by preventing 80S translation initiation complex formation [Citation83]. Indeed, it was shown that UPF1 increases death of mutant TDP-43-transfected neurons when exogenous UPF1 expression exceeds endogenous levels by 10 fold [Citation80]. As a result, NMD hyperactivation and inhibition may both be deleterious to neurons in the long term.
Mitochondrial functions
Mitochondrial abnormalities and defective energy metabolism are prevalent features reported in ALS patients and animal models [Citation84]. For example, morphological observations with familial ALS patient cells have shown loss of mitochondrial cristae integrity [Citation85,Citation86]. In addition, cells expressing certain ALS mutant genes show increased mitochondrial reactive oxygen species (ROS), reduced ATP synthesis and altered mitochondrial dynamics [Citation86–88]. Mechanistic studies have suggested that mitochondrial defects induced by ALS mutations can be attributed to abnormal interactions between mis-localized proteins and mitochondrial components and unwanted stress response activation [Citation87,Citation87–91]. Mutant SOD1, for instance, was found associated with the mitochondrial outer membrane and to promote apoptosis via aggregating with the anti-apoptotic mitochondrial protein Bcl-2 in human and mouse spinal cord [Citation19].
Pathological sequestration of RBPs and resulting effects on mRNA metabolism are now thought to be an important contributing factor to neuronal toxicity. Amongst the challenges faced by researchers in this area are the promiscuous binding properties of RBPs and, since protein aggregates appear to sequester many RNAs, it is difficult to associate (a) specific group(s) of mRNAs with ALS pathogenesis. With the aid of high-throughput sequencing and peptide synthesis technology, sequestration of mRNAs has been recently linked to mitochondrial dysfunction in ALS [Citation89–91]. It was shown that ALS mutant TDP-43 suppresses mitochondrial-encoded ND3 and ND6 protein expression by aberrantly binding to their mRNAs. Blocking TDP-43 translocation to mitochondria using synthetic peptides restored ND3 and ND6 protein levels and mitochondrial functions [Citation91]. Similar results were observed in ALS mutant FUS-expressing mouse neurons. Combinatorial analysis of ribosome profiling and RNA-IP revealed that mutant FUS binds to nuclear-encoded mitochondrial mRNAs and represses their translation. Perturbing the RNA binding ability of mutant FUS restored target mitochondrial protein levels and partially rescued neurotoxicity [Citation89]. It has also been recently shown that nuclear-encoded respiratory chain complex mRNAs are preferentially sequestered by insoluble FUS, which leads to reduced expression of the encoded proteins. Depletion of the FUS-targeted respiratory chain proteins using siRNAs is sufficient to bring about the same mitochondrial dysfunction as observed in cells expressing mutant FUS [Citation90]. Collectively, these studies suggest that disrupting expression of mRNAs encoding mitochondrial proteins may contribute to the abnormal energy metabolism in ALS patient cells.
Mutant RBPs can also disrupt mitochondrial functions by interacting with mitochondrial proteins or activating stress response pathways. It was shown that ALS mutant or excessive wild-type FUS localizes to mitochondria and interacts with ATP synthase F1 subunit beta (ATP5B) [Citation87]. The FUS–ATP5B interaction destabilizes mitochondrial complex V and impairs ATP synthesis. Similar to FUS, ALS mutant or excessive wild-type TDP-43 accumulates at mitochondria and suppresses complex I activity [Citation92]. Additionally, both FUS and TDP-43 activate the mitochondrial unfolded protein response (UPRmt) [Citation87,Citation92]. The UPRmt is one of the mitochondrial quality control pathways characterized by increased transcription of mitochondrial stress response genes to promote cell survival [Citation91]. However, prolonged UPRmt activation was reported to induce cytotoxicity in a C. elegans Parkinson’s disease model [Citation94]. Interestingly, downregulating the UPRmt-related gene LonP1 appeared to show opposite effects on neurodegeneration in FUS and TDP-43 transgenic flies. While knocking down LonP1 partially rescued FUS-induced retinal degeneration, the same knockdown exacerbated the degeneration phenotype and mitochondrial damage in TDP-43 expressing flies [Citation87,Citation92]. While the basis for this difference is not known, investigating the outcomes of prolonged UPRmt in models expressing different ALS mutant genes may further our understanding of the role of the mitochondrial stress response in ALS disease progression.
Mitochondria play pivotal roles in ion homeostasis by interacting with other organelles. Approximately 5–20% of the mitochondrial surface associates with endoplasmic reticulum (ER) to form a mitochondria-associated ER membrane (MAM) [Citation95]. MAM facilitates ER-mitochondria crosstalk and exchange of materials such as calcium and lipids [Citation96,Citation97]. Mitochondria can uptake calcium released from the ER through MAM and thus regulate their mobilities and ATP synthesis [Citation97,Citation98]. The ER protein VAPB interacts with mitochondrial protein PTPIP51 to form an ER-mitochondria tethering complex [Citation96,Citation99]. Overexpression of wild-type TDP-43 or FUS, or mutant derivatives, has been shown to disrupt VAPB–PTPIP51 interaction, resulting in ER-mitochondria dissociation [Citation22,Citation99]. Losing MAM results in reduced mitochondrial calcium uptake and aberrant energy metabolism. Indeed, wild-type and mutant TDP-43 or FUS transfected cells inhibit mitochondrial calcium uptake. However, TDP-43 and FUS do not directly interact with the VAPB-PTPIP51 complex. Instead, TDP-43 and FUS overexpression activates glycogen synthase kinase GSK3ß, which was shown to be a regulator of ER-mitochondria association. Intriguingly, pharmacological inhibition of GSK3ß in TDP-43 and FUS expressing cells promotes VAPB–PTPIP51 interaction, ER-mitochondria association and mitochondrial calcium uptake [Citation22,Citation99]. Remaining questions include how TDP-43 and FUS activate GSK3ß, and whether such activation occurs in ALS patients.
Nucleocytoplasmic transport
Nucleocytoplasmic transport defects have been linked to neurodegenerative diseases including ALS. It has been shown that protein aggregates or RNA repeat expansions induce nucleocytoplasmic redistribution of proteins and RNAs. TDP-43 protein aggregation is one of the most prevalent pathological features in ALS, detected in >90% of all ALS cases [Citation30]. Therefore, nucleocytoplasmic transport disruption in ALS patient cells is not unexpected. Indeed, as a proof-of-principle study, HEK293T cells expressing TDP-43 at sufficient levels to form cytoplasmic aggregates display a block to mRNA nuclear export [Citation20]. Suggesting a possible mechanism, the THO mRNA nuclear export complex subunit THOC2 was found to mis-localize to the cytoplasm, although THOC2 did not co-localize with the TDP-43 cytoplasmic aggregates [Citation20]. A later study revealed that components of the nucleocytoplasmic transport machinery were enriched in TDP-43 aggregates, including nucleoporins from the nuclear pore complex (NPC) and nuclear export factors. Human cells with TDP-43 protein aggregates display defects in the NPC, mRNA export and nuclear protein import, as well as irregular nuclear membrane morphology [Citation56]. Finally, DPRs translated from C9ORF72 expansion repeats also disrupt nucleocytoplasmic transport, by binding crucial proteins involved in the transport pathway including RanGAP1 and importin ß [Citation50,Citation100]. RanGAP1 is a GTPase-activating protein that facilitates efficient release of transport receptors and cargoes [Citation101,Citation102]. It was shown that RanGAP1 co-localizes with poly-GA aggregates, one of the DPRs derived from C9 RAN translation, in adeno-associated virus transduced mice [Citation100]. Arginine-containing DPRs, poly-PR and poly-GR, were found to interact with importin ß and to prevent cargo loading as well as nuclear import in mouse neurons and HeLa cells. Nonetheless, this process does not require formation of DPR aggregates [Citation50].
While RBP pathology disrupts nucleocytoplasmic transport, deleterious mutations in NPC subunits were also discovered in ALS patients. The GLE1 gene encodes a nucleoporin existing as two isoforms, GLE1A and GLE1B [Citation103]. The major isoform, GLE1B, is important for mRNA export and is primarily localized in the NPC by interacting with nucleoporin hCG1 [Citation104]. A nonsense mutation c.209 C > A and an intronic splice site mutation c.1965–2A>C in GLE1 were found in a sporadic and a familial ALS patient, respectively [Citation105]. The 69-aa truncated GLE1 protein S70X encoded by the nonsense mutation allele was undetectable in patient lymphoblastoid cells. The splice site allele produces a less stable GLE1 derivative with a novel C-terminus that fails to interact with hCG1 and thus leads to its absence in the NPC. Lymphoblastoid cell lines derived from ALS patients carrying either mutation also display lower levels of GLE1 mRNA. These data suggest that GLE1 haploinsufficiency may cause ALS [Citation105]. In fact, a role for GLE1 was suggested in MN development. Autosomal recessive mutations in GLE1 were shown to cause lethal congenital contracture syndrome 1 (LCCS1) and lethal arthrogryposis with anterior horn cell disease (LAAHD) [Citation106]. Both diseases are characterized by underdevelopment of spinal cord MNs in fetuses [Citation107,Citation108]. In addition to human studies, conditional ablation of nucleoporin Nup358 in transgenic mouse MNs was reported to disrupt a wide range of nucleocytoplasmic transport events [Citation109]. The Nup358-deficient mice displayed defects in MN functions and lipid metabolism [Citation109]. Although GLE1 and Nup358 have not been shown to be ALS-causative, loss of nucleocytoplasmic homeostasis appears to render MNs susceptible to degeneration.
Autophagy
Autophagy is the process by which misfolded proteins and damaged organelles are degraded. It begins with nucleation of autophagy-related protein-containing complexes following phagophore expansion, then subsequently progresses to autophagosome formation, autolysosome formation and substrate degradation [Citation110]. Several autophagic genes are tightly associated with ALS, such as optineurin (OPTN), sequestosome-1/p62 (SQSTM1/p62) and TANK-binding kinase 1 (TBK1) [Citation111–114]. Optineurin and p62 are autophagy adaptors that bind to polyubiquitinated substrates (e.g. protein aggregates, damaged mitochondria) and initiate autophagosome formation [Citation115]. TBK1 promotes substrate recruitment to autophagosomes by phosphorylating autophagy adaptors and enhances adaptor binding to polyubiquitin [Citation116,Citation117]. ALS mutations in OPTN and p62 suppress binding of the mutant proteins to substrates and result in accumulation of protein aggregates and damaged organelles [Citation118,Citation119]. Mutations in TBK1 suppress the protein’s kinase activity and compromise autophagy adaptor phosphorylation, which possibly reduces substrate recruitment to autophagosomes [Citation120]. Furthermore, LCD-containing RBPs such as TDP-43 accumulate in autophagy-compromised cells [Citation118], and autophagy induction promotes TDP-43 aggregate turnover and survival of neuronal cells [Citation121].
While mutations in autophagic genes cause ALS, dysregulation of certain other ALS genes reveals their unexpected roles in autophagy regulation. For example, loss of TDP-43 in flies blocks autophagosome-lysosome fusion and induces neurotoxicity [Citation122]. Activation of mTORC1 signaling ameliorates neurodegeneration in the TDP-43 depleted flies. Therefore, TDP-43 appears to be crucial to cellular proteostasis through autophagy regulation. FUS also appears to play a role in autophagy, as FUS dysregulation also inhibits autophagy. N2a cells overexpressing wild-type or ALS mutant FUS show autophagosome and p62 accumulation, suggesting that autophagy flux is inhibited by elevated wild-type FUS or mutant FUS expression [Citation21,Citation123]. Citation126 The C9ORF72 protein has also emerged as an autophagy regulator, complexing with p62, ULK1, SMCR8 and WDR41 to function in autophagy initiation, substrate recognition and autophagosome-lysosome fusion [Citation124,Citation125]. In addition to sequestering RBPs as described above, the C9ORF72 hexanucleotide repeats both suppress C9ORF72 protein levels and also produce toxic DPRs [Citation51,Citation124,Citation125]. Reduced C9ORF72 protein levels impair autophagy [Citation125,Citation128,Citation129], which in turn leads to toxic DPR accumulation and contributes to cell death [Citation127]. SOD1 is an antioxidant enzyme converting superoxide to hydrogen peroxide and oxygen [Citation130]. However, it was reported in SOD1 ALS mice that mutant SOD1 impairs mitophagy by disrupting dynein-endosome coupling and leads to accumulation of damaged mitochondria [Citation131].
Senataxin (SETX), an RNA:DNA helicase associated with both type 4 ALS (ALS4) and ataxia with oculomotor apraxia type 2 (AOA2), was shown recently to function in autophagy regulation [Citation132]. SETX was previously shown to function with the nuclear exosome to resolve R loops [Citation133,Citation134]. Preventing R-loop accumulation is especially important for maintaining genomic stability during transcription termination and transcription-replication fork collision [Citation134,Citation135]. In addition to R-loop resolution, SETX also affects expression of a number of genes through alternative RNA processing and transcriptional efficiency [Citation132]. SETX knockdown leads to substantial changes in autophagy gene expression, which is strongly correlated with the perturbed autophagy phenotype observed in SETX-knockdown and AOA2 patient cells.
DNA damage response
Maintenance of genomic stability in neurons is vital since neurons cannot be replaced by cell division [Citation136]. Many lines of evidence have shown that ALS mutant genes compromise DNA repair pathways and increases ROS production, which can ultimately lead to cell death [Citation137]. An elevated DNA damage response (DDR) has been observed in the central nervous system of ALS patients and transgenic models [Citation138,Citation139]. Although enhanced DDR may be a consequence of toxic protein accumulation [Citation140], several ALS-associated proteins also function in DDR signaling and genomic stability maintenance.
FUS is as described above an RBP, but it is also directly involved in the early stages of DDR signaling. Loss of nuclear FUS due to ALS-associated NLS mutations has been shown to impair proper DDR signaling [Citation26,Citation141]. Direct interaction of nuclear FUS and the histone deacetylase HDAC1 is required for DNA double-strand break (DSB) repair through non-homologous end joining (NHEJ) and homologous recombination [Citation26]. Cells expressing ALS mutant FUS show reduced efficiency in both NHEJ and homologous recombination-mediated DNA repair [Citation26]. FUS recruitment to DNA double/single strand breaks is mainly mediated by Poly(ADP-ribose) polymerase 1 (PARP-1). PARP-1 catalyzes protein poly ADP-ribosylation (PARylation) at DNA damage sites to recruit downstream DDR factors [Citation25,Citation142]. It was shown that FUS complexes with PARP-1, the DNA repair protein XRCC1 and the DNA ligase LIGIII at DNA damage sites in response to oxidative stress [Citation25]. Through direct interactions, FUS activates LIGIII activity for single-strand break repair. Loss of nuclear FUS by expressing ALS mutant derivatives or CRISPR knockout in human cells results in inefficient DNA ligation [Citation25]. PARP-dependent DDR signaling also modulates FUS nucleocytoplasmic distribution [Citation143,Citation144]. Wild-type FUS displays drastic cytosolic localization and aggregation following PARP-1 inhibition, whereas inhibition of the PAR-degrading enzyme PAR glycohydrolase (PARG) retains mutant FUS in the nucleus [Citation143]. Intriguingly, a subsequent study showed that although PARG inhibition releases FUS from DNA damage sites, PAR inhibition increases FUS cytoplasmic translocation [Citation144]. Since the two studies used different cell systems (neuron versus HeLa), and FUS shuttling in response to osmotic stress is cell type-dependent [Citation145], it is possible that FUS shuttling in response to DNA damage also varies between cell types.
A role for TDP-43 in DDR was also reported. It was shown that TDP-43 is recruited to DSB sites to facilitate NHEJ-mediated DNA repair. Co-IP experiments performed in iPSC-derived neural progenitor cells demonstrated that TDP-43 associates with the DSB repair XRCC4/LIG4 complex, but not with the single-strand break repair complex XRCC1/LIGIII, suggesting that TDP-43 is specifically involved in DSB repair [Citation146]. Nuclear loss of TDP-43 due to ALS mutations or aggregation compromises NHEJ and correlates with increased DNA damage in ALS patients [Citation146]. However, it is unclear whether TDP-43 recruitment to DSB sites is a PARP-1-mediated process.
In addition to impairment of DDR caused by nuclear FUS/TDP-43 loss-of-function, ALS mutations can also compromise genomic integrity through toxic gain-of-function. Five DPRs (poly-GA, -GR, -PR, -PA and -GP) are translated from the C9ORF72 RNA expansion by RAN translation [Citation147]. Among them, poly-GR, -PR and -GA are suggested to be neurotoxic (see above) and to inhibit DNA repair [Citation147,Citation148]. The inhibition is correlated with abnormal binding of DPRs to DNA repair factors. In cultured human cells, poly-PR was shown to bind nucleophosmin NPM1, a nucleolar protein involved in DNA repair, and thereby prevent its translocation to DNA damage sites [Citation148]. In C9-ALS patients, the DSB sensor phospho-ATM was detected in poly-GA cytoplasmic aggregates, suggesting inhibition of phospho-ATM recruitment to nuclear DSB sites, thereby compromising DDR signaling [Citation140]. How poly-GR participates in DDR signaling impairment is unclear. Nonetheless, it has been shown that poly-GR interacts with mitochondrial ribosomal proteins and leads to increased oxidative stress [Citation149]. Poly-GR expression also inhibits NHEJ at DSBs in cultured human cells [Citation148]. It is likely that poly-GR damages neuronal DNA by increasing ROS while inhibiting NHEJ, thus creating a vicious cycle.
Conclusions
An increasing number of cellular processes that are affected in ALS have been discovered. In keeping with the large number of genes that can be mutated in ALS, the mechanisms that trigger MN cell death are diverse. ALS mutations can induce both loss- and gain-of-function by profoundly changing protein, and RNA, properties. Accelerated phase separation can lead to excessive aggregate formation. The aggregated proteins and RNAs are therefore sequestered and prevented from functioning at their normal sites. Unresolved aggregation further activates prolonged stress response pathways that could be detrimental to cells in the long term. Mis-localized mutant proteins and toxic peptides obtain novel functions via interacting with new partners and cellular components. The discussed mechanisms by which TARDBP (), FUS () and C9ORF72 () mutations can compromise essential cellular processes are illustrated in the indicated figures and summarized in .
Figure 1. TDP-43 dysregulation compromises a range of essential cellular processes through gain- and/or loss-of-function. Gain-of-function: TDP-43 cytoplasmic aggregates sequester multiple nucleoporin components of the nuclear pore complex (NPC), leading to abnormal nuclear membrane morphology and defective nucleocytoplasmic transport. Cytoplasmic TDP-43 also localizes to mitochondria, binds to mRNAs encoding respiratory chain complex I core subunits ND3 and ND6, and represses their translation. Reduced respiratory chain complex protein expression likely results in energy deficiency. TDP-43 promotes ER-mitochondria dissociation by disrupting VAPB–PTPIP51 interactions and thus decreases mitochondrial calcium uptake. Loss-of-function: Mutant TDP-43 loses interactions with XRCC4/LIG4 complex at DSB sites due to cytoplasmic mis-localization and compromises NHEJ. Notably, loss of TDP-43 leads to cryptic exons' inclusion into mRNAs that causes frameshifts and NMD. TDP-43 appears to mediate autophagosome-lysosome fusion. Loss of TDP-43 blocks the fusion process and impairs autolysosome formation
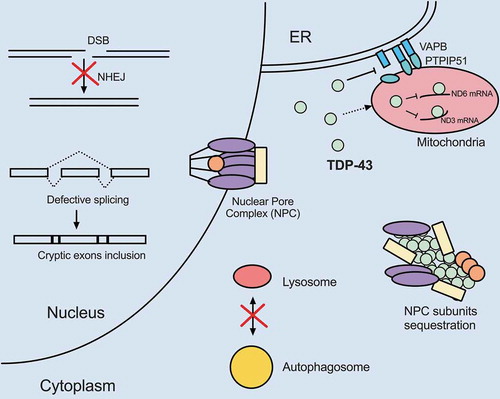
Figure 2. FUS dysregulation compromises nuclear and cytoplasmic processes shared with TDP-43. Overexpression of wild-type or expression of mutant FUS appear to disrupt pathways similar to TDP-43 dysregulation. FUS localizes to mitochondria and disrupts respiratory chain complex V assembly and results in energy deficiency. FUS gain-of-function also disrupts VAPB–PTPIP51 interactions and promotes ER-mitochondria dissociation. FUS plays physiological roles in DSB and SSB repair. Therefore, loss of nuclear FUS due to cytoplasmic mis-localization compromises DDR and increases DNA damage. FUS also participates in splicing, especially autoregulation of its own transcript level. Loss of nuclear FUS impairs autoregulation and results in increased FUS mRNA production. Mutant FUS cytoplasmic aggregates sequester spliceosome, translation and NMD factors, leading to mis-splicing of introns, pre-mature translational termination and hyper NMD. Additionally, FUS aggregates sequester numerous mRNAs including nuclear-encoded mitochondrial mRNAs and represses their translation. Deficiency in these mitochondrial components further leads to mitochondrial impairment. Finally, overexpression of wild-type or expression of mutant FUS inhibits autophagosome-lysosome fusion
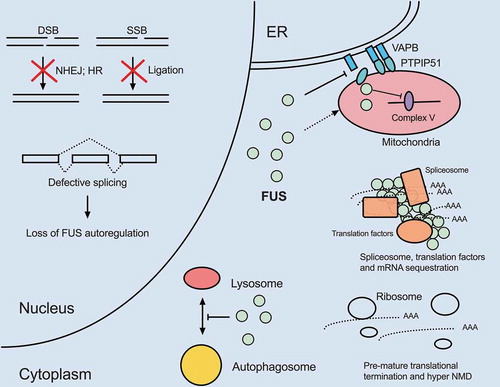
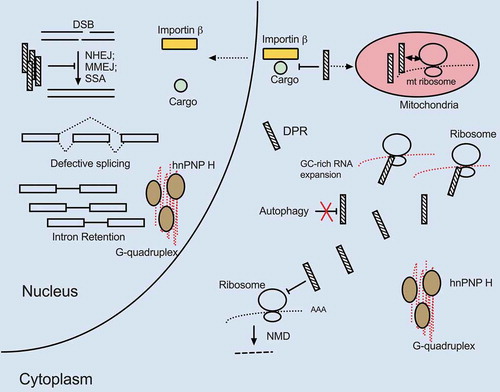
Figure 3. Mutant C9ORF72 triple hits. The GC expansion mutation within a C9ORF72 intron causes highly structured RNA G-quadruplex formation, DPR production through RAN translation and reduced C9ORF72 protein expression. All three processes are deleterious to cells and increase the likelihood of MN death. G quadruplex-containing C9 RNA sequesters hnRNP H, resulting in widespread transcripts mis-splicing, including of transcripts encoding proteasome subunits essential for protein degradation. The GC-rich intronic expansion can be translated via RAN translation, resulting in toxic DPR expression. DPRs inhibit DSB repair, NMD and prevent cargo binding of importin-ß as well as induce mitochondrial dysfunction by localizing to mitochondria and interacting with mitochondrial ribosome subunits. The GC-rich expansion in C9ORF72 inhibits transcription and can lead to overall reduction in C9ORF72 protein levels. Since C9ORF72 protein is important for autophagy, cells with lower C9ORF72 protein levels may further accumulate protein aggregates, damaged organelles and toxic DPRs
There are only two clinically approved ALS therapies developed since 1995. But progress in obtaining mechanistic insights into the disease has facilitated therapy development in recent years. The recently approved small molecule drug edaravone (traded as Radicava) is an antioxidant that can ameliorate neuronal oxidative stress [Citation151]. Rapamycin, a potent mTOR inhibitor that may activate autophagy, is under clinical investigation [Citation152]. Antisense gene therapy targeting C9ORF72 RNA repeats also enters early phase clinical study [Citation153]. Lipoamide is a small molecule recently reported to modulate the phase-separation behavior of FUS, which has the potential to be effective in reducing aggregate-mediated toxicity in ALS patients [Citation154]. We look forward to more new therapies in the coming years while our understanding of ALS mechanisms is increasingly strengthened.
Acknowledgments
Y.L.T. is supported by Taiwan Government Scholarship to Study Abroad. Work in the authors’ lab was supported by National Institutes of Health (NIH) grant R35 GM118136 awarded to J.L.M. Author contributions: Y.L.T. and J.L.M. wrote the manuscript.
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
References
- Brooks BR, Miller RG, Swash M, et al. El escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Sci. 2000;1:293–299.
- Brotman RG, Moreno-Escobar MC, Joseph J, et al. Amyotrophic lateral sclerosis. In: StatPearls. Treasure Island (FL): StatPearls Publishing; June 12, 2020.
- Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3(1):17085.
- Valko K, Ciesla L. Amyotrophic lateral sclerosis. Prog Med Chem. 2019;58:63–117.
- Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. 2007;369(9578):2031–2041.
- Niedermeyer S, Murn M, Choi PJ. Respiratory failure in amyotrophic lateral sclerosis. Chest. 2019;155(2):401–408.
- Laferriere F, Polymenidou M. Advances and challenges in understanding the multifaceted pathogenesis of amyotrophic lateral sclerosis. Swiss Med Wkly. 2015;145:w14054.
- Alsultan AA, Waller R, Heath PR, et al. The genetics of amyotrophic lateral sclerosis: current insights. Degener Neurol Neuromuscul Dis. 2016;6:49–64.
- Fay MM, Anderson PJ, Ivanov P. ALS/FTD-associated C9ORF72 repeat RNA promotes phase transitions in vitro and in cells. Cell Rep. 2017;21(12):3573–3584.
- Gopal PP, Nirschl JJ, Klinman E, et al. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc Natl Acad Sci U S A. 2017;114(12):E2466–E2475.
- Johnson BS, Snead D, Lee JJ, et al. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–20339.
- Patel A, Lee HO, Jawerth L, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162(5):1066–1077.
- Carija A, Navarro S, De Groot NS, et al. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biol. 2017;12:699–711.
- Gill C, Phelan JP, Hatzipetros T et al SOD1-positive aggregate accumulation in the CNS predicts slower disease progression and increased longevity in a mutant SOD1 mouse model of ALS. Sci Rep 2019;9(1):6724.
- Zhu C, Beck MV, Griffith JD, et al. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2018;115(18):4661–4665.
- Cozzolino M, Pesaresi MG, Amori I, et al. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid Redox Signal. 2009;11(7):1547–1558.
- Kamelgarn M, Chen J, Kuang L, et al. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc Natl Acad Sci U S A. 2018;115(51):E11904–E11913.
- Mandrioli J, Mediani L, Alberti S, et al. ALS and FTD: where RNA metabolism meets protein quality control. Semin Cell Dev Biol. 2020;99:183–192.
- Pasinelli P, Belford ME, Lennon N, et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43(1):19–30.
- Woerner AC, Frottin F, Hornburg D, et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science. 2016a;351(6269):173–176. .
- Soo KY, Sultana J, King AE, et al. ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discov. 2015;1(1):15030.
- Stoica R, Paillusson S, Gomez-Suaga P, et al. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016;17(9):1326–1342. .
- Veglianese P, Lo Coco D, Bao Cutrona M, et al. Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol Cell Neurosci. 2006;31(2):218–231.
- Guerrero EN, Mitra J, Wang H, et al. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum Mol Genet. 2019;28(15):2459–2476.
- Wang H, Guo W, Mitra J, et al. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat Commun. 2018a;9(1):3683.
- Wang WY, Pan L, Su SC, et al. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci. 2013b;16(10):1383–1391.
- Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14(9):544–558.
- Conlon EG, Manley JL. RNA-binding proteins in neurodegeneration: mechanisms in aggregate. Genes Dev. 2017;31(15):1509–1528.
- Kim G, Gautier O, Tassoni-Tsuchida E, et al. ALS genetics: gains, losses, and implications for future therapies. Neuron. 2020;108(5):822–842.
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438.
- Taylor JP, Brown RH Jr., Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206.
- Shorter J, Taylor JP. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis. 2013;1(1):e25200.
- Maharana S, Wang J, Papadopoulos DK, et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360(6391):918–921. .
- Dormann D, Rodde R, Edbauer D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. Embo J. 2010;29(16):2841–2857. .
- Hofweber M, Hutten S, Bourgeois B, et al. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell. 2018;173(3):706–719 e713. .
- Yoshizawa T, Ali R, Jiou J, et al. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell. 2018;173(3):693–705 e622. .
- Ito D, Seki M, Tsunoda Y, et al. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2011;69(1):152–162.
- Chen X, Wu X, Wu H, et al. Phase separation at the synapse. Nat Neurosci. 2020;23(3):301–310.
- Guillen-Boixet J, Kopach A, Holehouse AS, et al. RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell. 2020;181(2):346–361 e317. .
- Hubstenberger A, Noble SL, Cameron C, et al. Translation repressors, an RNA helicase, and developmental cues control RNP phase transitions during early development. Dev Cell. 2013;27(2):161–173.
- Takahara T, Maeda T. Transient sequestration of TORC1 into stress granules during heat stress. Mol Cell. 2012;47(2):242–252.
- Wang Y, Hu SB, Wang MR, et al. Genome-wide screening of NEAT1 regulators reveals cross-regulation between paraspeckles and mitochondria. Nat Cell Biol. 2018b;20(10):1145–1158.
- Mollet S, Cougot N, Wilczynska A, et al. Translationally repressed mRNA transiently cycles through stress granules during stress. Mol Biol Cell. 2008;19(10):4469–4479.
- Zhang J, Okabe K, Tani T, et al. Dynamic association-dissociation and harboring of endogenous mRNAs in stress granules. J Cell Sci. 2011;124(23):4087–4095.
- Mahboubi H, Stochaj U. Cytoplasmic stress granules: dynamic modulators of cell signaling and disease. Biochim Biophys Acta Mol Basis Dis. 2017;1863(4):884–895.
- Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20(11):649–666.
- Bom APDA, Rangel LP, Costa DCF, et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils IMPLICATIONS FOR CANCER. J Biol Chem. 2012;287(33):28152–28162.
- Wolozin B. Regulated protein aggregation: stress granules and neurodegeneration. Mol Neurodegener. 2012;7(1):56.
- Conlon EG, Lu L, Sharma A, et al. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife. 2016;5. DOI:10.7554/eLife.17820
- Hayes LR, Duan L, Bowen K, et al. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. Elife. 2020;9. DOI:10.7554/eLife.51685
- Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–1186. .
- Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. .
- Zhang K, Donnelly CJ, Haeusler AR, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. .
- Cruts M, Gijselinck I, Van Langenhove T, et al. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36(8):450–459.
- Prado LGR, Bicalho ICS, Magalhaes D, et al. C9ORF72 and the FTD-ALS spectrum: a systematic review of neuroimaging studies. Dement Neuropsychol. 2015;9(4):413–421.
- Chou CC, Zhang Y, Umoh ME, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21(2):228–239. .
- Hoell JI, Larsson E, Runge S, et al. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. 2011;18(12):1428–1431.
- Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol. 2017;18(7):437–451.
- Prudencio M, Belzil VV, Batra R, et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci. 2015;18(8):1175–1182. .
- Sun S, Ling SC, Qiu J, et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat Commun. 2015;6(1):6171. .
- Umoh ME, Fournier C, Li Y, et al. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology. 2016;87(10):1024–1030.
- Wang Q, Conlon EG, Manley JL, et al. Widespread intron retention impairs protein homeostasis in C9orf72 ALS brains. Genome Res. 2020;30(12):1705–1715.
- Conlon EG, Fagegaltier D, Agius P,,, et al. Unexpected similarities between C9ORF72 and sporadic forms of ALS/FTD suggest a common disease mechanism. Elife. 2018;7. 10.7554/eLife.37754
- Reber S, Stettler J, Filosa G, et al. Minor intron splicing is regulated by FUS and affected by ALS-associated FUS mutants. Embo J. 2016;35(14):1504–1521. .
- Gerbino V, Carri MT, Cozzolino M, et al. Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol Dis. 2013;55:120–128.
- Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–625.
- Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18(1):18–30.
- Li WC, You B, Hoque M, et al. Systematic profiling of poly(A) plus transcripts modulated by core 3 ‘ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015;11(4):e1005166.
- Nazim M, Masuda A, Rahman MA, et al. Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms. Nucleic Acids Res. 2017;45(3):1455–1468.
- Masuda A, Takeda J, Okuno T, et al. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev. 2015;29(10):1045–1057.
- Rot G, Wang Z, Huppertz I, et al. High-resolution RNA maps suggest common principles of splicing and polyadenylation regulation by TDP-43. Cell Rep. 2017;19(5):1056–1067.
- Tian B, Manley JL. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci. 2013;38(6):312–320.
- Tank EM, Figueroa-Romero C, Hinder LM, et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun. 2018;9(1):2845.
- Zhou Y, Liu S, Liu G, et al. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013;9(10):e1003895.
- Avendano-Vazquez SE, Dhir A, Bembich S, et al. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012;26(15):1679–1684.
- Mitchell JC, McGoldrick P, Vance C, et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013;125(2):273–288.
- Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858–3863.
- Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76(1):51–74.
- Ortega JA, Daley EL, Kour S, et al. Nucleocytoplasmic proteomic analysis uncovers eRF1 and nonsense-mediated decay as modifiers of ALS/FTD C9orf72 toxicity. Neuron. 2020;106(1):90–107 e113.
- Sun Y, Eshov A, Zhou J, et al. C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat Commun. 2020;11(1):3354.
- Xu W, Bao P, Jiang X, et al. Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain. 2019;142(5):1349–1364.
- Barmada SJ, Ju S, Arjun A, et al. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc Natl Acad Sci U S A. 2015;112(25):7821–7826.
- Isken O, Kim YK, Hosoda N, et al. Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell. 2008;133(2):314–327.
- Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2019;710:132933.
- Deng J, Yang M, Chen Y, et al. FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genet. 2015;11(9):e1005357.
- Onesto E, Colombrita C, Gumina V, et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol Commun. 2016;4(1):47.
- Deng J, Wang P, Chen X, et al. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc Natl Acad Sci U S A. 2018;115(41):E9678–E9686.
- Wang W, Li L, Lin WL, et al. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. 2013a;22(23):4706–4719.
- Nakaya T, Maragkakis M. Amyotrophic lateral sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci Rep. 2018;8(1):15575.
- Tsai Y-L, Coady TH, Lu L, et al. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020;34(11–12):785–805.
- Wang W, Wang L, Lu J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22(8):869–878.
- Wang P, Deng J, Dong J, et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019;15(5):e1007947.
- Shpilka T, Haynes CM. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol. 2018;19(2):109–120.
- Martinez BA, Petersen DA, Gaeta AL, et al. Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of parkinson’s disease. J Neurosci. 2017;37(46):11085–11100.
- Rizzuto R, Pinton P, Carrington W, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280(5370):1763–1766.
- Kornmann B. The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol. 2013;25(4):443–448.
- Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13(10):607–625.
- Saotome M, Safiulina D, Szabadkai G, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the miro GTPase. Proc Natl Acad Sci U S A. 2008;105(52):20728–20733.
- Stoica R, De Vos KJ, Paillusson S, et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun. 2014;5(1):3996. .
- Zhang YJ, Gendron TF, Grima JC, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19(5):668–677.
- Bischoff FR, Klebe C, Kretschmer J, et al. RanGAP1 induces GTPase activity of nuclear ras-related ran. Proc Natl Acad Sci U S A. 1994;91(7):2587–2591.
- Cole CN, Hammell CM. Nucleocytoplasmic transport: driving and directing transport. Curr Biol. 1998;8(11):R368–372.
- Kendirgi F, Barry DM, Griffis ER, et al. An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J Cell Biol. 2003;160(7):1029–1040.
- Kendirgi F, Rexer DJ, Alcázar-Román AR, et al. Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: a conserved mechanism in the export of Hsp70 mRNA. Mol Biol Cell. 2005;16(9):4304–4315.
- Kaneb HM, Folkmann AW, Belzil VV, et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24(5):1363–1373. .
- Nousiainen HO, Kestila M, Pakkasjarvi N, et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40(2):155–157.
- Herva R, Conradi NG, Kalimo H, et al. A syndrome of multiple congenital contractures: neuropathological analysis on five fetal cases. Am J Med Genet. 1988;29(1):67–76.
- Vuopala K, Ignatius J, Herva R. Lethal arthrogryposis with anterior horn cell disease. Hum Pathol. 1995;26(1):12–19.
- Cho KI, Yoon D, Qiu S, et al. Loss of Ranbp2 in motoneurons causes disruption of nucleocytoplasmic and chemokine signaling, proteostasis of hnRNPH3 and Mmp28, and development of amyotrophic lateral sclerosis-like syndromes. Dis Model Mech. 2017;10(5):559–579.
- Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–593.
- Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–1441.
- Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–1446.
- Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–636. .
- Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226.
- Bansal M, Moharir SC, Swarup G. Autophagy receptor optineurin promotes autophagosome formation by potentiating LC3-II production and phagophore maturation. Commun Integr Biol. 2018;11(2):1–4.
- Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10(1):5.
- Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113(15):4039–4044.
- Deng Z, Lim J, Wang Q, et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2019;16(5):917–931.
- Wong YC, Holzbaur ELF . Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42): E4439–E4448.
- De Majo M, Topp SD, Smith BN, et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol Aging. 2018;71:266 e261–266 e210.
- Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10(8):677–685.
- Xia Q, Wang H, Hao Z, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. Embo J. 2016;35(2):121–142. .
- Ling SC, Dastidar SG, Tokunaga S, et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. Elife. 2019;8. DOI:10.7554/eLife.40811.
- Chitiprolu M, Jagow C, Tremblay V, et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun. 2018;9(1):2794. .
- Sullivan PM, Zhou X, Robins AM, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016a;4(1):51.
- Ash PE, Bieniek KF, Gendron TF, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646.
- Boivin M, Pfister V, Gaucherot A, et al. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. Embo J. 2020;39(4). DOI:10.15252/embj.2018100574
- Sellier C, Campanari ML, Julie Corbier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. Embo J. 2016;35(12):1276–1297.
- Yang M, Liang C, Swaminathan K, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016;2(9):e1601167.
- Sea K, Sohn SH, Durazo A, et al. Insights into the role of the unusual disulfide bond in copper-zinc superoxide dismutase. J Biol Chem. 2015;290(4):2405–2418
- Xie Y, Zhou B, Lin MY, et al. Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS mice. Neuron. 2015;87(2):355–370.
- Richard P, Feng S, Tsai YL, et al. SETX (senataxin), the helicase mutated in AOA2 and ALS4, functions in autophagy regulation. Autophagy. 2020:1–18. 10.1080/15548627.2020.1796292
- Richard P, Feng S, Manley JL. A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev. 2013;27(20):2227–2232.
- Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell. 2011;42(6):794–805.
- Brambati A, Colosio A, Zardoni L, et al. Replication and transcription on a collision course: eukaryotic regulation mechanisms and implications for DNA stability. Front Genet. 2015;6:166.
- Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83(2):266–282.
- Konopka A, Atkin JD. The emerging role of DNA damage in the pathogenesis of the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Int J Mol Sci. 2018;19(10):3137.
- Farg MA, Konopka A, Soo KY, et al. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet. 2017;26(15):2882–2896.
- Qiu H, Lee S, Shang Y, et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. 2014;124(3):981–999.
- Nihei Y, Mori K, Werner G, et al. Poly-glycine-alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3. Acta Neuropathol. 2020;139(1):99–118. .
- Wang H, Hegde ML. New mechanisms of DNA repair defects in fused in sarcoma-associated neurodegeneration: stage set for DNA repair-based therapeutics? J Exp Neurosci. 2019;13:1179069519856358.
- Rulten SL, Rotheray A, Green RL, et al. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014;42(1):307–314.
- Naumann M, Pal A, Goswami A, et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun. 2018;9(1):335. .
- Singatulina AS, Hamon L, Sukhanova MV, et al. PARP-1 activation directs FUS to DNA damage sites to form PARG-reversible compartments enriched in damaged DNA. Cell Rep. 2019;27(6):1809–1821 e1805.
- Hock EM, Maniecka Z, Hruska-Plochan M, et al. Hypertonic stress causes cytoplasmic translocation of neuronal, but not astrocytic, FUS due to impaired transportin function. Cell Rep. 2018;24(4):987–1000 e1007.
- Mitra J, Guerrero EN, Hegde PM, et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A. 2019;116(10):4696–4705.
- Freibaum BD, Taylor JP. The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci. 2017;10:35.
- Andrade NS, Ramic M, Esanov R, et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol Neurodegener. 2020;15(1):13.
- Lopez-Gonzalez R, Lu Y, Gendron TF, et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016;92(2):383–391.
- Freibaum BD, Lu Y, Lopez-Gonzalez R, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–133.
- Takei K, Watanabe K, Yuki S, et al. Edaravone and its clinical development for a amyotrophic lateral sclerosis. Amyotroph Lat Scl Fr. 2017;18:5–10.
- Mandrioli J, D’Amico R, Zucchi E, et al. Rapamycin treatment for amyotrophic lateral sclerosis protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine (Baltimore). 2018;97(24):e11119.
- Ly CV, Miller TM. Emerging antisense oligonucleotide and viral therapies for amyotrophic lateral sclerosis. Curr Opin Neurol. 2018;31(5):648–654.
- Wheeler RJ, Lee HO, Poser I, et al. Small molecules for modulating protein driven liquid-liquid phase separa- 1345 tion in treating neurodegenerative disease. bioRxiv. 2019;721001. DOI:10.1101/721001.