
ABSTRACT
Tau accumulation is a core component of Alzheimer’s disease and other neurodegenerative tauopathies. While tau’s impact on neurons is a major area of research, the effect of extracellular tau on astrocytes is largely unknown. This article summarizes our recent studies showing that astrocyte senescence plays a critical role in neurodegenerative diseases and integrates extracellular tau into the regulatory loop of senescent astrocyte-mediated neurotoxicity. Human astrocytes in vitro undergoing senescence were shown to acquire the inflammatory senescence-associated secretory phenotype (SASP) and toxicity to neurons, which may recapitulate aging- and disease-associated neurodegeneration. Here, we show that human astrocytes exposed to extracellular tau in vitro also undergo cellular senescence and acquire a neurotoxic SASP (e.g. IL-6 secretion), with oxidative stress response (indicated by upregulated NRF2 target genes) and a possible activation of inflammasome (indicated by upregulated ASC and IL-1β). These findings suggest that senescent astrocytes induced by various conditions and insults, including tau exposure, may represent a therapeutic target to inhibit or delay the progression of neurodegenerative diseases. We also discuss the pathological activity of extracellular tau in microglia and astrocytes, the disease relevance and diversity of tau forms, therapeutics targeting senescence in neurodegeneration, and the roles of p53 and its isoforms in astrocyte-mediated neurotoxicity and neuroprotection.
Introduction
Aging is the predominant risk factor for nearly all major neurodegenerative diseases [Citation1]. According to the U.S. Census Bureau, the number of people 65 and older is expected to double to 95 million by 2060. As there are currently few effective therapeutic interventions to stop or slow age-related neurodegenerative disorders, understanding the mechanisms of disease progression is critical for developing novel therapies. One such mechanism currently being investigated is cellular senescence (CS), an irreversible or sustained cell proliferation arrest [Citation2,Citation3]. After a limited number of replicative cell divisions, known as the Hayflick limit, critically shortened telomeres initiate replicative CS. Cells can also enter premature or stress-induced senescence upon the accumulation of non-telomeric DNA damage induced by a various cellular stressors [Citation4,Citation5].
Senescent cells exhibit a phenotypic shift, known as the senescence-associated secretory phenotype (SASP), characterized by increased production and secretion of a variety of proteins [Citation6]. The cytokine profile of SASP varies based on factors such as the inducer of senescence and the senescent cell type, but frequently SASP includes secretion of matrix metalloproteinases; reactive oxygen species, and cytokines such as IL-1β, TNFα, and IL-6 [Citation6]. Senescent cells are cleared by the immune system, but may accumulate and contribute to chronic inflammation in aged individuals or in patients with disease [Citation2,Citation7,Citation8]. Through SASP, senescent cells contribute to the chronic sterile inflammation observed in aging known as “inflammaging,” [Citation3] and can induce senescence in a paracrine manner to nearby cells [Citation9]. This inflammatory environment contributes to the progression of neurodegeneration through activating nearby glia and inducing apoptosis in neuronal cells [Citation2,Citation7,Citation8]. Critically, clearance of senescent cells in mouse models, either by senolytics or cell programing, can extend lifespan and ameliorate cognitive decline [Citation10–14], emphasizing the potential of senescence-targeted therapies.
In addition to the adoption of SASP, senescent cells are characterized by upregulation of cell cycle inhibitors such as p53, p16INK4A, p21WAF [Citation4,Citation15–17], expression of the hydrolytic enzyme senescence-associated beta galactosidase (SA-ß-gal) [Citation18], and increased accumulation of DNA damage [Citation5]. Our laboratory has identified astrocytes as the major senescent cell type in patients with Alzheimer’s disease (AD) or amyotrophic lateral sclerosis (ALS) in patients receiving cranial radiation, and in aged individuals suggesting that senescent astrocytes accumulate during physiological brain aging and in patients with neurodegenerative disease [Citation7,Citation8]. Astrocytes are one of the most abundant cell types in the brain, and are critical for providing neurotrophic support and facilitating synaptic signaling via the tripartite synapse [Citation19]. These functions are compromised in aged astrocytes undergoing cellular senescence [Citation20]. For example, we demonstrated that senescent astrocytes promote non-cell autonomous neurotoxicity via SASP when co-cultured with mature neurons, motor neurons, neural progenitor cells (NPCs), or neural stem cells (NSCs) [Citation7,Citation8] ()). Treatment with a neutralizing IL-6 antibody ameliorates neuronal cell death, underscoring the role of SASP in causatively promoting neurotoxicity [Citation8] ()). Such astrocyte-mediated neurotoxicity does not require cell-cell contact and occurs in a paracrine manner when neurons are separated by a non-cell-permissive transwell membrane [Citation8]. In addition, other studies have shown that senescent astrocytes contribute to dysregulation of glutamate metabolism, a common feature of neurodegenerative diseases including tauopathies [Citation21–23], which may lead to neuronal excitotoxicity and cell death [Citation24]. Importantly, inhibiting senescence in astrocytes reduces SASP and prevents astrocyte-mediated neurotoxicity in vitro [Citation7,Citation8] ()) while clearance of senescent cells in vivo prevents progression of neurodegeneration and cognitive decline [Citation10,Citation11,Citation25,Citation26]. These studies demonstrate the potential therapeutic value of targeting senescent astrocytes and underscore the importance of identifying triggers of astrocyte senescence in neurodegenerative disease.
Figure 1. Astrocyte senescence and SASP in neurotoxicity and tauopathy. (a) Summary of our previous findings [Citation7,Citation8] of various triggers of senescence and SASP acquisition in human astrocytes. Using a trans-well co-culture system, we determined that senescent astrocytes exert neurotoxic effects on neurons via upregulation of inflammatory SASP cytokines and loss of neurotrophic factors. Inhibition of SASP by an IL-6 neutralizing antibody prevented astrocyte-mediated neurotoxicity. Conversely, a NGF neutralizing antibody diminished neuroprotective effects of non-senescent astrocytes. (b) Summary of tau propagation in neurodegeneration and our findings on tau-induced astrocyte senescence and SASP (from data shown below in and ). Physiological release of tau and/or pathological lysis of neurons with NFTs could contribute to the accumulation of extracellular tau. Three forms of extracellular tau are used for our experiments: monomers, oligomers (PFF) and their mixture to simulate the formation of larger aggregates. These tau treatments resulted in a phenotypic shift in human astrocytes with an upregulation of IL-6, IL-1β, TNFα, IL-8, and NOS2 (data shown in )
![Figure 1. Astrocyte senescence and SASP in neurotoxicity and tauopathy. (a) Summary of our previous findings [Citation7,Citation8] of various triggers of senescence and SASP acquisition in human astrocytes. Using a trans-well co-culture system, we determined that senescent astrocytes exert neurotoxic effects on neurons via upregulation of inflammatory SASP cytokines and loss of neurotrophic factors. Inhibition of SASP by an IL-6 neutralizing antibody prevented astrocyte-mediated neurotoxicity. Conversely, a NGF neutralizing antibody diminished neuroprotective effects of non-senescent astrocytes. (b) Summary of tau propagation in neurodegeneration and our findings on tau-induced astrocyte senescence and SASP (from data shown below in Figure 2 and Figure 3). Physiological release of tau and/or pathological lysis of neurons with NFTs could contribute to the accumulation of extracellular tau. Three forms of extracellular tau are used for our experiments: monomers, oligomers (PFF) and their mixture to simulate the formation of larger aggregates. These tau treatments resulted in a phenotypic shift in human astrocytes with an upregulation of IL-6, IL-1β, TNFα, IL-8, and NOS2 (data shown in Figure 3)](/cms/asset/62e5424f-1a99-4e50-a400-f8748d5aa2c2/kccy_a_1909260_f0001_c.jpg)
Figure 2. Extracellular tau induces cellular senescence in human astrocytes. (a) Schematic of experimental timeline. Human astrocytes were treated with tau monomers, PFF or combination of both at concentration of 0.1 µg/mL (total tau) for 24 hours and collected immediately or after 3 d. (b) Quantification (percentage) and (c) representative image of SA-β-gal positive cells in untreated control versus astrocytes incubated with tau after 3 d. Significance was determined by unpaired two-tailed Student’s t test (n = 3). (d) Fold change of p21WAF1 mRNA expression after 3 d normalized to GAPDH and presented relative to control. Significance was determined by paired two-tailed Student’s t test (n = 4). (e) Western blots of p16INK4A protein levels. Quantification of p16INK4A levels after 3 d was performed by normalization to GAPDH levels. Statistical significance was determined by paired two-tailed Student’s t test (n = 3). Representative blots are shown on the right. *P ≤ 0.05 and **P ≤ 0.01
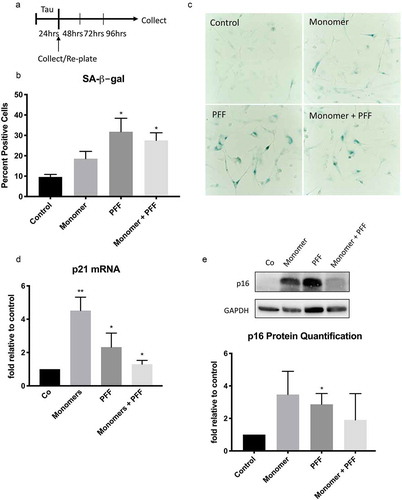
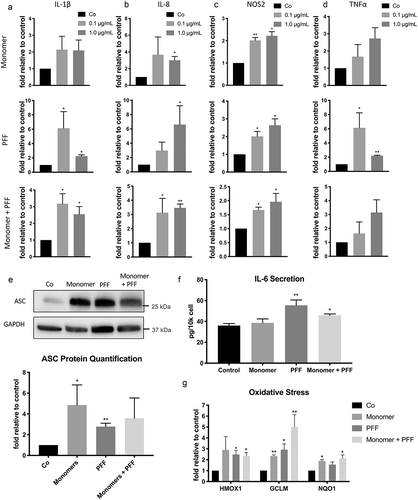
Figure 3. Extracellular tau induces SASP in human astrocytes. Human astrocytes were treated with tau monomers, PFF or combination of both at concentration of 0.1 or 1 µg/mL (total tau) and collected as shown in Figure 1A. All results shown here were from the cells collected after 3 d (Supplementary Figure 1 shows the data at 24 hours). Fold changes of mRNA expression of IL-1β (a), IL-8 (b), NOS2 (c) and TNFα (d) were normalized to GAPDH and presented relative to untreated control. Statistical significance was determined by paired two-tailed student’s t test (n = 3). (e) Representative western blots (top) and quantitative data (bottom) of ASC protein levels in astrocytes treated with 0.1 µg/mL. ASC levels were normalized to GAPDH levels. Statistical significance was determined by paired two-tailed student’s t test (n = 4). (f) Quantification of secreted IL-6 protein in culture media from astrocytes treated with 0.1 µg/mL. Significance was determined by unpaired two-tailed student’s t test (n = 4). (g) Fold changes of mRNA expression of oxidative stress-responsive NRF2 target genes (HMOX1, GCLM, and NQO1) in astrocytes treated with 0.1 µg/mL normalized to GAPDH and presented relative to untreated control. Statistical significance was determined by paired two-tailed student’s t test (n = 4). *P ≤ 0.05 and **P ≤ 0.01
Aging is associated with increased accumulation of senescent astrocytes [Citation8,Citation20]; however, the induction of CS can be accelerated by various endogenous and exogenous stressors. Previously, our laboratory has examined premature or stress-induced senescence in irradiated astrocytes. This is associated with increased DNA-damage and enhanced production of SASP-associated cytokines IL-6, IL-1β, and IL-8 [Citation7] ()). Additional disease-specific stressors may also induce astrocyte senescence. For example, paraquat toxin and amyloid-beta protein induce astrocyte senescence in in vitro models of Parkinson’s disease (PD) and AD, respectively [Citation25,Citation27]. Another critical feature of AD is the accumulation of tau protein. In the healthy brain, tau is found primarily within mature neurons where it stabilizes microtubules and promotes axonal transport. Accumulation of both intracellular tau aggregates and extracellular tau is core components of neurologic diseases including AD and other tauopathies ()). Although the direct impact of tau on the astrocyte phenotype is not well understood, senescent cells, including astrocytes, are observed in tau-associated diseases [Citation8,Citation12,Citation26] suggesting that tau may promote astrocyte senescence and dysfunction. Critically, transplantation of healthy astrocytes has been shown to improve disease progression in a tau mouse model [Citation28] underscoring the importance of astrocytes in tau-associated neurologic disease. To investigate this further, in this study we examined the impact of extracellular tau on primary human astrocytes. There is a myriad of different types and aggregation states of tau observed in neurodegenerative diseases. We chose to investigate the wild-type truncated tau fragment dGAE (AA297-391) as monomers, pre-formed fibrils (PFF), and a combination of both. Since the dGAE fragment is part of the core paired helical filaments (PHF) and does not require a mutation to become pathogenic, the results of this study are likely to be applicable to a broad range of dementia patients [Citation29–31]. Here we report our findings characterizing a direct role of extracellular tau in the induction of astrocyte senescence and SASP thereby identifying a novel mechanism of astrocyte senescence and a potential therapeutic target in tau-associated neurodegeneration.
Methods
Primary cell cultures
Primary human astrocytes were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured at 37°C, 5% CO2 in astrocyte medium supplemented with 2% fetal bovine serum, growth supplement, and 1% penicillin/streptomycin all obtained from ScienCell Research Laboratories. Cells were passaged at a split ratio of 1:2 when confluent, according to the protocol available at https://www.sciencellonline.com/PS/1800.pdf. All experiments used the cells at early passage (i.e. passage number 10 or earlier).
Cell treatments
Human astrocytes were treated for 24 hours with either Active Human Recombinant Truncated Tau Fragment (AA297-391) (dGAE) Protein Monomers (SPR-4414), Active Human Recombinant Truncated Tau Fragment (AA297-391) (dGAE) Protein Preformed Fibrils (Type 1) (SPR-461) or both monomers and pre-formed fibrils (PFF) at a final concentration of 0.1 μg/mL or 1 μg/mL in astrocyte media. These monomers and PFF were purchased from StressMarq Biosciences Inc. (Victoria, British Columbia). Cells on 6-well plates were treated with tau at 70–80% confluency. After 24 hours of treatment, cells were trypsinized and then either collected for RNA extraction or re-plated at 10,000 cells/cm2 on 6-well plates with fresh media containing no tau and cultured for further 3 d (96 hours in total). The latter cells after 3-d recovery were examined for SA-β-gal and collected for RNA extraction and preparation of protein lysates. Cell culture media after 3-d recovery were used for ELISA quantification of secreted IL-6.
SA-β-gal staining
SA-β-gal staining was performed according to the protocol of the Senescence-β-Galactosidase Staining Kit (Cell Signaling Technology, Danvers, MA, USA). At least 200 cells were counted in each treatment group from three different experiments.
IL-6 quantification
Quantification IL-6 in media was assessed via Human IL-6 ELISA kit according to manufacturer instructions (Sigma Aldrich; St. Louis, MO).
Immunoblotting
Cells and tissues were lysed in RIPA buffer. Protein concentration was measured using the Bradford assay method. NuPAGE 4 × loading buffer was added to all lysates and then boiled for 5 min. Then, 80 μg of protein was loaded onto a Tris-glycine gel (Thermo Fisher Scientific, Waltham, MA, USA) for electrophoresis. Proteins were then transferred onto a PVDF membrane. Membranes were blocked in 1:1 mixture of SuperBlock (Thermo Fisher Scientific) and Tris-Buffered Saline (125 mM Tris-HCl, pH 7.6, and 200 mM NaCl) containing 0.1% Tween-20 (TBS-Tween-20). Membranes were incubated in primary antibodies overnight at 4°C, and washed three times in TBS-Tween-20. Membranes were then incubated in secondary antibody for 1 h at room temperature and the signal was visualized using SuperSignal (Thermo Fisher Scientific) developing reagent and visualized on ChemiDoc Imaging Systems (Bio-Rad, Hercules, CA, USA). ImageLab software (Bio-Rad) was used to quantify signal intensities. Primary antibodies and dilution are GAPDH (Millipore MAB374) at 1:500, p16INK4A (Cell Signaling Technology 80,772) at 1:1000, and ASC (Santa Cruz sc-514,414) at 1:100 dilution.
RNA extraction and cDNA preparation
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Crawley, UK) according to the manufacturer’s instructions. Cells were homogenized and lysate mixed 1:1 with 70% ethanol and centrifuged through an RNeasy Mini Spin column. RNA was eluted with RNase-free water. The abundance and quality of the resulting RNA were assessed using a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). RNA samples were diluted so that 2000 ng total RNA could be used for a 5-μl reverse-transcription reaction. cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific).
Quantitative real-time polymerase chain reaction (qRT-PCR)
For the quantitative analysis of mRNA expression, the Tecan Sunrise 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA) was used with Taqman primers from Life Technologies (Thermo Fisher Scientific). Each reaction was performed in at least triplicate using 2 μl of cDNA in a final volume of 5 μl. The following thermal cycle was used for all samples: 95°C for 10 min; 40 cycles of 95°C for 30 s, primer-specific annealing temperatures for 40 s, and 72°C for 40 s. The expression level of each target gene analyzed based on the ΔΔCt method and the results were expressed as relative expression normalized to GAPDH. Taqman primer assays for p21 (Hs00355782_m1), NOS2 (Hs01075529_m1), IL-8 (Hs00174103_m1), IL-1β (Hs01555410_m1), TNFα (Hs00174128_m1), HMOX1 (Hs01110250_m1), GCLM (Hs00978072_m1), NQO1 (Hs01045993_g1), and GAPDH (Hs02758991_g1) were purchased from Life Technologies (Thermo Fisher Scientific).
Statistics
Data are presented as mean ± SEM of at least three independent experiments. Comparisons were made using two-sided, paired and unpaired Student’s t-test. Differences were considered significant at *P ≤ 0.05, **P ≤ 0.01 or n.s. (not significant).
Results
Extracellular tau induces senescence in human astrocytes
Human astrocytes were treated for 24 hours with a final concentration of 0.1 μg/mL of dGAE (tau) monomers, preformed fibrils, or a combination of both. Since limited work has been done with extracellular tau, and even less so with dGAE, we determined tau dosage based on physiological levels of tau in cerebrospinal fluid and relied on previous literature of in vitro experiments with extracellular tau [Citation32,Citation33]. After incubation with tau, cells were collected as pellets or re-plated with fresh media and allowed to recover for 3 d ()). Increased activity of senescence-associated β-galactosidase (SA-β-gal), a lysosomal hydrolytic enzyme, is a routinely used marker of cell senescence [Citation18]. After a 3-d recovery period, we observed an increase in the percent of SA-β-gal positive astrocytes exposed to tau, with no significant cell death, indicating tau-induced cellular senescence (, c)). To further validate the induction of cellular senescence, we examined expression of cell cycle inhibitors p21WAF1 [Citation17] and p16INK4A [Citation16], which are known to be upregulated in senescent cells. After 3 d, astrocytes treated with tau had increased expression of p21WAF1 ()) and p16INK4A ()), further supporting our conclusion that tau induces cellular senescence in human astrocytes.
Astrocytes acquire SASP in response to extracellular tau
While it has been shown that extracellular tau triggers a pro-inflammatory reaction in microglia [Citation34], the effects of tau on astrocytes is poorly defined. We examined the inflammatory response of astrocytes exposed to either 0.1 μg/mL or 1.0 μg/mL concentrations of tau monomers, preformed fibrils, or a combination of both. All three treatment groups had increased expression of IL-1β and IL-8 mRNA at 24 hours which remained elevated after 3 d (); Supplemental Figure 1(a, b)). We observed a delayed increase of NOS2 expression in astrocytes incubated with the tau treatments (), Supplemental Figure 1(c)), suggesting an increase in oxidative stress following the initial inflammatory reaction. An increase in NOS2 is also suggestive of oxidative damage which may be associated with DNA damage [Citation35,Citation36]. Additionally, there was delayed upregulation of TNFα after 3 d (), Supplemental Figure 1(d)). Oxidative damage is also associated with activation of the nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3 (NLRP3) which recruits the apoptosis-associated speck-like protein containing a CARD (ASC) [Citation37]. Activation of the NLRP3-ASC inflammasome is associated with increased production of IL-1β, and is observed in tauopathy mouse models and the brain tissue of AD patients [Citation38]. Consistently, we observed the elevated ASC protein ()) and IL-1β mRNA ()), indicating that extracellular tau promotes the activation of the NLRP3-ASC inflammasome in astrocytes. Further confirming that astrocytes exposed to tau exhibit a chronic inflammatory state, we observed an increase in secreted IL-6 in culture medium of tau exposed astrocytes after the 3-d recovery period ()). The increased secretion of IL-6 and the persistent upregulation of IL-1β, IL-8, TNFα, and NOS2 indicate the acquisition of SASP ( and )). Since oxidative damage is associated with both activation of the NRLP3 inflammasome and SASP, we investigated the expression of genes commonly upregulated in cells to combat oxidative stress. HMOX1, GCLM, and NQO1 are included in the NRF2 system, which plays a major role in cellular defenses against mitochondrial oxidative stress [Citation39]. We observed increased expression of HMOX1, GCLM, and NQO1 in tau-exposed astrocytes, further supporting our hypothesis that extracellular tau induces oxidative stress ()).
Discussion/review
Tau accumulation in the brain is a key event in a number of neurodegenerative diseases, collectively defined as tauopathies. This includes diseases such as AD, PD, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), chronic traumatic encephalopathy (CTE), frontotemporal dementia (FTD), and aging-related tau astrogliopathy (ARTAG). In healthy neurons, tau stabilizes microtubules, but upon phosphorylation or pathological truncation it breaks away from the microtubule to form the intracellular aggregates observed in neurodegenerative disease (), ). Pathological tau aggregates are found primarily within neurons in AD. However, intracellular tau in glia has also been observed in AD and to a greater extent in astrocytes of other tauopathies such as CBD and ARTAG [Citation40]. Although tau research often focuses on its activity as an intracellular aggregate, recent evidence suggests extracellular tau is more neurotoxic [Citation41], participates in the spreading of tau misfolding [Citation42,Citation43], and promotes the propagation of inflammation [Citation44]. In addition, extracellular tau accumulation is an early event in neurodegeneration [Citation45] and its concentration in the cerebral spinal fluid of AD patients correlates to progression of cognitive decline [Citation46]. These findings suggest a critical functional role for extracellular tau in neurodegenerative disease.
Figure 4. Astrocyte senescence as a central player and therapeutic target in neurodegeneration. Astrocyte senescence is a triggered by a number of stressors leading to neurodegeneration (shown in red). Key initiators of the p53-p21 senescence pathway are DNA damage and oxidative stress. In our previous findings, we showed how senescent astrocytes contribute to neurodegeneration through SASP neurotoxicity and loss of neurotrophic support [Citation7,Citation8] (Figure 1A). SASP promoted inflammation and tau activation of microglia further contribute to the cytotoxic environment and the propagation of tau pathology. Senescent astrocytes also fail to regulate glutamate homeostasis and thereby exacerbate neuronal excitotoxicity. There are several therapeutic avenues targeting astrocyte senescence (listed in green) toward the treatment of neurodegenerative disorders
![Figure 4. Astrocyte senescence as a central player and therapeutic target in neurodegeneration. Astrocyte senescence is a triggered by a number of stressors leading to neurodegeneration (shown in red). Key initiators of the p53-p21 senescence pathway are DNA damage and oxidative stress. In our previous findings, we showed how senescent astrocytes contribute to neurodegeneration through SASP neurotoxicity and loss of neurotrophic support [Citation7,Citation8] (Figure 1A). SASP promoted inflammation and tau activation of microglia further contribute to the cytotoxic environment and the propagation of tau pathology. Senescent astrocytes also fail to regulate glutamate homeostasis and thereby exacerbate neuronal excitotoxicity. There are several therapeutic avenues targeting astrocyte senescence (listed in green) toward the treatment of neurodegenerative disorders](/cms/asset/bc881c8f-e0d1-427a-9f41-f3e798b4ae8a/kccy_a_1909260_f0004_c.jpg)
Several theories about the origin of extracellular tau exist. It was originally believed that upon lysis, neurons containing NFTs release tau into the extracellular environment spreading the pathology [Citation41] ()). However, new evidence shows that tau release is a physiological process independent of cell death and possibly regulated by neuronal activity [Citation47] ()). It is hypothesized that pathological tau seeds are released from mature neurons and subsequently taken up by other cells including astrocytes, which are well positioned at the synaptic terminal to capture extracellular tau [Citation48].
The tau protein has 6 natural isoforms with over 80 phosphorylation sites in addition to mutations and truncations associated with disease [Citation49,Citation50]. The structure of tau aggregation filaments is conserved among patients within a disease, but is distinct between tauopathies [Citation51–54]. These pathological tau “strains,” have different aggregation and seeding potency, and exhibit cell-type specificity [Citation55]. Differing cellular responses to the structures of tau could possibly explain the heterogenous clinical presentations of tauopathies [Citation56]. Most studies employ the use of a mutated form of tau, typically P301S or P301L. These mutations are seen in familial cases of FTD and lead to tangle-like pathology in transgenic mouse models [Citation57,Citation58]. However, mutations in tau are, in fact, quite rare, accounting for approximately 5% of cases [Citation59]. Thus, it is possible that the results from previous studies investigating the microglial response to extracellular mutant tau might not accurately recapitulate what is occurring in the majority of tauopathy patients. We decided to use a wild-type dGAE, a truncated core tau fragment (AA297-391) found in AD paired helical filaments (PHF) [Citation30,Citation31,Citation60] and containing the ordered sequences that have been resolved by cryo‐EM in AD [Citation52]. Whereas other tau fragments require heparin to induce aggregation in vitro and result in conformations differing from those observed in AD [Citation61], this fragment self-assembles in vitro into PHF resembling those observed in AD brains when monomers are combined with preformed fibrils (PFF) [Citation29,Citation62,Citation63]. These studies suggest that the dGAE fragment of tau more closely recapitulates the pathology of tauopathies in vitro
Most knowledge on extracellular tau is focused on neuronal interactions and microglia, with very few studies directly examining astrocyte involvement with extracellular tau. Extracellular tau contributes to the increase in inflammation observed in dementia by eliciting an inflammatory response in microglia [Citation34,Citation64,Citation65] (). This is achieved in part by activating NLRP3 [Citation44] (). NLRP3 inflammasome activation has been observed in tauopathy mouse models [Citation38] and is associated with senescence and SASP [Citation66]. Here we show that similar to tau-exposed microglia and replicative-exhausted or irradiated astrocytes [Citation7,Citation8], human astrocytes have an inflammatory reaction to extracellular tau, activate the NLRP3 inflammasome, and exhibit SASP (), , ). The preformed fibrils used in this study are similar to the endogenous oligomer form of tau ()). This early aggregation state is thought to be the most toxic form of the protein rather than NFTs or monomers [Citation67,Citation68], and propagates intercellular tau misfolding through trans-cellular movement [Citation43]. Exposure to preformed fibrils of tau elicited the greatest increase in inflammatory cytokines (), suggesting that astrocytes are more sensitive to this aggregation state of tau. Previous studies have shown that dGAE monomers and preformed fibrils aggregate together in vitro forming larger PHF [Citation29,Citation62,Citation63]. The weaker response elicited by the combination of monomers and preformed fibrils in some cases may further support the hypothesis of the smaller tau oligomers being more toxic possibly through increased cell uptake of this smaller tau species, but not the larger aggregates [Citation43,Citation67,Citation68]. The differing response of astrocytes to specific tau aggregation states is consistent with previous studies demonstrating similar specificity in the inflammatory response of tau-exposed microglia and neurons [Citation34,Citation41,Citation43,Citation44,Citation69].
Previously, we and others have shown increased numbers of senescent cells, including senescent astrocytes, in brain tissues from patients with neurodegenerative tauopathies [Citation8,Citation25,Citation27,Citation70]. Brain cell types that have been reported to undergo cellular senescence in neurodegenerative diseases include endothelial cells, microglia, and astrocytes [Citation2,Citation8,Citation71,Citation72]. Of these, senescent microglia in the brain tissue of AD patients are co-localized to degenerating tau structures suggesting that tau may promote CS in additional cell types [Citation71]. In the present study, we demonstrate a direct role of extracellular tau in the induction of astrocyte senescence ().
Evidence of tau pathology promoting senescence has also been observed in animal studies. Bussian et al. reported an accumulation of p16INK4A-positive senescent microglia and astrocytes in a MAPTP3012PS19 mouse model of tau-dependent neurodegeneration [Citation10]. In another mouse model of tau-associated dementia, rTg4510, Musi and colleagues detected enhanced brain expression of Cdkna2, the gene encoding p16INK4A, at the onset of appearance of neurofibrillary tangles compared to age-matched controls [Citation26]. In both mouse models, clearance of the senescent cells prevented the development of NFTs and improved cognitive decline, suggesting that senescent cells may promote tau pathology. Similar benefits were observed following senolytic treatment of the senescence-accelerated mouse (SAM) model which exhibits impaired cognition, spontaneous amyloidosis, and tau hyperphosphorylation [Citation11,Citation73–75]. Finally, in a mouse model study of PD, clearance of senescent cells mitigates neurodegeneration [Citation25]. Taken together, these studies suggest that senescence is not only a consequence of tau accumulation but integral for the development of tau pathology and neurocognitive decline.
Increased DNA damage and impaired DNA damage response (DDR) promote neurodegeneration [Citation76]. In healthy cells, endogenous tau localized to the nucleus has a protective DNA function. However, prefibrillar tau oligomers have been shown to alter the protective function of nonpathogenic tau in neurons [Citation77]. Tau may directly cause DNA instability [Citation78] or indirectly induce DNA damage through oxidative injury [Citation79,Citation80] (). Indicators of oxidative stress are increased early in the course of neurologic diseases, including tauopathies [Citation35,Citation81–83], and proceeds the formation of NFTs and tau hyperphosphorylation [Citation84]. In this study, we observed an upregulation of NOS2, and the NRF2 target genes HMOX1, GCLM, and NQO1 in astrocytes treated with tau (). This supports previous studies suggesting that oxidative stress is both a consequence and an enhancer of tau toxicity, possibly leading to DNA damage and senescence [Citation81] (). Following the accumulation of DNA damage, p53 activation leads to the upregulation of senescence pathway target genes [Citation85]. In this study, we observed an increased expression of p21WAF1, a transcriptional target of p53 ()), suggesting a role for p53 in tau-induced astrocyte senescence (). Consistent with this in vitro finding, elevated p53 activity is a common feature of neurodegenerative diseases, particularly in white matter glial cells distributed in brain regions undergoing degeneration [Citation86,Citation87].
As mentioned, elimination of senescent cells themselves or their pro-inflammatory effects by senolytics or senomorphics (anti-SASP therapies), respectively, further underscores the importance of cellular senescence in neurodegeneration (). Senolytic drugs, such as propranolol and sildenafil, had been identified as potential therapeutics which might provide benefit in neurodegenerative diseases including AD [Citation11,Citation12,Citation25]. Another therapeutic approach targeting cellular senescence in neurodegeneration is reprograming senescent, neurotoxic astrocytes to proliferative, neuroprotective astrocytes. Among multiple physiological protein isoforms of p53, an N-terminally truncated isoform Δ133p53α functions as an endogenous repressor of astrocyte senescence by dominant-negatively inhibiting a subset of p53 target genes involved in senescence, while a C-terminally modified isoform p53β cooperates with p53 to accelerate astrocyte senescence [Citation7,Citation8,Citation88,Citation89]. Our immunohistochemical studies revealed that Δ133p53α is diminished in the brain tissue of aged individuals and in patients with neurodegenerative disease [Citation7,Citation8]. Critically, reconstituting Δ133p53α expression in replicative and radiation-induced senescent astrocytes ameliorates astrocyte-mediated neurotoxicity and restores their neuroprotective functions in co-culture and transwell experiments [Citation9,Citation10]. This Δ133p53α-induced phenotypic conversion of astrocytes is attributed to decreased secretion of several SASP factors including IL-6 and enhanced expression of neuroprotective factors such as IGF-1 and NGF [Citation7,Citation8]. These studies thus suggest that Δ133p53α may provide a therapeutic opportunity of functionally reprograming senescent astrocytes in neurodegenerative diseases (). Stem cell transplantations or infusions are also being investigated as another possible therapeutic mechanism to restore tissue homeostasis [Citation90] (). Finally, studies are investigating immunotherapies or tau-directed antibodies to promote the clearance of tau and prevent further neurodegeneration [Citation91] (). Given the data presented here (, ) and our previous studies, senolytics and SASP-targeting treatments may be of therapeutic benefit for a wide array of tauopathies.
Conclusion
There is a critical need to study neurodegenerative diseases in the context of aging and senescence. Although tau release is a normal physiological process [Citation47], accumulation of extracellular tau is an early event in neurodegeneration, possibly beginning decades before disease onset [Citation45] ()). While there is increasing evidence for the existence of a relationship between senescence and tau, it was previously unknown whether tau contributes directly to the induction of astrocyte senescence. Here we provide evidence of tau directly inducing astrocyte senescence and SASP (, ). Our findings suggest that tau can further propagate neurodegeneration through promoting astrocyte senescence and SASP () and )). This feedforward mechanism in the regulatory loop involving extracellular tau, astrocyte senescence, SASP and neuronal death could be accelerated by various endogenous and exogenous stressors. This could include genetic risk factors which lead to increased levels of tau and senescent glia [Citation1,Citation54,Citation59,Citation70], stressors promoting inflammation or DNA damage, aging-related changes, amyloid-β deposition, brain radiotherapy, and traumatic brain injury () [Citation92]. Future studies on eliminating and reprograming senescent astrocytes will hopefully bring about the development of novel therapies for neurodegenerative diseases.
Supplemental Material
Download PNG Image (221.8 KB){kind=link}
Acknowledgments
This research was supported by the Intramural Research Program of the NIH and NCI-CCR. Dr Beck is a Molecular Pathology Fellow in the NIH Comparative Biomedical Scientist Training Program supported by the National Cancer Institute in partnership with Purdue University. and are created with BioRender.com.
Disclosure statement
The authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Hou Y, Dan X, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–581.
- Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128(4):1208–1216.
- Olivieri F, Prattichizzo F, Grillari J, et al. Cellular senescence and inflammaging in age-related diseases. Mediators Inflamm. 2018;2018:2018.
- Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;1(5):a001883.
- Di Fagagna FDA. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer. 2008;8(7):512–522.
- Young AR, Narita M. SASP reflects senescence. EMBO Rep. 2009;10(3):228–230.
- Turnquist C, Beck JA, Horikawa I, et al. Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro Oncol. 2019;21(4):474–485.
- Turnquist C, Horikawa I, Foran E, et al. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016;23(9):1515–1528.
- Nelson G, Wordsworth J, Wang C, et al. A senescent cell bystander effect: senescence‐induced senescence. Aging Cell. 2012;11(2):345–349.
- Bussian TJ, Aziz A, Meyer CF, et al. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562(7728):578–582.
- Dobarro M, Orejana L, Aguirre N, et al. Propranolol restores cognitive deficits and improves amyloid and Tau pathologies in a senescence-accelerated mouse model. Neuropharmacology. 2013;64:137–144.
- Orejana L, Barros-Miñones L, Jordán J, et al. Sildenafil ameliorates cognitive deficits and tau pathology in a senescence-accelerated mouse model. Neurobiol Aging. 2012;33(3):625. e11-625. e20.
- Xu M, Pirtskhalava T, Farr JN, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246–1256.
- Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22(5):719–728.
- Mondal AM, Horikawa I, Pine SR, et al. p53 isoforms regulate aging-and tumor-associated replicative senescence in T lymphocytes. J Clin Invest. 2013;123(12):5247–5257.
- Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130(8):1715–1725.
- Roninson IB. Oncogenic functions of tumour suppressor p21Waf1/Cip1/Sdi1: association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002;179(1):1–14.
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, et al. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4(12):1798.
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35.
- Salminen A, Ojala J, Kaarniranta K, et al. Astrocytes in the aging brain express characteristics of senescence‐associated secretory phenotype. Eur J Neurosci. 2011;34(1):3–11.
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Archiv-Euro J Physiol. 2010;460(2):525–542.
- Dabir DV, et al. Impaired glutamate transport in a mouse model of tau pathology in astrocytes. J Neurosci. 2006;26(2):644–654.
- Hunsberger HC, Rudy CC, Batten SR, et al. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem. 2015;132(2):169–182.
- Limbad C, Oron TR, Alimirah F, et al. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PloS One. 2020;15(1):e0227887.
- Chinta SJ, Woods G, Demaria M, et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018;22(4):930–940.
- Musi N, Valentine JM, Sickora KR, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018;17(6):e12840.
- Bhat R, Crowe EP, Bitto A, et al. Astrocyte senescence as a component of Alzheimer’s disease. PloS One. 2012;7(9):e45069.
- Hampton DW, Webber DJ, Bilican B, et al. Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci. 2010;30(30):9973–9983.
- Al-Hilaly YK, Pollack SJ, Vadukul DM, et al. Alzheimer’s disease-like paired helical filament assembly from truncated tau protein is independent of disulfide crosslinking. J Mol Biol. 2017;429(23):3650–3665.
- Novak M, Kabat J, Wischik C. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. Embo J. 1993;12(1):365–370.
- Wischik C, Novak M, Thørgersen HC. et al., Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proceedings of the National Academy of Sciences, 1988;85( 12): 4506–4510.
- Blennow K, Wallin A, Ågren H, et al. tau protein in cerebrospinal fluid. Molecular and Chemical Neuropathology. 1995;26(3):231–245.
- Fá M, Puzzo D, Piacentini R, et al. Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep. 2016;6(1):19393.
- Perea JR, Ávila J, Bolós M. Dephosphorylated rather than hyperphosphorylated Tau triggers a pro-inflammatory profile in microglia through the p38 MAPK pathway. Exp Neurol. 2018;310:14–21.
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3(3):205–214.
- Sohn JJ, Schetter AJ, Yfantis HG, et al. Macrophages, nitric oxide and microRNAs are associated with DNA damage response pathway and senescence in inflammatory bowel disease. PLoS One. 2012;7(9):e44156.
- Jo E-K, Kim JK, Shin D-M, et al. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13(2):148–159.
- Ising C, Venegas C, Zhang S, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–673.
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284(20):13291–13295.
- Kovacs GG. Astroglia and tau: new perspectives. Front Aging Neurosci. 2020;12(12).
- Gómez-Ramos A, Díaz-Hernández M, Cuadros R, et al. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006;580(20):4842–4850.
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–12852.
- Kfoury N, Holmes BB, Jiang H, et al. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287(23):19440–19451.
- Stancu I-C, Cremers N, Vanrusselt H, et al. Aggregated Tau activates NLRP3–ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137(4):599–617.
- Maeda S, Sahara N, Saito Y, et al. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease. Neurosci Res. 2006;54(3):197–201.
- Andreasen N, Minthon L, Davidsson P, et al. Evaluation of CSF-tau and CSF-Aβ42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol. 2001;58(3):373–379.
- Pooler AM, Phillips EC, Lau DHW, et al. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14(4):389–394.
- Perea JR, Lopez E, Díez-Ballestros, et al. Extracellular monomeric tau is internalized by astrocytes. Front Neurosci. 2019;13:442.
- Avila J, Jiménez JS, Sayas CL, et al. Tau structures. Front Aging Neurosci. 2016;8:262.
- Stoothoff WH, Johnson GV. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta Mol Basis Dis. 2005;1739(2–3):280–297.
- Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21(10):428–433.
- Fitzpatrick AW, Falcon B, He S, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547(7662):185–190.
- Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561(7721):137–140.
- Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. 2019;568(7752):420–423.
- Narasimhan S, Guo JL, Changolkar L, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci. 2017;37(47):11406–11423.
- Sanders DW, Kaufman S, DeVos S, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82(6):1271–1288.
- Götz J, Chen F, Barmettler R, et al. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276(1):529–534.
- Allen B, Ingram E, Takao M, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22(21):9340–9351.
- Stanford PM, Brooks W, Teber E, et al. Frequency of tau mutations in familial and sporadic frontotemporal dementia and other tauopathies. J Neurol. 2004;251(9):1098–1104.
- Skrabana R, Kontsek P, Mederlyova A, et al. Folding of Alzheimer’s core PHF subunit revealed by monoclonal antibody 423. FEBS Lett. 2004;568(1–3):178–182.
- Zhang W, Falcon B, Murzin AG, et al. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. Elife. 2019;8:e43584.
- Al‐Hilaly YK, Foster BE, Biasetti L, et al. Tau (297‐391) forms filaments that structurally mimic the core of paired helical filaments in Alzheimer’s disease brain. FEBS Lett. 2019;2020;594(5):944-950.
- Harrington CR, Storey JMD, Clunas S, et al. Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer disease. J Biol Chem. 2015;290(17):10862–10875.
- Morales I, Jiménez JM, Mancilla M, et al. Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis. 2013;37(4):849–856.
- Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351.
- Osorio C, Kanukuntla T, Diaz E, et al. The post-amyloid era in Alzheimer’s disease: trust your gut feeling. Front Aging Neurosci. 2019;11:143.
- Gerson JE, Castillo-Carranza DL, Kayed R. Advances in therapeutics for neurodegenerative tauopathies: moving toward the specific targeting of the most toxic tau species. ACS Chem Neurosci. 2014;5(9):752–769.
- Ward SM, Himmelstein D, Lancia J, et al. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans. 2012;40(4):667–71.
- Evans LD, Wassmer T, Fraser G, et al. Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep. 2018;22(13):3612–3624.
- Tominaga T, Shimada R, Okada Y, et al. Senescence-associated-β-galactosidase staining following traumatic brain injury in the mouse cerebrum. PloS One. 2019;14(3):e0213673.
- Streit WJ, Braak H, Xue Q-S, et al. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118(4):475–485.
- Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005;28(4):202–208.
- Canudas AM, Gutierrez-Cuesta J, Rodríguez MI, et al. Hyperphosphorylation of microtubule-associated protein tau in senescence-accelerated mouse (SAM). Mech Ageing Dev. 2005;126(12):1300–1304.
- Kawamata T, Akiguchu I, Yagi H, et al. Neuropathological studies on strains of senescence-accelerated mice (SAM) with age-related deficits in learning and memory. Exp Gerontol. 1997;32(1–2):161–169.
- Takeshita S, Hosokawa M, Irino M, et al. Spontaneous age-associated amyloidosis in senescence-accelerated mouse (SAM). Mech Ageing Dev. 1982;20(1):13–23.
- Coppede F, Migliore L. DNA damage and repair in Alzheimer’s disease. Curr Alzheimer Res. 2009;6(1):36–47.
- Violet M, Chauderlier A, Delattre L, et al. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiol Dis. 2015;82:540–551.
- Padmaraju V, Indi SS, Rao KSJ. New evidences on Tau–DNA interactions and relevance to neurodegeneration. Neurochem Int. 2010;57(1):51–57.
- Cente M, Filipcik P, Pevalova M, et al. Expression of a truncated tau protein induces oxidative stress in a rodent model of tauopathy. Eur J Neurosci. 2006;24(4):1085–1090.
- Frost B, Hemberg M, Lewis J, et al. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17(3):357.
- Alavi Naini SM, Soussi-Yanicostas N, Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxidative medicine and cellular longevity, 2015. 2015.
- Albers DS, Augood SJ, Park LCH, et al. Frontal lobe dysfunction in progressive supranuclear palsy: evidence for oxidative stress and mitochondrial impairment. J Neurochem. 2000;74(2):878–881.
- Castellani R, Smith MA, Richey PL, et al. Evidence for oxidative stress in Pick disease and corticobasal degeneration. Brain Res. 1995;696(1–2):268–271.
- Dumont M, Stack C, Elipenahli C, et al. Behavioral deficit, oxidative stress, and mitochondrial dysfunction precede tau pathology in P301S transgenic mice. Faseb J. 2011;25(11):4063–4072.
- Gong L, Gong H, Pan X, et al. p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015;25(3):351–369.
- Kitamura Y, Shimohama S, Kamoshima W, et al. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun. 1997;232(2):418–421.
- Chang JR, Ghafouri M, Mukerjee R, et al. Role of p53 in neurodegenerative diseases. Neurodegen Dis. 2012;9(2):68–80.
- Fujita K, Mondal AM, Horikawa I, et al. p53 isoforms Δ133p53 and p53β are endogenous regulators of replicative cellular senescence. Nat Cell Biol. 2009;11(9):1135–1142.
- Horikawa I, Park K-Y, Isogaya K, et al. Δ133p53 represses p53-inducible senescence genes and enhances the generation of human induced pluripotent stem cells. Cell Death Differ. 2017;24(6):1017–1028.
- Duncan T, Valenzuela M. Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res Ther. 2017;8(1):111.
- Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer’s disease. Trends Mol Med. 2015;21(6):394–402.
- Turnquist C, Harris BT, Harris CC. Radiation-induced brain injury: current concepts and therapeutic strategies targeting neuroinflammation. Neurooncol Adv. 2020;2(1):vdaa057.