
ABSTRACT
Accurate and complete DNA replication and separation are essential for genetic information inheritance and organism maintenance. Errors in DNA duplication are the main source of genetic instability. Understanding DNA duplication regulation is the key to elucidate the mechanisms and find treatment strategies for human genetic disorders, especially cancer. The mechanistic target of rapamycin (mTOR) is a central regulator of cell growth and proliferation by integrating and processing extracellular and intracellular signals to monitor the well-being of cell physiology. mTOR signaling dysregulation is associated with many human diseases including cancer and diabetes. Emerging evidence has demonstrated that mTOR signaling plays a key role in DNA duplication. We herein review the current knowledge of mTOR signaling in the regulation of DNA replication origin licensing, replication fork progression, and stabilization.
Introduction
The mechanistic target of rapamycin (mTOR) is a conserved member of the PI3K-related kinase (PIKK) family, containing the conserved kinase, FAT, FATC, and HEAT domains [Citation1,Citation2]. mTOR was first identified in the budding yeast Saccharomyces cerevisiae by screening rapamycin-resistant mutants [Citation3]. The yeast genome encodes two mTOR kinases, mTOR1 and mTOR2 [Citation1,Citation4]. Yeast mTOR forms two distinct complexes, mTORC1 and mTORC2. mTORC1 complex is composed of either mTOR1 or mTOR2 together with LST8, KOG1, and TCO89. mTOR2 is essential and also forms a rapamycin-insensitive mTORC2 complex together with LST8, AVO1, AVO2, AVO3, and BIT61 [Citation5]. Except for yeast, all studied organisms have one mTOR kinase. In mammalian cells, mTORC1 is composed of mTOR, mLST8, PRAS40, Deptor, and Raptor while mTORC2 consists of mTOR, mLST8, Rictor, Deptor, hSin1, and Protor [Citation6,Citation7]. Recently, mTOR was found to interact with ETS (E26 transformation-specific) transcription factor ETV7 to form rapamycin insensitive mTORC3 [Citation8]. In addition, a fourth mTOR complex (mTORC4) was reported to be composed of mTOR, mLST8, DNA-PKcs, and mEAK7 [Citation9] ().
Figure 1. The identified protein complexes that are formed by mTOR kinase, and their proved or potential (labeled with “?” mark) roles and mechanisms in the regulation of physiology of normal cells or cancer cells. DDR, DNA damage response; CSC, cancer stem cell. “?” denotes an unidentified mechanism
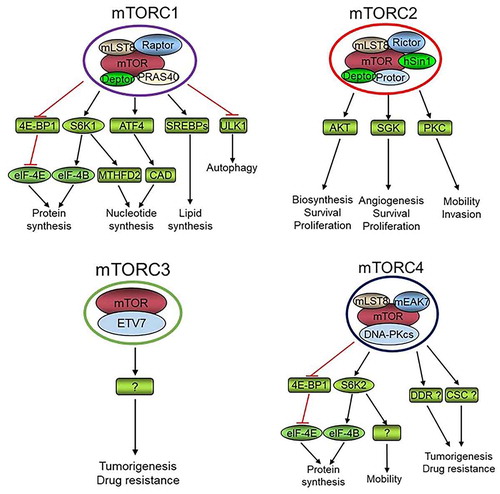
mTORC1 lies at the hub of intracellular and extracellular signal transduction networks via integrating and processing multiple signals, and dictates the rates of macromolecule synthesis and hence cell growth, proliferation, and survival [Citation6,Citation7,Citation10–13]. mTORC1 senses intracellular alteration of amino acids, energy, oxygen, and other stresses to control protein translation, autophagy, ribosome biogenesis, stress-induced transcription, and cell cycle transition through numerous downstream pathways. Cell growth and proliferation not only depend on nutrients but also on extracellular growth factors. Actually, the position and number of a specific type of cells in a tissue of an organism are mainly determined by growth factor signaling. The predominant growth-promoting signaling pathways are PI3K-AKT and RAS-MAPK, both of which activate mTORC1 and are frequently upregulated in cancers [Citation14]. Intracellular oxygen concentration and energy status are transmitted to mTORC1 through LKB1-AMPK or Redd1/2 and LKB1-AMPK, respectively [Citation12]. Wnt and p38MAPK also signal to mTORC1 network [Citation15,Citation16], while recently it was found that mTORC1 signaling suppresses Wnt/β-catenin signaling through downregulating the Wnt receptor FZD level [Citation17]. Inflammation promotes mTORC1 signaling via TNFα–IKKβ pathway [Citation18]. Moreover, cylinB/CDK1 increases the mTORC1 activity during G2/M phase of the cell cycle [Citation19]. Most of these signals concourse on mTORC1 via the upstream TSC1/2 complex, a GTPase activating protein (GAP) that converts GTP-Rheb to GDP-Rheb. Rheb is a Ras-like GTPase that promotes the activation of mTORC1 kinase [Citation11]. Amino acids are essential for mTORC1 activation through Rag instead of TSC1/2 complex [Citation20]. Thus, mTORC1 controls cell growth and proliferation and maintains tissue homeostasis via integrating and processing multiple signals [Citation1,Citation2,Citation6,Citation7,Citation12]. Increasing mTORC1 substrates have been found and thus more functions have been ascribed to mTORC1 [Citation6,Citation7]. The best understood function of mTORC1 is its promotion of protein translation by enhancing the phosphorylation of S6K1 and 4E-BP1 [Citation21–24]. eIF-4E is an essential component of eIF-4F, which initiates cap-dependent protein translation, while eIF-4E is suppressed by 4E-BP1. Activation of mTORC1 leads to phosphorylation of 4E-BP1 and release of eIF-4E resulting in increased cap-dependent protein translation [Citation25]. In addition, mTORC1 phosphorylates and activates S6K1 kinase, which in turn phosphorylates and activates eIF-4B to promote cap-dependent protein translation [Citation25]. Importantly, mTORC1 plays a key role in pyrimidine synthesis via phosphorylating S6K1 to phosphorylate CAD (Carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase) at Ser1859 [Citation26,Citation27] and purine production via ATF4/MTHFD2 axis [Citation28]. Moreover, mTORC1 promotes lipid biosynthesis by activating SREBPs [Citation29]. Furthermore, mTORC1 inhibits catabolism including suppressing autophagy by phosphorylating and inactivating ULK1, a key player in the early stages of autophagy [Citation30,Citation31]. Therefore, mTORC1 signaling increases cell anabolism and decreases catabolism to maintain cell physiology under ever-changing cell growth conditions.
There are several feedback loops in the mTORC1 pathways. An increased mTORC1-S6K1 activation leads to IRS phosphorylation and subsequent ubiquitin-proteasome-mediated degradation resulting in attenuation of PI3K-AKT-mTORC1 signaling [Citation32]. Increased mTORC1-S6K1 activity can also form a positive feedback loop to enhance mTORC1 signaling by directly phosphorylating mTOR at S2448 [Citation33,Citation34]. One of the main and conserved functions of mTORC2 is regulating the actin cytoskeleton organization, which is vital for cell mitosis, motility, and the maintenance of cell morphology [Citation1,Citation6,Citation35–37]. mTORC2 phosphorylates and activates the AGC family kinases AKT, PKC, and SGK1, which have shared and distinct substrates that regulate macromolecule biosynthesis; angiogenesis; and cell survival, proliferation, mobility, and invasion [Citation38–40]. The full activation of AKT depends on the phosphorylation of Ser308 by PDK1 and Thr473 by mTORC2. Therefore, increased mTORC2 activity accelerates PI3K-AKT-mTORC1 signaling and forms a second positive feedback loop [Citation11]. mTORC3 is rapamycin insensitive and its molecular functions are largely unknown. The currently available results suggest mTORC3 may play a role in tumorigenesis and/or anticancer drug resistance [Citation8]. Through mEAK-7 and other unknown mechanisms, mTORC4 may regulate cell proliferation and migration through S6K2 and 4E-BP1 [Citation9,Citation41]. Regarding the critical roles for DNA-PKcs in the regulation of DNA damage repair and maintenance of cancer stem cells (CSCs) [Citation42,Citation43], it is intriguing to anticipate that mTORC4 plays a role in tumorigenesis and anticancer drug resistance by controlling DNA damage response and the physiology of CSCs. Therefore, more functions of mTORC3 and mTORC4 will be elucidated, and we anticipate more mTOR complexes to be identified. This review addresses the emerging roles of mTOR signaling in the regulation of DNA replication origin licensing, replication fork progression, and stabilization.
mTOR signaling promotes DNA replication licensing
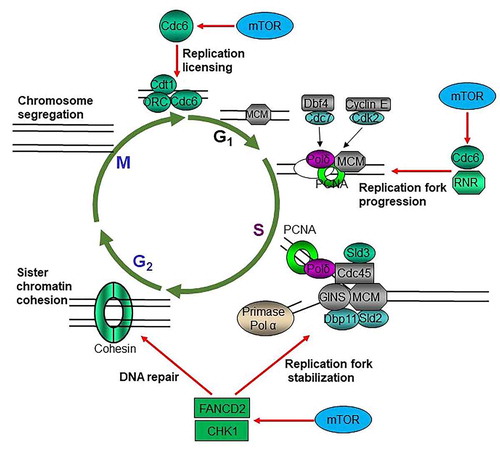
Once and only once per cell cycle for each DNA replication is essential for maintaining the integrity of genetic information [Citation44]. DNA replication origin activation is crucial for maintaining genomic integrity in all organisms and is tightly regulated to occur only once per cell cycle [Citation45]. A pre-replicative complex (pre-RC) forms at the origin of replication during late mitosis and early G1 phase. Formation of pre-RC is a critical step for the complete and faithful duplication of the genome to ensure that each daughter cell will carry the same genetic information as the parent cell [Citation46]. In most eukaryotes, a pre-RC is composed of six origin recognition complex proteins (ORC1–6), CDC6, CDT1, and a heterohexamer of MCM proteins (MCM2–7). CDC6 and CDT1 are licensing factors for DNA replication, and their deregulation results in impaired DNA replication thereby genome instability [Citation47]. Insufficient origin licensing during G1 phase, increased and/or ectopic licensing during G1, or re-licensing during S and G2 phases accounts for the oncogene-induced replication stress [Citation47–49]. Moreover, the aberrant expression of CDT1, CDC6, and ORC or abrogation of their regulation leads to re-replication of the genome leading to genome instability [Citation50–52]. Most components of DNA replication machineries are overexpressed in the majority of cancers that are associated with the poor overall survival (OS) and/or disease-free survival (DFS) of many cancer patients (TCGA data). mTOR signaling is the predominant growth-promoting signaling pathway, which enhances CDKs activity and increases cell cycle progression. mTOR is required for the progression of G1 phase, especially the early G1 phase both in yeast and mammalian cells [Citation1,Citation53]. Recent studies have suggested that mTOR signaling may promote DNA replication origin licensing through upregulating CDC6 [Citation54,Citation55] (). CDC6 is essential for the loading of MCM2–7 complex during DNA replication [Citation47]. It has been reported that inhibition of mTOR kinase results in dramatic reduction of MCM2–7 components as well as PCNA on chromatins, and both mTORC1-S6K1 and mTORC2 were found to be required for the regulation of CDC6. One of the mechanisms was supposed to be mTOR signaling suppression of miR-3178, which targets CDC6 mRNA [Citation54]. Thus, mTOR may promote the loading and maintenance MCM2–7 complexes on chromatins by positively regulating CDC6. The discovery of deregulation of the pre-RC component CDC6 by mTOR signaling might reveal the mechanism of the promotion of genome instability and heterogeneity of cancer cells by the RAS/PI3K signaling pathway.
Figure 2. mTOR promotes cycle progression through multiple mechanisms. mTOR promotes replication licensing by enhancing MCM2–7 loading via regulating CDC6; mTOR accelerates DNA replication progression through upregulating RNR to provide dNTPs and increasing CDC6; mTOR maintains replication fork stability via sustaining CHK1 and FANCD2
mTOR signaling promotes DNA replication progression
During G1 to S phase transition, the pre-RC complex is phosphorylated and activated by Dbf4 dependent Cdc7 kinase and cyclin E dependent CDK2 kinase to start the one-way and no-return cell cycle progression [Citation46,Citation56]. Deoxyribonucleotides (dNTPs) are the building blocks of DNA and essential for DNA replication and repair. Increased or imbalanced dNTPs lead to genome instability, while decreased dNTP level impairs cell survival [Citation57–59]. Balanced dNTPs pool is essential for the accurate and efficient DNA synthesis for replication and repair, defects of which lead to cell death or genome instability [Citation58–60]. Rapid proliferating cancer cells encounter frequent metabolic stress due to the transient and long-term lack of nutrients, oxygen, and growth factors [Citation61,Citation62]; however, the mechanisms by which cancer cells maintain dNTPs level under ever-changing microenvironment to meet the rapid proliferation and massive DNA damage repair are not fully understood. Moreover, in response to DNA damage and replication stress, there is a 6–8-fold increase of intracellular dNTPs in yeast through several mechanisms [Citation63–67], which is essential for cells to survive DNA damage via translesion DNA synthesis (because incompletely replicated DNA results in cell death) [Citation68]. Ribonucleotide reductase (RNR) catalyzes the rate-limiting step in the production of dNTPs from ribonucleotides, and its expression and activity are tightly controlled in all organisms under normal growth and stressful conditions [Citation57–59]. The mammalian RNR is composed of two identical RRM1 and two small catalytic subunits of either RRM2 or p53R2. p53R2 is induced by DNA damage (e.g. radiotherapy) and is regulated by p53 [Citation69]. Thus, the RNR activity is stimulated after DNA damage by p53. Both RRM1 and RRM2 are dynamically regulated during cell cycle progression [Citation57–60]. The activity of RNR is principally controlled by RRM2 levels in mammalian cells [Citation70]. It was reported that mTOR signaling enhances the cap-dependent protein translation and gene transcription of RRM1 and RRM2, and that p53 suppresses RRM1 and RRM2 via inhibiting mTORC1 [Citation71]. Moreover, mTOR maintains cell survival but at the cost of increased mutation rate in response to genotoxins by increasing the expression of RNR subunits in yeast [Citation72]. Regarding the fact that most cancers have lost p53 signaling mainly due to TP53 mutations and have obtained mTOR signaling elevation resulting from mutations of PTEN, PIK3CA, and receptor tyrosine kinases [Citation73,Citation74], positive regulation of RNR may be one of the main underlying mechanisms of the dependence of cancer cells on PI3K-AKT-mTOR signaling for survival [Citation75–77]. This also supports the long-standing idea that mTOR is an important target for anticancer drug development [Citation75–77]. In addition to replication licensing, CDC6 also plays multiple other roles in ensuring precise chromosome duplication [Citation78]. CDC6 is crucial for proper S-phase DNA replication progression [Citation79]. CDC6 can trigger a checkpoint response, which could ensure that all DNA is replicated before mitotic entry [Citation80]. Therefore, mTOR signaling promotes DNA replication progression by positively regulating RNR as well as CDC6 under normal growth conditions and in response to genotoxic stress ().
mTOR signaling maintains DNA replication fork stability during replication stress
The physiological behavior of a cell is ultimately dictated by the codes of its genome. However, the genomes of all living cells are under constant attacks by physical, chemical, and biological agents, all of which can induce genetic alterations. In order to survive and maintain genome integrity, all organisms have been endowed with genome surveillance systems during evolution [Citation56]. A genome surveillance system is a signal transduction cascade composed of signals, sensors, transducers, and effectors [Citation81]. In metazoans, genome surveillance is mainly performed by ATM-CHK2 and ATR-CHK1 checkpoints. DNA double-stranded breaks (DSBs) produced by ionizing radiation or DNA metabolism are sensed by MRE11-RAD50-NBS1 complex (MRN), which recruits activated monomer ATM to damage sites [Citation82]. ATM phosphorylates MRN complex and histone H2AX to amplify the signals. Following recruitment to DSBs, a plethora of substrates including CHK2, MDM2, and p53 are phosphorylated by ATM with the help of mediators including MDC1, 53BP1, and BRCA1 [Citation56,Citation83–86]. Single-stranded breaks (SSBs), much more common DNA damage intermediates produced by different kinds of genotoxins, are coated by replicating factor A (RPA). ATRIP-ATR complex then binds to this RPA-coated nucleofilament and phosphorylates CHK1 with the help of TOPBP1, RAD9-RAD1-HUS1 (9–1–1 complex), and RAD17-RFC clam loader [Citation87,Citation88]. Activated CHK2 and CHK1 phosphorylate numerous downstream effectors to amplify and relay the signals to exert the DNA damage responses (DDRs) such as cell cycle arrest, senescence, or apoptosis [Citation42,Citation56,Citation81,Citation89,Citation90]. ATR-CHK1 is at the heart of DDR [Citation91]. Although ATR-CHK1 checkpoint has many functions, including during DSB repair, interstrand crosslink repair, and meiosis, as well as at telomeres and in response to mechanical and osmotic stresses, the main function of the ATR-CHK1 checkpoint is in the replication stress response (RSR) [Citation91]. ATR orchestrates multiple branches of RSR by signaling to arrest cells at S and G2/M phases, stabilizing stalled replication forks, inhibiting origin firing, and increasing dNTPs biosynthesis [Citation91–94]. From yeast to mammalian cells, the stabilization of stalled replication forks is regulated by CHK1 (yeast Rad53), which makes CHK1 essential for cell survival in all eukaryotes [Citation90]. CHK1 is a conserved major caretaker of genome stability and cell survival. Once fired, DNA replication proceeds with no return. Uncontrolled DNA replication leads to DNA replication stress, which activates ATR-CHK1 replication checkpoint to stabilize stalled replication fork, inhibit DNA replication late origin firing, and arrest cell cycle progression. It has been reported that transient inhibition of mTOR kinase leads to CHK1 checkpoint activation [Citation54], while long-term mTOR signaling suppression results in the decrease of CHK1 level [Citation95,Citation96]. Consistently, mTOR inhibition results in replication fork collapse under DNA lesions and replication stress in yeast (Shen et al., unpublished) [Citation72]. One of the mechanisms is the positive control of the E2F transcription factor by mTOR signaling to promote CHK1 gene transcription [Citation95,Citation96]. This is consistent with the earlier findings that the RB-E2F axis regulates CHK1 expression [Citation97], and mTORC1 signaling promotes the activity of CDKs [Citation98–100]. Therefore, mTOR signaling is required to maintain genome stability in part by maintaining CHK1 checkpoint to stabilize replication fork under replication stress.
The Fanconi Anemia (FA) signaling pathway maintains genome integrity by promoting DNA damage repair through translesion DNA synthesis (TLS), nucleotide excision repair (NER), and homologous recombination (HR) [Citation101,Citation102]. FA signaling is activated by different kinds of genotoxins and important for the activation of the ATM-CHK2 and ATR-CHK1 checkpoints. FANCD2 is a key player in FA signaling. In response to DNA damage, activation of the FA core E3 ubiquitin ligase complex leads to monoubiquitination of FANCI and FANCD2, which are recruited to DNA damage sites to promote DNA repair [Citation103,Citation104]. We previously reported that FANCD2 is required for the timely ATM-CHK2 activation in the early steps of FA signaling-mediated repair of inter cross-link induced DNA lesions [Citation105]. Mechanistically, mTOR positively controls FANCD2 gene expression via either mTORC1-S6K1 or mTORC2-NFκB signaling in a cell type dependent manner [Citation105,Citation106]. Thus, it is possible that promotion of FANCD2 dependent activation of the ATM checkpoint in the early response to DNA damage is one of the mechanisms by which mTOR signaling promotes genome stability under normal growth condition and cell survival in response to genotoxins. In addition, accumulating evidence has shown an important role for FANCD2 in the maintenance of replication fork stability. Under replication stress, mononubiquitinated FANCD2 is recruited to stalled replication forks to stabilized forks, restart stalled replication forks, and suppress the firing of new and dormant origins [Citation107–109]. Taken together, mTOR signaling maintains replication fork stability under replication stress by upregulating both CHK1 and FANCD2 ().
Closing remarks
mTOR kinase forms multiple protein complexes and is a central integrator and processor of extracellular and intracellular signals. Emerging evidence has shown that mTOR signaling plays an important role in the regulation of DNA duplication at several stages of the cell cycle. First, mTOR controls DNA replication origin licensing by regulating CDC6. Second, mTOR promotes DNA replication fork progression via sustaining ribonucleotide reductase and CDC6 levels. Third, mTOR maintains replication fork stabilization through increasing CHK1 and FANCD2 expressions. A report suggests that mTOR may be recruited to stalled replication forks during replication stress [Citation110]. It is important to elucidate the functions of chromatin-associated mTOR and the mechanisms of the recruitment of mTOR to chromatin in response to DNA lesions. After DNA replication, the sister chromatins are bound together by cohesin through late S, G2, and early M phases, detached during prometaphase, and separated during anaphase of the cell cycle. Moreover, mTORC1 activity was found to be upregulated by cyclin B/CDK1 during G2/M phase of the cell cycle [Citation19], and mTOR inhibition leads to defects in chromosome segregation during M phase [Citation111]. It remains to be determined whether and how mTOR signaling regulates sister chromatins cohesion during and after DNA replication, detachment from prophase to prometaphase, and separation from metaphase to telophase. Moreover, DNA-PKcs are important in the response and repair of DNA DSBs via non-homologous end joining (NHEJ) [Citation112]. Regarding the fact that more than 90% of DSBs are repaired by NHEJ, it will be interesting to explore whether mTOR kinase interacts with DNA-PCcs at DSBs sites and directly participates in the procedures of NHEJ. Considering that the mTOR signaling pathway is dysregulated in most cancers and DNA replications stress is the main source of cancer cell genome instability, elucidating the roles and mechanisms for mTOR in the regulation of DNA duplication may be the key to understand tumorigenesis and find cancer treatment strategies.
Acknowledgments
This work was supported by the American Cancer Society (IRG–67–003–47), National Institute of Health (R01CA246553), Major Research Project of Science Technology Department of Zhejiang Province (2021C03124), and Wu Jieping Medical Foundation (320.6750.19092-12).
Disclosure statement
The authors declare no competing interests.
Additional information
Funding
References
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484.
- Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103(2):253–262.
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–909.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976.
- Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10(3):457–468.
- Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63–71.
- Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Bio. 2020;21(4):183–203.
- Harwood FC, Geltink RIK, O’Hara BP, et al. ETV7 is an essential component of a rapamycin-insensitive mTOR complex in cancer. Sci Adv. 2018;4(9):eaar3938.
- Nguyen JT, Haidar FS, Fox AL, et al. mEAK-7 forms an alternative mTOR complex with DNA-PKcs in human cancer. Iscience. 2019;17:190-+.
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17(6):596–603.
- Soulard A, Hall MN. SnapShot: mTOR signaling. Cell. 2007;129(2):434.
- Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21(2):209–218.
- Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science. 2011;334(6056):678–683.
- Zhang HL, Xing XW, Liu Y, et al. Comprehensive network analysis of different subtypes of molecular disorders in lung cancer. Oncologie. 2020;22(2):107–116.
- Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–968.
- Li Y, Inoki K, Vacratsis P, et al. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J Biol Chem. 2003;278(16):13663–13671.
- Zeng H, Lu B, Zamponi R, et al. mTORC1 signaling suppresses Wnt/beta-catenin signaling through DVL-dependent regulation of Wnt receptor FZD level. Proc Natl Acad Sci U S A. 2018;115(44):E10362–E10369.
- Lee DF, Kuo HP, Chen CT, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130(3):440–455.
- Astrinidis A, Senapedis W, Coleman TR, et al. Cell cycle-regulated phosphorylation of hamartin, the product of the tuberous sclerosis complex 1 gene, by cyclin-dependent kinase 1/cyclin B. J Biol Chem. 2003;278(51):51372–51379.
- Kim E, Goraksha-Hicks P, Li L, et al. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10(8):935–945.
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22.
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–734.
- Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4(5):335–348.
- Biterge Sut B, Ikinci Keles A. Prognostic and predictive significance of eukaryotic elongation factor 1D (eEF1D) in breast cancer: a potential marker of response to endocrine therapy. Oncologie. 2020;22(3):147–154.
- Bjornsti MA, Houghton PJ. Lost in translation: dysregulation of cap-dependent translation and cancer. Cancer Cell. 2004;5(6):519–523.
- Ben-Sahra I, Howell JJ, Asara JM, et al. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323–1328.
- Robitaille AM, Christen S, Shimobayashi M, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339(6125):1320–1323.
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, et al. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351(6274):728–733.
- Bakan I, Laplante M. Connecting mTORC1 signaling to SREBP-1 activation. Curr Opin Lipidol. 2012;23(3):226–234.
- Grasso D, Renna FJ, Vaccaro MI. Initial steps in mammalian autophagosome biogenesis. Front Cell Dev Biol. 2018;6. DOI:10.3389/fcell.2018.00146
- Dossou AS, Basu A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers (Basel). 2019;11(10):1422.
- O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66(3):1500–1508.
- Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem. 2005;280(27):25485–25490.
- Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005;280(28):26089–26093.
- Pearce LR, Huang X, Boudeau J, et al. Identification of protor as a novel rictor-binding component of mTOR complex-2. Biochem J. 2007;405(3):513–522.
- Frias MA, Thoreen CC, Jaffe JD, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16(18):1865–1870.
- Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–U1130.
- Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101.
- Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302.
- Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008;416:375–385.
- Nguyen JT, Ray C, Fox AL, et al. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci Adv. 2018;4(5):eaao5838.
- Pilie PG, Tang C, Mills GB, et al. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16(2):81–104.
- Wang YL, Xu HN, Liu TR, et al. Temporal DNA-PK activation drives genomic instability and therapy resistance in glioma stem cells. Jci Insight. 2018;3(3). DOI:10.1172/jci.insight.98096.
- Diffley JFX. Quality control in the initiation of eukaryotic DNA replication. Philos T R Soc B. 2011;366(1584):3545–3553.
- Aguilera A, Garcia-Muse T. Causes of genome instability. Annu Rev Genet. 2013;47:1–32.
- Masai H, Matsumoto S, You ZY, et al. Eukaryotic Chromosome DNA Replication: where, When, and How?. Annual Review of Biochemistry. 2005;307(1):89–130.
- Petropoulos M, Champeris Tsaniras S, Taraviras S, et al. Replication Licensing Aberrations, Replication Stress, and Genomic Instability. Trends Biochem Sci. 2019;44(9):752–764.
- Jones RM, Mortusewicz O, Afzal I, et al. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene. 2013;32(32):3744–3753.
- Kotsantis P, Silva LM, Irmscher S, et al. Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun. 2016;7. DOI:10.1038/ncomms13087
- Vaziri C, Saxena S, Jeon Y, et al. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11(4):997–1008.
- Melixetian M, Ballabeni A, Masiero L, et al. Loss of Geminin induces rereplication in the presence of functional p53. J Cell Biol. 2004;165(4):473–482.
- Klotz-Noack K, McIntosh D, Schurch N, et al. Re-replication induced by geminin depletion occurs from G2 and is enhanced by checkpoint activation. J Cell Sci. 2012;125(10):2436–2445.
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35.
- Wu X, Li S, Hu X, et al. mTOR signaling upregulates CDC6 via suppressing miR-3178 and promotes the loading of DNA replication helicase. Sci Rep. 2019;9(1):9805.
- Muller D, Shin S, Goullet De Rugy T, et al. eIF4A inhibition circumvents uncontrolled DNA replication mediated by 4E-BP1 loss in pancreatic cancer. JCI Insight. 2019;4(21). DOI:10.1172/jci.insight.121951.
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85.
- Aye Y, Li M, Long MJ, et al. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015;34(16):2011–2021.
- Mathews CK. Deoxyribonucleotides as genetic and metabolic regulators. Faseb J. 2014;28(9):3832–3840.
- Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706.
- Kolberg M, Strand KR, Graff P, et al. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta. 2004;1699(1–2):1–34.
- Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8(3):180–192.
- Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40(2):323–332.
- Chabes A, Georgieva B, Domkin V, et al. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell. 2003;112(3):391–401.
- Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell. 1998;94(5):595–605.
- Zhao X, Rothstein R. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc Natl Acad Sci U S A. 2002;99(6):3746–3751.
- Lee YD, Elledge SJ. Control of ribonucleotide reductase localization through an anchoring mechanism involving Wtm1. Genes Dev. 2006;20(3):334–344.
- Lee YD, Wang J, Stubbe J, et al. Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol Cell. 2008;32(1):70–80.
- Prakash S, Prakash L. Translesion DNA synthesis in eukaryotes: a one- or two-polymerase affair. Genes Dev. 2002;16(15):1872–1883.
- Yousefi B, Samadi N, Ahmadi Y. Akt and p53R2, partners that dictate the progression and invasiveness of cancer. Adv Protein Chem Struct. 2014;22:24–29.
- D’Angiolella V, Donato V, Forrester FM, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149(5):1023–1034.
- He Z, Hu X, Liu W, et al. P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget. 2017;8(25):41422–41431.
- Shen C, Lancaster CS, Shi B, et al. TOR signaling is a determinant of cell survival in response to DNA damage. Mol Cell Biol. 2007;27(20):7007–7017.
- Liu W, Zhou Y, Reske SN, et al. PTEN mutation: many birds with one stone in tumorigenesis. Anticancer Res. 2008;28(6A):3613–3619.
- Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18(12):744–757.
- Tamborero D, Gonzalez-Perez A, Perez-Llamas C, et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. 2013;3:2650.
- Lamberti G, Brighi N, Maggio I, et al. The role of mTOR in neuroendocrine tumors: future cornerstone of a winning strategy? Int J Mol Sci. 2018;19(3):747.
- Fruman DA, Chiu H, Hopkins BD, et al. The PI3K pathway in human disease. Cell. 2017;170(4):605–635.
- Oehlmann M, Score AJ, Blow JJ. The role of Cdc6 in ensuring complete genome licensing and S phase checkpoint activation. J Cell Biol. 2004;165(2):181–190.
- Lau E, Zhu C, Abraham RT, et al. The functional role of Cdc6 in S-G2/M in mammalian cells. EMBO Rep. 2006;7(4):425–430.
- Clay-Farrace L, Pelizon C, Santamaria D, et al. Human replication protein Cdc6 prevents mitosis through a checkpoint mechanism that implicates Chk1. Embo J. 2003;22(3):704–712.
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323.
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506.
- Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–235.
- Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–870.
- Bakkenist CJ, Kastan MB. Chromatin perturbations during the DNA damage response in higher eukaryotes. Adv Protein Chem Struct. 2015;36:8–12.
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Bio. 2013;14(4):197–210.
- Wang X, Zou L, Lu T, et al. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol Cell. 2006;23(3):331–341.
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300(5625):1542–1548.
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–745.
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9(8):616–627.
- Saldivar JC, Cortez D, Cimprich KA. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol. 2017;18(10):622–636.
- Sundar R, Brown J, Ingles Russo A, et al. Targeting ATR in cancer medicine. Curr Probl Cancer. 2017;41(4):302–315.
- Lecona E, Fernandez-Capetillo O. Targeting ATR in cancer. Nat Rev Cancer. 2018;18(9):586–595.
- Rundle S, Bradbury A, Drew Y, et al. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers (Basel). 2017;9(5):41.
- Zhou X, Liu W, Hu X, et al. Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Sci Rep. 2017;7(1):1535.
- Selvarajah J, Elia A, Carroll VA, et al. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget. 2015;6(1):427–440.
- Chinnam M, Goodrich DW. Rb1, development, and cancer. Curr Top Dev Biol. 2011;94:129–169.
- Averous J, Fonseca BD, Proud CG. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene. 2008;27(8):1106–1113.
- Fingar DC, Richardson CJ, Tee AR, et al. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24(1):200–216.
- Oka K, Ohya-Shimada W, Mizuno S, et al. Up-regulation of cyclin-E-1 via proline-mTOR pathway is responsible for HGF-mediated G(1)/S progression in the primary culture of rat hepatocytes. Biochem Biophys Res Commun. 2013;435(1):120–125.
- Sumpter R, Levine B. Emerging functions of the Fanconi anemia pathway at a glance. J Cell Sci. 2017;130(16):2657–2662.
- Niraj J, Farkkila A, D’Andrea AD. The fanconi anemia pathway in cancer. Annu Rev Canc Biol. 2019;3:457–478.
- Joo W, Xu GZ, Persky NS, et al. Structure of the FANCI-FANCD2 complex: insights into the fanconi anemia DNA repair pathway. Science. 2011;333(6040):312–316.
- Knipscheer P, Raschle M, Smogorzewska A, et al. The fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326(5960):1698–1701.
- Shen CX, Oswald D, Phelps D, et al. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res. 2013;73(11):3393–3401.
- Guo F, Li J, Du W, et al. mTOR regulates DNA damage response through NF-kappaB-mediated FANCD2 pathway in hematopoietic cells. Leukemia. 2013;27(10):2040–2046.
- Chaudhury I, Sareen A, Raghunandan M, et al. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013;41(13):6444–6459.
- Lossaint G, Larroque M, Ribeyre C, et al. FANCD2 binds MCM proteins and controls replisome function upon activation of S phase checkpoint signaling. Mol Cell. 2013;51(5):678–690.
- Michl J, Zimmer J, Buffa FM, et al. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat Struct Mol Biol. 2016;23(8):755-+.
- Dungrawala H, Rose KL, Bhat KP, et al. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell. 2015;59(6):998–1010.
- Shen CX, Bjornsti MA. TOR signaling maintains nucleolar architecture and timely exit from mitosis. Cancer Res. 2009;69(9).
- Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–143.