
ABSTRACT
The DNA damage response (DDR) consists of multiple specialized pathways that recognize different insults sustained by DNA and repairs them where possible to avoid the accumulation of mutations. While loss of activity of genes in the DDR has been extensively associated with cancer predisposition and progression, in recent years it has become evident that there is a relationship between the DDR and cellular metabolism. The activity of the metabolic pathways can influence the DDR by regulating the availability of substrates required for the repair process and the function of its players. Additionally, proteins of the DDR can regulate the metabolic flux through the major pathways such as glycolysis, tricarboxylic acid cycle (TCA) and pentose phosphate pathway (PPP) and the production of reactive oxygen species (ROS). This newly discovered connection bears great importance in the biology of cancer and represents a new therapeutic opportunity. Here we describe the nature of the relationship between DDR and metabolism and its potential application in the treatment of cancer. Keywords: DNA repair, metabolism, mitochondria
KEYWORDS:
Introduction
DNA is constantly challenged by endogenous and exogenous sources of damage that threatens its integrity. Endogenous sources of damage include replication stress, telomere shortening and reactive oxygen species (ROS) produced by cellular metabolism, whilst exogenous sources can consist of UV light, ionizing radiation (IR) and various chemical agents such as alkylating agents. DNA can be damaged by the introduction of either single-strand breaks (SSB) or double–strand breaks (DSB). A sophisticated and complex system of DNA repair pathways has evolved to detect and repair the damage, including base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), homologous recombination (HR) and non–homologous end joining (NHEJ) [Citation1]. Unsurprisingly, mutations in many different DNA damage response (DDR) proteins have been associated with several cancers [Citation2]. Loss of DDR can result in a higher mutagenesis rate and an increase in genomic instability, which contributes to cancer promotion and progression [Citation3–5].
In addition to DDR–associated mutations and defects, cancer cells are also characterized by a complete rewiring of their metabolic machinery, necessary in order to meet the increased energy demand associated with rapid proliferation [Citation6,Citation7] . The first description of this unique metabolic signature was made by Otto Warburg through the observation that cancer cells preferentially utilize glucose for energy production, even in the presence of normal oxygen concentrations, a phenotype which is now known as aerobic glycolysis or the Warburg effect [Citation8,Citation9]. Cancer cells also feature a deregulated amino–acid metabolism and rely on alternative metabolic intermediates to produce the required biomass and to regulate signals required for proliferation and survival [Citation10–13].
A growing body of evidence suggests that the connection between cancer metabolism and the DDR could be exploited therapeutically. Certain metabolites, such as glutamine and aspartate, are required for the de novo synthesis of nucleotides, which influences both the replication of DNA, and the repair process [Citation14–16]. Similarly, the production of ROS during cellular metabolism can increase oxidative DNA damage and influence the activity of the DDR [Citation17,Citation18]. The link between metabolism and DDR is not unilateral, indeed the activation of the DDR can also modify metabolic activity in order to supply the DDR with substrates required for its function. The DDR kinase Ataxia–Telangiectasia Mutated (ATM), for example, can regulate the activity of the pentose phosphate pathway (PPP), in order to boost the biosynthesis of nucleotides [Citation19,Citation20].
The interplay between DDR and cellular metabolism represents a new therapeutic opportunity in the fight against cancer. The observation that cellular metabolism influences the activity of the DDR by regulating the availability of substrates, and that the DDR itself can in turn regulate the metabolic pathways in a concerted network, provides many putative targets that could sensitize cancer cells to already known DDR–targeting drugs or lead to the discovery of new therapeutics targeting the metabolic regulation of the DDR.
In this review, we discuss the recent literature describing the relationship between DDR and metabolism and speculate on the clinical application and the possible future development of the field.
ATM
ATM is a serine/threonine kinase recruited and activated upon induction of DNA DSBs [Citation21–23]. It mediates the activation of a signaling cascade which leads to the initiation of a DNA damage checkpoint in order to induce cell cycle arrest to enable DNA repair or trigger apoptosis, depending on the extent of the damage. The role of ATM in the regulation of the DDR is very well established, including the link between loss of function mutations and tumorigenesis. Nonetheless, in recent years, researchers have pointed out a role for ATM in the regulation of energy metabolism and oxidative stress, highlighting the versatility of this protein.
One of the first evidence of ATM’s metabolic function demonstrated that ATM could protect against metabolic syndrome and atherosclerosis [Citation24]. Specifically, in Apolipoprotein E (ApoE)−/- mice (a mouse model of atherosclerosis) fed a high fat diet, ATM deficiency was responsible for increased adiposity, increased blood pressure and increased glucose intolerance and insulin resistance, all features of metabolic syndrome. ATM has also been shown to be important in the regulation of the anti-oxidative stress response. In 2010, Alexander et al. found that ATM could regulate the activity of mTORC1 in response to ROS-induced stress. They demonstrated that ATM is activated by ROS and in turn triggers a signaling cascade, mediated by an LKB1/AMPK/TSC2 axis, which allows inhibition of mTORC1 [Citation25]. Notably, ATM’s activity as a sensor of redox status (therefore of oxidative stress) was independent of its activation by DNA DSB [Citation26]. Previous studies have shown that the altered ROS production in ATM null cells was a consequence of mitochondrial dysfunction [Citation27,Citation28], such that loss of ATM in thymocytes was associated with an increased number of mitochondria accompanied by an augmented production of mitochondrial ROS (mROS), decreased Complex I activity and increased oxygen consumption rate (OCR) [Citation29]. The role of ATM in regulating cellular metabolism also represents a bridge between replication stress and metabolic reprogramming during senescence. ATM loss in ribonucleotide reductase M2 (RRM2)-deficient cells, prevented the senescent phenotype normally induced by replication stress [Citation20], promoting glucose and glutamine uptake in a c-MYC dependent manner.
Subsequent studies have revealed that ATM regulates the pentose phosphate pathway (PPP) in order to promote NADPH and nucleotide production [Citation19]. Specifically, ATM activation by DNA DSBs increased the activity of glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of the oxidative branch of the PPP, which is the main source of NADPH, an important antioxidant cofactor [Citation30]. More recently, a role for ATM in the regulation of glycolysis has been uncovered [Citation31], whereby increased glycolytic activity in cancer-associated fibroblasts (CAFs) was regulated by ATM and was instrumental to the promotion of cellular migration and invasion in vitro in a lactate-dependent manner. The authors found that during hypoxia, ATM, in a DSB-independent manner, can directly phosphorylate the glucose transporter GLUT1, thus promoting its translocation to the plasma membrane. Consistently, in vivo loss of ATM in CAFs was able to reduce the growth and the number of metastasis, an effect that could be rescued by administration of lactate [Citation31].
Collectively, these reports put ATM at the center of a signaling network with far-reaching consequences in the regulation of cellular metabolism, spanning from the sensing of oxidative stress and the regulation of PPP, to the regulation of mitochondrial metabolism and the promotion of glycolytic flux.
DNA-PK
ATM is not the only kinase of the DDR involved in the regulation of metabolism. DNA-dependent protein kinase (DNA-PK) is a nuclear serine/threonine kinase involved in the sensing of DNA DBS and has a role in the non-homologous end-joining (NHEJ) repair pathway [Citation32,Citation33]. It was previously shown to be critical for the transcriptional regulation of lipogenesis [Citation34], where Upstream stimulatory factor 1 (USF-1) phosphorylation by DNA-PK is a necessary event for the transcriptional activation of the fatty acid synthase (FAS) gene. DNA-PK dephosphorylation by the protein phosphatase PP1 activates DNA-PK. Loss of DNA-PK, both in vitro and in vivo, strongly attenuates both phosphorylation and acetylation of USF-1, leading to reduced activation of FAS gene and de novo lipogenesis [Citation34]. DNA-PK can also regulate the activation of AMP-activated protein kinase (AMPK) in response to glucose deprivation [Citation35]. AMPK has many different metabolic functions, spanning from glucose uptake and energy production to mitochondrial biogenesis [Citation36]. The authors found that the two kinases can interact and DNA-PK-deficient cells fail to phosphorylate AMPK during glucose deprivation [Citation35]. DNA-PK was also shown to be at the center of a genetic program responsible for the metabolic decline, associated with aging [Citation37]. It was shown that in aged muscle, DNA DSBs are more abundant and DNA-PK activation was increased. Analysis of aged muscle from mice bearing a skeletal muscle-specific ablation of DNA-PK, showed increased mitochondrial AMPK activity, in comparison to DNA-PK proficient tissue. Inhibition of DNA–PK was also protective toward weight gain in mice on a high fat diet (HFD), which also displayed a better glucose tolerance and insulin sensitivity, demonstrating that DNA–PK is a critical regulator of metabolic, mitochondrial and physical decline occurring during aging [Citation37].
p53
The protein p53, encoded by the gene TP53, is a transcription factor and a tumor suppressor critically involved in the DDR and cell cycle regulation. p53 expression is rapidly increased upon induction of DNA damage or in response to other stresses [Citation38]. According to the extent of the damage, p53 can induce cell cycle arrest and the repair of the DNA; however, if the damage is too severe or sustained, p53 can induce apoptosis or senescence [Citation39–41]. In the last few decades, it has become clear that p53 sits at the center of a network which integrates the regulation of the DDR, cell cycle and apoptosis, with cellular metabolism. In 2004, it was demonstrated that p53 can bind to the promoter of two important glucose transporters, GLUT1 and GLUT4, downregulating their expression [Citation42]. Subsequently, a novel p53-induced gene was identified, TP53-induced glycolysis and apoptosis regulator (TIGAR), which directly links p53 to the regulation of glycolysis and ROS production [Citation43]. TIGAR was shown to inhibit the glycolytic activity and the production of ROS. Consistently, loss of TIGAR resulted in p53-induced cell death [Citation43].
The restriction of the glycolytic flux mediated by p53, leads to an increased flux of carbon through the PPP. This diversion is instrumental to the generation of NADPH, which is essential to produce reduced glutathione and to decrease the levels of ROS [Citation43]. Glutathione is another by product of serine metabolism and is a cellular antioxidant that helps regulate NADPH/NADP+ levels [Citation44,Citation45]. In response to DNA damage, p53 represses glycolysis to re-route glucose through the PPP, thus providing the cells with the nucleotide precursors necessary to repair the damage [Citation46]. Nonetheless, p53 is also able to inhibit the flux of the PPP, by directly binding to and blocking the activity of glucose-6-phosphate dehydrogenase (G6PD) [Citation47]. Inhibition of G6PD and of the overall activity of the PPP, results in reduced glucose consumption, decreased production of NADPH and accumulation of lipids, which altogether lead to a reduced biosynthesis of macromolecules [Citation47]. This phenotype bears great importance in the context of tumor growth, since p53 is inactivated in many cancers and therefore cannot exert its control on the biosynthetic activity, thus allowing cancer cells to increase consumption of glucose and production of macromolecules necessary to sustain growth. The dual and apparently contrasting role of p53 in regulating the PPP, could perhaps be explained by the capacity of p53 to adjust its function based on the context: in presence of oxidative stress or DNA damage, p53 can increase the activity of the PPP, reducing the glycolytic rate, in order to allow the generation of NADPH and nucleotides; conversely, in other conditions, p53 controls the consumption of glucose and biosynthesis levels by inactivating G6PD, a function that, when lost in cancer, allows the increase of macromolecules to sustain growth, consistent with the role of p53 as a tumor suppressor.
p53 was also found to regulate mitochondrial respiration [Citation48]. p53 null cells showed a reduced OCR with increased lactate production. This phenotype was due to the reduced expression of synthesis of cytochrome c oxidase 2 (SCO2), which is required for the correct assembly of the cytochrome c oxidase complex during mitochondrial respiration [Citation49]. Loss of p53, resulting in reduced mitochondrial respiration, rerouted cellular metabolism toward glycolysis (as demonstrated by increased production of lactate) without altering the total amount of ATP, recapitulating the Warburg effect. This was the first evidence that loss of p53 renders cells less dependent on oxygen, a notion which bears important consequences for cancer growth and progression [Citation48]. In 2018, two novel transcription factors, p53 inhibitor of TIGAR activation (PITA) and p53 inhibitor of SCO2 activation (PISA) were identified as regulators of the p53-mediated control of metabolism [Citation50]. PITA and PISA inhibit the p53-mediated transcription of TIGAR and SCO2, respectively. PITA increased glycolytic flux by augmenting 6-phosphofructokinase 1 (PFK1) activity, while PISA reduced cytochrome c oxidase activity and the mitochondrial respiration [Citation50].
p53 has also been shown to be involved in the regulation of amino-acid metabolism, particularly glutamine [Citation51] and serine [Citation52]. In the case of glutamine, p53 regulates the expression of phosphate-activated mitochondrial glutaminase (GLS2) which converts glutamine to glutamate, to generate glutathione and lower ROS levels, protecting DNA from oxidative damage, in wild-type (wt) p53 cells [Citation51]. Cancer cells react to serine deprivation by increasing serine synthesis and reducing glycolytic flux at the advantage of the TCA cycle [Citation52]. p53 activation during serine starvation, promoted the use of serine for the synthesis of glutathione, in order to preserve the cell from ROS. p53−/- cells displayed a more marked reduction in proliferation and loss of viability during serine deprivation, both in vitro and in vivo, suggesting that serine depletion could be a potential strategy for treating p53−/- tumors [Citation52]. More recently, p53 was shown to increase the expression of SLC1A3, an aspartate/glutamate transporter, during glutamine deprivation to enable cancer cells to maintain the activity of the TCA cycle and the electron transport chain (ETC), and to promote glutamate, glutamine and nucleotide synthesis in order to rescue cell viability. p53−/- cells failed to maintain the activity of their TCA cycle during glutamine deprivation, both in vitro and in vivo [Citation53].
Additionally, p53 was found to be required for the switch from OXPHOS to glycolysis in hepatocellular carcinoma (HCC) cells in a PUMA-dependent manner. PUMA can inhibit the activity of the mitochondrial pyruvate carrier (MPC) by preventing its oligomerization, therefore reducing the mitochondrial uptake of pyruvate and overall, the activity of OXPHOS, promoting a switch toward a glycolytic metabolism [Citation54]. This report demonstrates the versatility of p53 in the regulation of cellular metabolism, describing a novel oncogenic function in suppressing the mitochondrial metabolism to increase glycolytic flux and sustain tumorigenesis.
The role of p53 in the regulation of cellular metabolism is intricate and complex. The observation that loss of p53 recapitulates the Warburg effect, may explain why losing p53 activity can be advantageous for cancer cells, improving their metabolic performance in hypoxic environments. According to this model, p53 possesses further tumor suppressive abilities, in addition to its role in cell cycle and apoptosis. On the other end, some cancers may gain greater advantage by retaining p53 function, in order to remain viable upon deprivation of nutrients such as serine and glutamine. In this scenario, p53 would instead support tumor growth, balancing the metabolic activity of cells according to the nature of the stress they must deal with. Further research is necessary to fully understand the pleiotropic role of p53, but it is evident that its function in the regulation of metabolism varies considerably, according to context and tissue.
ATM, DNA-PK and p53 are amongst the best known and most studied components of the DDR, and by virtue of their importance, there has been a constant growth of literature investigating their emerging role in metabolism. Furthermore, proteins in DNA repair pathways involved in the detection and repair of more specific type of lesions, such as BER, NER, MMR and HR, have also been associated with a metabolic phenotype, highlighting how the DDR as a whole is a metabolic process able to participate in the regulation of cellular metabolism.
Base excision repair
DNA damage which arises from oxidation, deamination and alkylation causing DNA double helix distortions is repaired via the base excision repair (BER) pathway [Citation55]. X-ray repair cross-complementing protein 1 (XRCC1) coordinates BER repair in mammalian cells [Citation56] and it has been shown that cells deficient in XRCC1 display significant changes to pathways such as cellular serine biosynthesis, one-carbon metabolism, amino acid uptake and synthesis, and regulation of cellular redox status [Citation57]. XRCC1 depletion triggered the upregulation of phosphoserine aminotransferase 1 (PSAT1), phosphoglycerate dehydrogenase (PHGDH), and phosphoserine phosphatase (PSPH), three key enzymes involved in serine biosynthesis. Furthermore, upregulation of one-carbon metabolism upon induction of SSBs was activated as a result of suboptimal BER. Similar to XRCC1, loss of other BER proteins including polynucleotide kinase/phosphatase (PNKP) and tyrosyl-DNA phosphodiesterase 1 (TDP1) resulted in an increased expression of PSAT1 and PHGDH [Citation57] signifying a metabolic remodeling of cells in response to BER loss.
Markkanen et al. demonstrated that upon XRCC1 depletion, the enzyme bifunctional methylenetetrahydrofolate dehydrogenase (MTHFD2) which is involved in the tetrahydrofolate salvage pathway was significantly upregulated [Citation57]. They also observed an increase in the expression of several amino acid transporters, in addition to an increase in intracellular amino acid synthesis. XRCC1 depletion also led to an increase in intracellular levels of glutathione. Interestingly a number of other enzymes were also differentially regulated upon XRCC1 depletion, including Glutathione Peroxidase 1 (GPX1). A similar phenotype to that observed upon MMR loss [Citation57,Citation58]. Although loss of XRCC1 activates the DDR, Markkanen et al. acknowledge that there are key differences between the classical DDR and the changes observed upon XRCC1 knockdown. In response to acute DNA damage, the canonical DDR response drives short-term adaptions in the cell to initiate repair, whereas XRCC1 knockdown cells have had to adapt to long-term activation of the DDR. As a result, it is possible that this prolonged stimulation of the DDR may be the driving force behind the metabolic reprogramming and consequently the observed gene expression changes in these BER-deficient cells. Nonetheless, although the precise mechanism by which XRCC1 may influence the expression of these metabolic genes is yet to be fully elucidated, their work clearly indicates that deficiency in BER creates favorable conditions for the malignant transformation of cells [Citation57].
Poly [ADP-ribose] polymerase 1 (PARP1) is a key molecule in BER among other DNA repair pathways. In response to DNA damage, PARP1 uses oxidized NAD (NAD+) as a substrate to ADP(ribosyl)ate both itself and target proteins and initiate repair. In addition to acting as a DNA damage sensor, PARP1 plays a role in transcriptional regulation, cell death, angiogenesis and metabolism. PARP1 drives the majority of NAD+ catabolic activity in cells upon recognition of DNA damage. NAD+ is an important cofactor in metabolism and its reduction to NADH is essential for glycolysis and several steps of the TCA cycle. Murata et al. uncovered a novel pro-survival metabolic response to DNA damage regulated by PARP1. They used fluorescence lifetime imaging technology (FLIM) to examine real-time metabolic changes in response to DNA damage and demonstrated that PARP1 activation drives a metabolic shift from glycolysis to OXPHOS in order to replenish ATP and promote cell survival [Citation59] ().
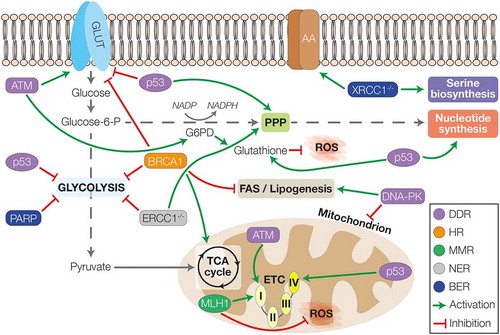
Figure 1. The interplay between DDR and cellular metabolism. The players of the different DDR pathways can differentially regulate the energetic metabolism. ATM was shown to upregulate the expression of glucose transporters and to promote the activity of G6PD and the PPP, leading also to the synthesis of nucleotide and production of glutathione to counteract ROS. ATM can also promote mitochondrial metabolism by increasing the activity of complex I. p53 instead, can inhibit glucose uptake and glycolysis, favouring the activity of PPP. Furthermore, p53 can induce the synthesis of nucleotide and glutathione and increase mitochondrial activity promoting the assembly of complex IV. DNA-PK is able to induce lipogenesis and FAS whilst downregulating the number of mitochondria during ageing. Amongst HR genes, BRCA1 can inhibit the activity of glycolysis to favour mitochondrial metabolism through the TCA cycle; whilst in the context of MMR, MLH1 is important for the regulation of complex I activity and the inhibition of ROS production. Loss of NER activity (ERCC1-/-) leads to inhibition of glycolysis favouring the re-routing of glucose towards PPP. Finally, in the context of BER, loss of XRCC1 is associated with increased expression of AA-transporters and upregulation of serine biosynthesis; whilst increased PARP activity leads to compromising of glycolysis. GLUT – glucose transporters; AA – amino acids transporters; G6PD – glucose-6-phosphate dehydrogenase; PPP – pentose phosphate pathway; ROS – reactive oxygen species; TCA cycle – tricarboxylic acid cycle; ETC – electron transport chain; DDR – DNA damage response; HR – homologous recombination; MMR – mismatch repair; NER – nucleotide excision repair; BER – base excision repair
Moreover, studies have shown that increased PARP activity correlated with a depletion in NAD+ availability corresponding with a reduction in Sirtuin (SIRT) activity which is important for the maintenance of metabolic homeostasis [Citation60]. SIRT1 and PARP1 compete for the NAD+ pool. By sensing NAD+/NADH ratios and nicotinamide levels, SIRT1, a member of the sirtuin family, controls the acetylation status and activity of a number of enzymes and transcriptional regulators [Citation61]. Upon DNA damage, PARP activation has been shown to inhibit SIRT1 activity and influence cellular metabolism. Bai et al. highlight the interplay between SIRT1 and PARP1 when they demonstrate that PARP1-deficient mice display a phenotype comparable to that seen by SIRT1 activation. This is characterized by increased mitochondrial content, increased energy expenditure and protection against metabolic disease [Citation62]. Inhibition of PARP resulted in a significant enrichment of several common metabolic pathways including protein synthesis, nitrogen metabolism and taurine metabolism [Citation63]. Many of the metabolic phenotypes observed were cell line dependent however, which highlights the variable metabolic response cancer cells undergo. This demonstrates the need to better understand the metabolic architecture of specific cancer subtypes.
Several studies have also described the association between XRCC1 and PARP1 [Citation64,Citation65]. PARP1 binds to intermediates of the BER pathway and becomes activated for poly(ADP-ribose) (PAR) synthesis. PAR subsequently mediates recruitment and functions of key BER factors such as XRCC1 which in turn binds to and recruits DNA polymerase β (pol β) [Citation64]. A complex series of reactions occur upon activation of the BER pathway and absence of one of the key players may significantly reduce the efficiency of the overall pathway, consequently increasing genomic instability. As such, it is possible that the metabolic reprogramming Markkanen et al. observe in BER-deficient cells cannot be attributed to XRCC1 loss alone but is a result of accumulating molecular changes in the BER pathway occurring upon XRCC1 loss within these cells.
Nucleotide excision repair
The nucleotide excision repair (NER) pathway predominantly removes bulky DNA adducts which distort the normal structure of the DNA helix, usually caused by ultraviolet radiation, chemicals or ROS [Citation66]. NER involves more than 30 genes, and functions to eliminate base pair-disrupting lesions operating via two pathways; global NER (GG-NER) and transcription coupled NER (TCR). Milanese et al., use NER-deficient mice (ERCC1-defective (TCR) and Xpg-knockout mice (GG-NER)) to demonstrate that the combination of defects in TCR and GG-NER, result in transcription stalling which drives a metabolic rewiring in cells [Citation67]. This rewiring was controlled by an ATP-mediated allosteric mechanism, which enhanced the ability of NER-deficient cells to respond to stress. In their model, they demonstrated that defective NER caused reduced transcription and results in elevated levels of ATP. The surplus of ATP, generated as a result of transcription arrest, inhibited glycolysis and favors glucose entering the PPP by inhibiting phosphofructokinase 1 (PFK). This catalyzed the conversion of fructose-6-phosphate to 1,6-bisphosphate, the key regulatory step in glycolysis. Increased levels of ATP promote glucose rerouting through the PPP upon inhibition of PFK, which favors the production of reducing equivalents in the form of NADPH. A consequence of this rewired metabolism in favor of PPP over glycolysis was an increased defense against oxidation, required to counteract stress caused by DNA damage. Their data demonstrated that a persistent reduction of transcription over time was required to rewire glucose metabolism and increase the reductive capacity of NER–defective cells, proposing a connection between defective DNA repair, metabolism and aging. It is possible that transcription stalling and consequent increase in ATP levels is driving the observed metabolic phenotype by altering the expression of certain transcription factors, irrespective of NER pathway status. Although the precise mechanism underpinning the relationship between altered levels of ATP, DNA damage and metabolism has yet to be fully elucidated, Milanese et al. indicate that the metabolic rewiring precedes the transcriptional response [Citation67].
DNA mismatch repair pathway
The DNA MMR pathway plays a key role in maintaining genomic stability, primarily repairing base–base mismatches and insertion/deletion loops which arise during DNA replication. The function of the DNA MMR pathway is carried out predominantly by four key proteins; MSH2, MSH6, MLH1 and PMS2, which are highly conserved across species [Citation68]. Although the main role of the MMR pathway is in the repair of replication errors, more recently there is evidence for MMR capability in the repair of oxidative DNA damage to mitochondrial DNA (mtDNA) [Citation69–73]. We have demonstrated that inhibition of a number of mitochondrial genes, including POLG and PINK1, results in an accumulation of oxidative 8–oxoG lesions specially in mtDNA that can induce synthetic lethality in MLH1–deficient cells [Citation74–78]. Additionally, we and others have identified the presence of MLH1, but not MSH2, in the mitochondria [Citation74,Citation79]. Furthermore, loss of MMR was associated with an increased number of mismatches in the non-coding D-loop in the mtDNA of retinal endothelial cells, which was associated with decreased respiration and increased apoptosis [Citation71]. This phenotype could be rescued by the addition of MLH1, but not MSH2. More recently, we have shown that loss of MLH1 results in a mitochondrial phenotype characterized by a reduction in activity of respiratory chain complex I, a reduced mtDNA copy number and consequently reduced oxidative phosphorylation (OXPHOS) in cancer cells [Citation58]. In addition, a significant decrease in basal OCR and spare respiratory capacity (SRC), which reflects the cells ability to respond to increased energy demands, was observed in the MLH1-deficient cells compared to the MLH1-proficient cells [Citation58]. We hypothesize that oxidative phosphorylation is impaired in MLH1-deficient cells, due to a decreased Complex I activity. This is supported by the reduction in gene expression levels of mitochondrial encoded subunits, NADH-ubiquinone oxidoreductase chain 2 (MT-ND2) and NADH-ubiquinone oxidoreductase chain 5 (MT-ND5). Moreover, MLH1-deficient cells displayed a reduced anti-oxidant response with a significant reduction in both Nuclear factor erythroid 2-related factor 2 (NRF2) and GPX1 at mRNA and protein level [Citation58], resulting in an increased sensitivity to ROS inducing agents and Complex I inhibition.
Homologous recombination
The main HR protein found to be involved in energy metabolism is Breast Cancer 1 early onset (BRCA1). BRCA1 is a tumor suppressor gene and its inherited mutations are associated with increased risk of breast and ovarian cancer [Citation80,Citation81] due to its significant role in DNA DSB repair [Citation82].
BRCA1’s ability to interact with acetyl-CoA carboxylase α (ACCA), an enzyme involved in fatty acid (FA) synthesis, was the first evidence of a potential metabolic role [Citation83]. Furthermore, downregulation of BRCA1 increased lipogenesis in breast cancer cells [Citation84]. The authors showed that BRCA1 can interact with the phosphorylated ACCA, preventing its activation and acting as a regulator of FA synthesis; therefore, loss of BRCA1 resulted in upregulation of lipogenesis, allowing ACCA to be dephosphorylated and activated [Citation84]. Later studies demonstrated that re-expression of BRCA1 led to a strong inhibition of glycolysis and activation of the TCA cycle and OXPHOS. Reconstitution of BRCA1 was responsible for a global transcriptional change resulting in downregulation of glycolytic enzymes and concurrent upregulation of antioxidant response and TCA cycle genes [Citation85]. BRCA1 mutated cells were also shown to have higher levels of HIF1α and upregulated adenylate kinase A4 (AK4) (required for the regulation of AMPK and the balance of adenosine nucleotides), alterations which are consistent with a switch toward aerobic glycolysis [Citation86].
BRCA1 was also reported to regulate mitochondrial metabolism by regulating the activity of nicotinamide N-methyltransferase (NNMT) [Citation87]. BRCA1 depletion induced a reduction in OCR and ATP production without affecting glycolytic activity. Consistent with this mitochondrial phenotype, BRCA1-deficient cells were sensitive to mitochondrial metabolic targeting agents, such as VLX600 (which inhibits mitochondrial respiration), tigecycline (which inhibits mitochondrial protein translation) and WZB117 (which blocks GLUT1 and reduces ATP levels). Interestingly, BRCA2- or RAD51-depletion were not able to achieve the same phenotype, highlighting a BRCA1-specific function rather than a product of HR loss [Citation87].
Furthermore, genetic ablation of BRCA1 in cardiomyocytes was shown to be responsible for a significant alteration of glucose and FA metabolism [Citation88]. Cardiomyocyte-specific BRCA1 loss resulted in a significant reduction of acetyl-CoA decarboxylase 2 (ACC2) and malonyl-CoA decarboxylase (MCD), both events that lead to a reduced fatty acid oxidation. Additionally, the peroxisome proliferator-activated receptor (PPAR) α, an important regulator of lipid and energy metabolism, was downregulated in BRCA1−/- cardiomyocytes. CD36, GLUT-4 and carnitine palmitoyltransferase 1 (CPT1) expression was also significantly reduced. Collectively, these metabolic alterations led to an overall reduction in ATP production, due to the impaired glucose and fatty acids utilization in the absence of BRCA1 [Citation88].
Interestingly, BRCA1 also functions in the maintenance of energy homeostasis in skeletal muscle [Citation89,Citation90]. Physical exercise increased the interaction between BRCA1 and phosphorylated ACC. Knock-down of BRCA1 in human myotubes resulted in accumulation of lipids, reduced Akt activation and glucose uptake in response to insulin, as well as a decreased OCR [Citation89]. Conversely, BRCA1 loss in skeletal muscle in vivo, resulted in reduced accumulation of lipid droplets, overall decreasing the animals’ weight gain. BRCA1 knock-out mice demonstrated a reduced mitochondrial respiration rate and enlarged mitochondria, with a reduced tolerance to exercise and an increased whole-body energy expenditure [Citation90]. Taken together, BRCA1 appears to have a central role in regulating energy metabolism in skeletal muscle, specifically by balancing lipid accumulation and consumption.
The ability of BRCA1 to modulate the expression of metabolic genes appears to be a novel function, parallel but independent from the canonical BRCA1 activity in HR, as demonstrated by the fact that loss of BRCA2 and RAD51, whilst disrupting HR, did not affect cell metabolism [Citation87]. Although the molecular mechanism by which BRCA1 modulates the metabolic activity is still unclear, and likely to be tissue-specific, there is evidence pointing toward a ‘direct” effect. BRCA1 can directly bind important players in the metabolic homeostasis, such as ACCA [Citation83,Citation84], HIF1a [Citation91] and Akt [Citation92], regulating either their stability or activation (by phosphorylation), ultimately regulating the expression of downstream metabolic genes. Therefore, to date, the role of BRCA1 in the regulation of metabolism appears to be independent from its involvement in the repair of DNA.
DNA repair in the mitochondrion
Like nuclear DNA, mitochondrial DNA (mtDNA) is also subject to damage, particularly oxidative damage, given its localization within the mitochondrion and its proximity to the ROS-producing oxidative phosphorylation. MtDNA was found to be more susceptible than nuclear DNA to ROS-induced damage, most likely due to the lack of protective histones [Citation93]. Many inherited disorders are due to mutations in mitochondrial genes [Citation94,Citation95], hence the growing interest in the process of mtDNA repair. In contrast to nuclear DNA repair, there is a more limited repertoire of DNA repair pathways in the mitochondria. Originally thought to be limited to short–patch BER, accumulating studies have identified the presence of additional repair mechanisms including long–patch BER, MMR, HR and NHEJ, whilst NER is thought to be limited to nuclear DNA repair [Citation96–98]. The most active repair pathway in the mitochondria, is by far BER. Several glycosylases have been found to localize in this organelle and to participate in the repair of mtDNA (extensively reviewed in [Citation99] and [Citation100]). Mitochondrial-targeting of 8-oxoguanine DNA glycosylase (OGG1) was found to be sufficient to protect mice from diet–induced obesity, type II diabetes and adipose inflammation by increasing whole-body energy expenditure and mitochondrial respiration in the adipose tissue [Citation101]. Recently, DNA polymerase beta (polβ) was identified in mitochondrial protein extracts of mammalian origin (human and mouse), where is able to directly interact with proteins involved in the maintenance of mtDNA [Citation102]. Consistently, loss of polβ results in accumulation of mutations in mtDNA and mitochondrial dysfunction, demonstrating that the correct maintenance of mtDNA is a requirement for a balanced mitochondrial homeostasis [Citation102]. Despite some components of the MMR and other repair pathways have been reported in the mitochondria, it is still unclear how they contribute to the mtDNA maintenance and the overall mitochondrial function. A significantly more detailed discussion about the DNA repair pathways found active in the mitochondria, and their difference from the nuclear mechanism, is reviewed in [Citation103]).
Targeting the metabolic phenotype to treat cancer
Increasing evidence indicates that cancer is a “metabolic disease”, influenced by complex interactions between tumors and their microenvironments. The knowledge that healthy cells and cancer cells utilize distinct energy generating pathways, could serve as a potential therapeutic target and there is growing interest in developing techniques to target metabolism for immunotherapy [Citation8,Citation104]. Identifying the unique metabolic dependencies of specific cancer cells, may enable the noninvasive recognition of tumors, making it possible to directly target their essential pathways in order to prevent tumor progression [Citation13,Citation104,Citation105].
In this review, we discuss the link between defective DDR and cancer metabolism, as illustrated in . Cancer cells with defects in the DDR as demonstrated here often display altered metabolic phenotypes, which may be exploited therapeutically. The knowledge that BRCA1 plays a key role in regulating mitochondrial metabolism could be exploited when developing new therapies based on the use of drugs that inhibit energy metabolism [Citation87]. It is worth considering that these drugs which are either in clinical trials or already FDA approved, could also be useful for treating other DDR deficient cancers that display a similar metabolic phenotype. Several studies have also demonstrated the role of ATM in mitochondrial function, where loss of the protein is associated with an increased production of mitochondrial ROS [Citation27–29], a phenotype that could be exploited to sensitize cancer cells to ROS-inducing agents. Interestingly, metformin, a common drug used for the treatment of diabetes, may have anti-cancer properties, in DDR deficient cancers [Citation106]. Metformin inhibits Complex I activity, reducing oxygen consumption and ATP production, and is being assessed alone and in combination with other anti-cancer agents, for the treatment of a range of cancer types [Citation107]. Significantly, we have recently shown that MLH1 loss results in an increased sensitivity to Complex I inhibition and therefore MLH1-deficient patients may benefit from Metformin treatment. In addition, due to a decreased antioxidant response MLH1-deficient cells were increasingly sensitive to parthenolide, a ROS-inducing agent [Citation58]. Molina et al. also describe the discovery of IACS-010759, a small molecule inhibitor of Complex I, which could similarly benefit these patients. Treatment with this inhibitor inhibited proliferation and induced apoptosis in several cancer models reliant on OXPHOS. Their work highlights that some tumors rely heavily on OXPHOS for the production of macromolecules, in addition to ATP synthesis, similar to glycolysis. IACS-010759 is currently being evaluated in phase 1 clinical trials for relapsed/refractory AML and solid tumors (NCT03291938) [Citation108].
Moreover, Lahiguera et al. demonstrate the importance of OXPHOS inhibitors in treating BRCA1/2 mutated cancers and other tumors affected by defects in HR. Homologous recombination-defective (HRD) cells are more sensitive to metformin and NAD+ concentration changes as they rely on oxidative metabolism to supply NAD+ and ATP for PARP1-mediated DNA repair [Citation109]. Metformin alters the redox state and ATP levels of HRD cells reducing the capacity of these cells to repair damaged DNA. Lahiguera et al. also highlighted that a high glycolytic metabolism reduces the effect of PARP inhibitors like olaparib on tumor cells by reducing the capacity of the cells to block PARP activity and poly-ADP protein ribosylation. Their results highlight that that PARP inhibitors require active OXPHOS metabolism to influence cell viability and by blocking glycolysis they resensitized the tumors to olaparib. As such, their results suggest that metabolic profiling tumors could indicate response to olaparib and provide a new way of selecting patients for PARP inhibitor treatment [Citation109].
Furthermore, it may be possible to target the PPP for the treatment of DDR-deficient tumors. Loss of ERCC1, XPG and ATM alter their metabolic dependencies, inhibiting glycolysis and causing them to rely on the PPP to enhance the production of NADPH [Citation67]. Reprogramming NADPH homeostasis is key in cancer progression and could be exploited therapeutically. Recent research indicates that inhibition of G6PD, a key enzyme in the PPP, shows promise in reducing tumor growth and metastasis. It has been shown that inhibition of PPP with polydatin, an inhibitor of G6PD, has a potential anticancer effect on cancer cells [Citation110,Citation111]. Given that PPP is pivotal in enabling cancer cells to combat oxidative stress particularly in cells with impaired NER, it would be interesting to investigate whether PPP inhibition could be an effective strategy in tumors where an impaired NER drives a similar phenotype.
In this review, we have highlighted that alterations in many components of numerous DDR pathways can influence glycolysis and this may be targeted therapeutically. For instance, CAFs with an increased activation of ATM, display a higher glycolytic activity [Citation31], BRCA1 deficient cells preferentially depend on glycolysis [Citation85,Citation86], and p53 drives increased glycolysis in HCC cells [Citation54]. There are several glycolytic inhibitors in preclinical or early stage clinical trials showing promise in suppressing tumor progression, which inhibit different stages of the pathway from glucose uptake to specific glycolytic enzymes [Citation112] (). Although preliminary results from direct inhibition of glycolysis are promising, there are major concerns regarding toxicity and off target effects due to the expression of the target proteins in healthy cells. Nonetheless, there are several promising glucose transporter (GLUT) inhibitors, such as phloretin and WZB117, and glycolytic enzyme inhibitors, such as lonidamine (inhibits HK2), koningic acid (inhibits GAPDH), oxamate (inhibits LDH-A) and dichloroacetate (inhibits PDK) that have proven effective in suppressing cancer cell growth in in vitro and in vivo models [Citation113]. It would be interesting to investigate the use of glycolytic inhibitors in combination with additional cancer chemotherapeutics to create a novel targeted therapy for the treatment of DDR impaired cancers. For example, the phosphoinositide 3-kinase (PI3K)-AKT signaling network regulates multiple steps in glucose metabolism through post-translation regulation of metabolic enzymes and transcriptional control of metabolic processes [Citation114]. For instance, AKT promotes glucose uptake through GLUT1 and GLUT4, controls key steps in glycolysis through phosphorylation and activation of specific glycolytic enzymes and stimulates the activity of PFK1 [Citation114]. There are a number of PI3K inhibitors approved by the FDA including, but not limited to, Alpelisib [Citation115] for breast cancer treatment and Idelalisib for chronic lymphocytic leukemia treatment (CLL) [Citation116], as well as many more undergoing preclinical trials [Citation117] that could be repurposed for targeting DDR impaired cancers. Therefore, PI3K inhibitors could be considered as a potential therapeutic option for targeting PI3K mutant and DDR-deficient cancers with altered dependencies on glucose metabolism.
Table 1. Therapeutic agents targeting metabolism for cancer treatment; either approved, in clinical trials, or undergoing preclinical research
Furthermore, the knowledge that cancer cells enhance TCA cycle function for the production of energy and biosynthetic precursors, has made this pathway a potential therapeutic target for cancer. In this review we have highlighted that DDR pathway proteins ATM, BRCA1 and p53 play a role in regulating TCA cycle metabolism, and thus we speculate that inhibitors perturbing aberrant TCA function could be a promising treatment approach for DDR impaired tumors. One method to disrupt the TCA cycle is through the inhibition of glutaminolysis, which provides glutamine as a fuel source for the TCA cycle for many tumors. CB-839, which inhibits glutaminase (GLS), is currently being investigated clinically as a monotherapy and in combination with other drugs, and shows great potential in blocking tumor growth [Citation118]. An additional therapeutic target is the α-ketoglutarate dehydrogenase complex (KGDHC), the rate-limiting enzyme in the TCA cycle. CPI-613, a lipoate analog that inhibits KGDHC and induces ROS, is undergoing phase I and II clinical trials as a monotherapy and in combination with other chemotherapeutics and is showing therapeutic promise [Citation118].
Reprogramming of fatty acid (FA) synthesis and lipid metabolism represents another aspect of metabolism frequently reprogrammed in DDR-deficient cancer cells. Whilst healthy cells depend on the uptake of FAs from exogenous sources to fulfill requirements, cancer cells can reactivate FA synthesis regardless of lipid concentration in the circulation [Citation119]. In cancer cells, FAs act as fundamental substrates required for energy storage, membrane synthesis, generation of signaling molecules and lipid droplet formation. DDR proteins DNA-PK and BRCA1 can influence lipogenesis in cancer cells, therefore targeting FA synthesis could offer an additional therapeutic target for tumors with this altered metabolic pathway. Key enzymes involved in lipogenesis and lipolysis are often upregulated in cancer cells thus making them attractive therapeutic targets. To date, efforts to target FA metabolism in cancer have been largely focussed on FAS inhibition [Citation120]. There are a number of FAS inhibitors being investigated in preclinical and clinical trials, a promising example of which is TVB-2640 which binds to and blocks FAS, inducing apoptosis, reducing cell signaling and inhibiting cell proliferation. TVB-2640 is currently undergoing clinical trials in combination with other chemotherapeutics in a range of cancer types [Citation121]. Another method to inhibit FA synthesis in cancer cells is to inhibit ACC which catalyzes the rate-limiting carboxylation reaction of acetyl-coA to -CoA. The ACC inhibitor, ND-646 has shown promising results in non-small cell lung cancer mouse models alone and in combination with carboplatin [Citation122,Citation123]. Other potential inhibitors targeting ATP-citrate lyase (ACLY), acetyl-coenzyme A synthetase (ACSS2), and stearoyl-CoA desaturase-1 (SCD) are undergoing preclinical and clinical investigations, however increasing evidence suggests that inhibiting single enzymes will not be sufficient when targeting FA metabolism as a cancer treatment [Citation120].
Finally, inhibition of nucleotide acid metabolism has been a means of targeting cancer cells for decades. One example is methotrexate which inhibits dihydrofolate reductase (DHFR), an enzyme necessary to synthesize tetrahydrofolates required to produce purines and pyrimidines and is one of the most common anti-cancer drugs used in the treatment of several malignancies [Citation124,Citation125]. A number of new inhibitors of the folate synthesis have been developed to be more selective and less toxic, which target either DHFR [Citation126] or other enzymes of the pathway [Citation127,Citation128]. Another common target is thymidylate synthase (TS), which can be inhibited with 5-fluorouracil, a nucleoside analog of uracil approved for use in several cancers [Citation129]. Gemcitabine is another anti-cancer nucleoside that inhibits the ribonucleotide reductase (RNR), preventing the generation of deoxyribonucleotides necessary for the synthesis and repair of DNA [Citation130]. Finally, inosine–5ʹ–monophosphate dehydrogenase (IMPDH), an enzyme critical in the synthesis of guanine, is often found to be overexpressed in cancer and therefore is being investigated as a potential therapeutic target [Citation131–134].
Conclusions
The DDR and cellular metabolism are fundamental biological processes; therefore, their intertwined relationship is a new, promising avenue in our understanding of both normal and more importantly cancer cell biology. The fact that an impaired DDR can give rise to a specific metabolic phenotype which reshapes the metabolic landscape of cancer cells (by modifying the flux of substrates and consequently the prevalence of some metabolic pathways over others), offers a unique opportunity to target cancer by exploiting the “metabolic vulnerabilities” exposed by the loss of specific DDR players. Whether it is the production of ROS or the reliance on specific metabolic routes, an impaired DDR provides a window of opportunity that is specific to cancer cells, potentially sparing healthy cells from such a clinical intervention. Significantly, it is becoming clear that many interactions between these processes remain unknown. To take full advantage of the link between DDR and metabolism, we need to understand in more detail the nature of this relationship, dissecting mechanisms which may be cancer type-specific and/or DDR pathway-specific, to achieve a broader picture that can inform targeted and safe therapeutic interventions. High-throughput functional genomic approaches will enable a better understanding of the precise vulnerabilities within the interplay of these fundamental processes. A more in-depth understanding of this emerging relationship is required to truly be able to understand and exploit resistance mechanisms and to design better rationale-based therapeutics for DNA repair-deficient patients.
Acknowledgments
We thank Barts Charity, Cancer Research UK and the Medical Research Council for supporting our research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Lombard DB, Chua KF, Mostoslavsky R, et al. DNA repair, genome stability, and aging. Cell. 2005;120(4):497–512.
- Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16(1):35–42.
- Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer. 2012;12(9):587–598.
- Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013;2(3):107–129.
- Wolters S, Schumacher B. Genome maintenance and transcription integrity in aging and disease. Front Genet. 2013;4:19.
- Cassim S, Vučetić M, Ždralević M, et al. Warburg and Beyond: the Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer.Cancers (Basel). 2020;12:5.
- Parks SK, Mueller-Klieser W, Pouysségur J Lactate and Acidity in the Cancer Microenvironment. Annual Review of Cancer Biology; 2020. p. 141–58.
- Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314.
- Cassim S, Raymond VA, Dehbidi-Assadzadeh L, et al. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle. 2018;17(7):903–916.
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95.
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47.
- Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(2):113.
- DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200.
- Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39(8):347–354.
- Rabinovich S, Adler L, Yizhak K, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature. 2015;527(7578):379–383.
- Kim J, Hu Z, Cai L, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. 2017;546(7656):168–172.
- Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–62.
- Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7(6):e2253.
- Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. Embo J. 2011;30(3):546–555.
- Aird KM, Worth AJ, Snyder NW, et al. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep. 2015;11(6):893–901.
- Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281(5383):1677–1679.
- Kim ST, Lim DS, Canman CE, et al. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274(53):37538–37543.
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506.
- Schneider JG, Finck BN, Ren J, et al. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4(5):377–389.
- Alexander A, Cai SL, Kim J, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107(9):4153–4158.
- Guo Z, Kozlov S, Lavin MF, et al. ATM activation by oxidative stress. Science. 2010;330(6003):517–521.
- Eaton JS, Lin ZP, Sartorelli AC, et al. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest. 2007;117(9):2723–2734.
- Ambrose M, Goldstine JV, Gatti RA. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet. 2007;16(18):2154–2164.
- Valentin-Vega YA, Maclean KH, Tait-Mulder J, et al. Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 2012;119(6):1490–1500.
- Jain M, Brenner DA, Cui L, et al. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res. 2003;93(2):e9–16.
- Sun K, Tang S, Hou Y, et al. Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine. 2019;41:370–383.
- Hartley KO, Gell D, Smith GC, et al. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell. 1995;82(5):849–856.
- Jackson SP. DNA-dependent protein kinase. Int J Biochem Cell Biol. 1997;29(7):935–938.
- Wong RH, Chang I, Hudak CS, et al. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009;136(6):1056–1072.
- Amatya PN, Kim HB, Park SJ, et al. A role of DNA-dependent protein kinase for the activation of AMP-activated protein kinase in response to glucose deprivation. Biochim Biophys Acta. 2012;1823(12):2099–2108.
- Ruderman NB, Carling D, Prentki M, et al. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123(7):2764–2772.
- Park SJ, Gavrilova O, Brown AL, et al. DNA-PK Promotes the Mitochondrial, Metabolic, and Physical Decline that Occurs During Aging. Cell Metab. 2017;25(5):1135–46.e7.
- Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene. 1999;18(53):7644–7655.
- Levine AJ, Hu W, The FZ. P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13(6):1027–1036.
- Riley T, Sontag E, Chen P, et al. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–412.
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310.
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64(7):2627–2633.
- Bensaad K, Tsuruta A, Selak MA, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120.
- Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217(7):2291–2298.
- Wu G, Fang YZ, Yang S, et al. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489–492.
- Franklin DA, He Y, Leslie PL, et al. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci Rep. 2016;6:38067.
- Jiang P, Du W, Wang X, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13(3):310–316.
- Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–1653.
- Papadopoulou LC, Sue CM, Davidson MM, et al. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23(3):333–337.
- Wang S, Peng Z, Yang L, et al. KRAB-type zinc-finger proteins PITA and PISA specifically regulate p53-dependent glycolysis and mitochondrial respiration. Cell Res. 2018;28(5):572–592.
- Suzuki S, Tanaka T, Poyurovsky MV, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A. 2010;107(16):7461–7466.
- Maddocks OD, Berkers CR, Mason SM, et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493(7433):542–546.
- Tajan M, Hock AK, Blagih J, et al. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab. 2018;28(5):721–36.e6.
- Kim J, Yu L, Chen W, et al. Wild-Type p53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell. 2019;35(2):191–203.e8.
- Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5(4):a012583–a.
- Sultana R, Abdel-Fatah T, Abbotts R, et al. Targeting XRCC1 Deficiency in Breast Cancer for Personalized Therapy. Cancer Res. 2013;73(5):1621.
- Markkanen E, Fischer R, Ledentcova M, et al. Cells deficient in base-excision repair reveal cancer hallmarks originating from adjustments to genetic instability. Nucleic Acids Res. 2015;43(7):3667–3679.
- Rashid S, Freitas MO, Cucchi D, et al. MLH1 deficiency leads to deregulated mitochondrial metabolism. Cell Death Dis. 2019;10(11):795.
- Murata MM, Kong X, Moncada E, et al. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol Biol Cell. 2019;30(20):2584–2597.
- Bai P, Cantó C, Oudart H, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13(4):461–468.
- Yu J, Auwerx J. The role of sirtuins in the control of metabolic homeostasis. Ann N Y Acad Sci. 2009;1173(1):E10–E9.
- Ke Y, Wang C, Zhang J, et al. The Role of PARPs in Inflammation-and Metabolic-Related Diseases: molecular Mechanisms and Beyond. Cells. 2019;8(9):1047.
- Bhute VJ, Palecek SP. Metabolic responses induced by DNA damage and poly (ADP-ribose) polymerase (PARP) inhibition in MCF-7 cells. Metabolomics. 2015;11(6):1779–1791.
- Masson M, Niedergang C, Schreiber V, et al. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18(6):3563–3571.
- Horton JK, Stefanick DF, Gassman NR, et al. Preventing oxidation of cellular XRCC1 affects PARP-mediated DNA damage responses. DNA Repair (Amst). 2013;12(9):774–785.
- Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst). 2015;36:13–18.
- Milanese C, Bombardieri CR, Sepe S, et al. DNA damage and transcription stress cause ATP-mediated redesign of metabolism and potentiation of anti-oxidant buffering. Nat Commun. 2019;10(1):4887.
- Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18(1):85–98.
- Brierley DJ, Martin SA.Oxidative Stress and the DNA Mismatch Repair Pathway. Antioxid Redox Signal. 2013;18(18):2420–2428.
- Russo MT, De Luca G, Degan P, et al. DNA repair strategies to combat the threat from 8-oxoguanine. Mutat Res. 2007;614(1):69–76.
- Mishra M, Kowluru RA. Retinal Mitochondrial DNA Mismatch Repair in the Development of Diabetic Retinopathy, and Its Continued Progression After Termination of Hyperglycemia. Invest Ophthalmol Vis Sci. 2014;55(10):6960–6967.
- Mason PA, Matheson EC, Hall AG, et al. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003;31(3):1052–1058.
- Fontana GA, Gahlon HL. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020;48(20):11244–11258.
- Martin SA, McCabe N, Mullarkey M, et al. DNA Polymerases as Potential Therapeutic Targets for Cancers Deficient in the DNA Mismatch Repair Proteins MSH2 or MLH1. Cancer Cell. 2010;17(3):235–248.
- Martin SA, McCarthy A, Barber LJ, et al. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol Med. 2009;1(6–7):323–337.
- Hewish M, Martin SA, Elliott R, et al. Cytosine-based nucleoside analogs are selectively lethal to DNA mismatch repair-deficient tumour cells by enhancing levels of intracellular oxidative stress. Br J Cancer. 2013;108(4):983–992.
- Guillotin D, Austin P, Begum R, et al. Drug-Repositioning Screens Identify Triamterene as a Selective Drug for the Treatment of DNA Mismatch Repair Deficient Cells. Clin Cancer Res. 2017;23(11):2880–2890.
- Martin SA, Hewish M, Sims D, et al. Parallel High-Throughput RNA Interference Screens Identify PINK1 as a Potential Therapeutic Target for the Treatment of DNA Mismatch Repair–Deficient Cancers. Cancer Res. 2011;71(5):1836–1848.
- Mootha VK, Bunkenborg J, Olsen JV, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115(5):629–640.
- Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71.
- Couch FJ, Weber BL. Mutations and polymorphisms in the familial early-onset breast cancer (BRCA1) gene. Breast Cancer Information Core. Hum Mutat. 1996;8(1):8–18.
- Starita LM, Parvin JD. The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol. 2003;15(3):345–350.
- Magnard C, Bachelier R, Vincent A, et al. BRCA1 interacts with acetyl-CoA carboxylase through its tandem of BRCT domains. Oncogene. 2002;21(44):6729–6739.
- Moreau K, Dizin E, Ray H, et al. BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. J Biol Chem. 2006;281(6):3172–3181.
- Privat M, Radosevic-Robin N, Aubel C, et al. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PLoS One. 2014;9(7):e102438.
- Concolino A, Olivo E, Tammè L, et al. Proteomics Analysis to Assess the Role of Mitochondria in BRCA1-Mediated Breast Tumorigenesis. Proteomes. 2018;6:2.
- Kanakkanthara A, Kurmi K, Ekstrom TL, et al. BRCA1 Deficiency Upregulates NNMT, Which Reprograms Metabolism and Sensitizes Ovarian Cancer Cells to Mitochondrial Metabolic Targeting Agents. Cancer Res. 2019;79(23):5920–5929.
- Singh KK, Shukla PC, Yanagawa B, et al. Regulating cardiac energy metabolism and bioenergetics by targeting the DNA damage repair protein BRCA1. J Thorac Cardiovasc Surg. 2013;146(3):702–709.
- Kc J, Ek G, Norrbom J, et al. BRCA1 is a novel regulator of metabolic function in skeletal muscle. J Lipid Res. 2014;55(4):668–680.
- Kc J, Tarpey MD, Ap V, et al. Induced Cre-mediated knockdown of Brca1 in skeletal muscle reduces mitochondrial respiration and prevents glucose intolerance in adult mice on a high-fat diet. Faseb J. 2018;32(6):3070–3084.
- Kang HJ, Kim HJ, Rih JK, et al. BRCA1 plays a role in the hypoxic response by regulating HIF-1alpha stability and by modulating vascular endothelial growth factor expression. J Biol Chem. 2006;281(19):13047–13056.
- Xiang T, Ohashi A, Huang Y, et al. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008;68(24):10040–10044.
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94(2):514–519.
- Druzhyna NM, Wilson GL, LeDoux SP. Mitochondrial DNA repair in aging and disease. Mech Ageing Dev. 2008;129(7–8):383–390.
- Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698.
- Boesch P, Weber-Lotfi F, Ibrahim N, et al. DNA repair in organelles: pathways, organization, regulation, relevance in disease and aging. Biochim Biophys Acta. 2011;1813(1):186–200.
- Liu P, Demple B. DNA repair in mammalian mitochondria: much more than we thought? Environ Mol Mutagen. 2010;51(5):417–426.
- Yang JL, Weissman L, Bohr VA, et al. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair (Amst). 2008;7(7):1110–1120.
- Prakash A, Base Excision DS. Repair in the Mitochondria. J Cell Biochem. 2015;116(8):1490–1499.
- Sharma P, Sampath SH. Mitochondrial DNA Integrity: role in Health and Disease. Cells. 2019;8(2):2.
- Komakula SSB, Tumova J, Kumaraswamy D, et al. The DNA Repair Protein OGG1 Protects Against Obesity by Altering Mitochondrial Energetics in White Adipose Tissue. Sci Rep. 2018;8(1):14886. .
- Sykora P, Kanno S, Akbari M, et al. DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol Cell Biol. 2017;37(16):16. .
- Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol. 2012;13(10):659–671.
- Cassim S, Pouyssegur PJ. Tumor Microenvironment: a Metabolic Player that Shapes the Immune Response. Int J Mol Sci. 2019;21(1):1.
- Coller HA. Is Cancer a Metabolic Disease? Am J Pathol. 2014;184(1):4–17.
- Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, et al. Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011;10(9):1499–1501.
- Wheaton WW, Weinberg SE, Hamanaka RB, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242.
- Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24(7):1036–1046. .
- Á L, Hyroššová P, Figueras A, et al. Tumors defective in homologous recombination rely on oxidative metabolism: relevance to treatments with PARP inhibitors. EMBO Mol Med. 2020;12(6):e11217.
- Jiang J, Chen Y, Dong T, et al. Polydatin inhibits hepatocellular carcinoma via the AKT/STAT3-FOXO1 signaling pathway. Oncol Lett. 2019;17(5):4505.
- Mele L, La Noce M, Paino F, et al. Glucose-6-phosphate dehydrogenase blockade potentiates tyrosine kinase inhibitor effect on breast cancer cells through autophagy perturbation. J Exp Clin Cancer Res. 2019;38(1):160.
- Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med. 2013;45(10):e45–e.
- Abdel-Wahab AF, Mahmoud W, Al-Harizy RM. Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol Res. 2019;150:104511.
- Hoxhaj G, Manning BD. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20(2):74–88.
- André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N Engl J Med. 2019;380(20):1929–1940.
- Markham A. . Idelalisib: first Global Approval. Drugs. 2014;74(14):1701–1707.
- Yang J, Nie J, Ma X, et al. PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26.
- Anderson NM, Mucka P, Kern JG, et al. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9(2):216–237.
- Chen M, Huang J. The expanded role of fatty acid metabolism in cancer: new aspects and targets. Precision Clin Med. 2019;2(3):183–191.
- Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122(1):4–22.
- Punekar S, Cho DC. Novel Therapeutics Affecting Metabolic Pathways. Am Soc Clin Oncol Educ Book.2019 (39):e79–e87.
- Li E-Q, Zhao W, Zhang C, et al. Synthesis and anti-cancer activity of ND-646 and its derivatives as acetyl-CoA carboxylase 1 inhibitors. Eur J Pharm Sci. 2019;137:105010.
- Svensson RU, Parker SJ, Eichner LJ, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22(10):1108–1119.
- Kim YI. Folate and colorectal cancer: an evidence-based critical review. Mol Nutr Food Res. 2007;51(3):267–292.
- Assaraf YG. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007;26(1):153–181.
- Krug LM, Azzoli CG, Kris MG, et al. 10-propargyl-10-deazaaminopterin: an antifolate with activity in patients with previously treated non-small cell lung cancer. Clin Cancer Res. 2003;9(6):2072–2078.
- Jackman AL, Taylor GA, Gibson W, et al. ICI D1694, a quinazoline antifolate thymidylate synthase inhibitor that is a potent inhibitor of L1210 tumor cell growth in vitro and in vivo: a new agent for clinical study. Cancer Res. 1991;51(20):5579–5586.
- Shih C, Chen VJ, Gossett LS, et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997;57(6):1116–1123.
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–338.
- Plunkett W, Huang P, Xu YZ, et al. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22(4 Suppl 11):3–10.
- Chen L, Pankiewicz KW. Recent development of IMP dehydrogenase inhibitors for the treatment of cancer. Curr Opin Drug Discovery Dev. 2007;10(4):403–412.
- Zou J, Han Z, Zhou L, et al. Elevated expression of IMPDH2 is associated with progression of kidney and bladder cancer. Med Oncol. 2015;32(1):373.
- Fellenberg J, Kunz P, Sähr H, et al. Overexpression of inosine 5ʹ-monophosphate dehydrogenase type II mediates chemoresistance to human osteosarcoma cells. PLoS One. 2010;5(8):e12179.
- Malek K, Boosalis MS, Waraska K, et al. Effects of the IMP-dehydrogenase inhibitor, Tiazofurin, in bcr-abl positive acute myelogenous leukemia. Part I. In vivo studies. Leuk Res. 2004;28(11):1125–1136.
- Golub D, Iyengar N, Dogra S, et al. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front Oncol. 2019;9:417.
- Heckman-Stoddard BM, DeCensi A, Sahasrabuddhe VV, et al. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia. 2017;60(9):1639–1647.