
ABSTRACT
Cyclin-dependent kinase (CDK) 1 complexed with cyclin B is a driver of mitosis, while CDK2 drives S phase entry and replicon initiation. CDK2 activity increases as cells progress through S phase, and its cyclin partner switches from cyclin E to cyclin A. Activation of CDK2 requires dephosphorylation of tyrosine-15 by CDC25A. DNA damage activates the checkpoint protein CHK1, which phosphorylates and degrades CDC25A to prevent activation of CDK2 and protect from cell cycle progression before damage is repaired. CHK1 inhibitors were developed to circumvent this arrest and enhance the efficacy of many cancer chemotherapeutic agents. CHK1 inhibition results in the accumulation of CDC25A and activation of CDK2. We demonstrate that inhibition of CDK2 or suppression of cyclin A also results in accumulation of CDC25A suggesting a feedback loop that prevents over activation of this pathway. The feedback inhibition of CDC25A targets phosphorylation of S88-CDC25A, which resides within a CDK consensus sequence. In contrast, it appears that CDK complexes with cyclin B (and possibly cyclin E) stabilize CDC25A in a feed-forward activation loop. While CDK2/cyclin A would normally be active at late S/G2, we propose that this feedback inhibitory loop prevents over activation of CDK2 in early S phase, while still leaving CDK2/cyclin E to catalyze replicon initiation. One importance of this observation is that a subset of cancer cell lines are very sensitive to CHK1 inhibition, which is mediated by CDK2/cyclin A activity in S phase cells. Hence, dysregulation of this feedback loop might facilitate sensitivity of the cells.
Introduction
Cell cycle progression is regulated by cyclin-dependent kinases (CDK) in complex with a cyclin. CDK2 in complex with either cyclin E or cyclin A is required for initiation and progression through S phase, while CDK1 in complex with cyclin A or cyclin B is required for mitosis [Citation1]. Importantly, CDK2 activity increases as cells progress through S and G2 phase [Citation2]. Several recent papers have demonstrated the critical role that this increasing CDK activity has in cell cycle progression. In a yeast model engineered to contain a single cyclin/CDK protein, the phosphorylation of different CDK substrates varied dramatically with the level of CDK activity: low-level CDK activity drove early cell cycle phosphorylation (i.e. S phase), whereas high-level CDK activity drove late cell cycle events (i.e. G2/M) [Citation3,Citation4]. These differences could also be discriminated using a CDK inhibitor whereby low concentrations were sufficient to reduce CDK activity below the threshold required for late phosphorylation events, whereas much higher concentrations were required to suppress CDK activity below the threshold for early S phase substrates. We recently reported a similar phenomenon in human cells where low-level CDK2 activity is required for replication origin firing and S phase progression, while high activity levels can lead to cytotoxicity if it occurs prematurely in S phase [Citation5,Citation6].
Our interest in CDK1/2 regulation arose from research on the DNA damage response pathway. DNA double-strand breaks or single-strand regions in DNA activate the ATM and ATR kinases which in turn activate CHK1 to prevent S and G2 progression, and provide time for repair and recovery. Inhibitors of CHK1 (CHK1i) were developed because they overcome cell cycle arrest and force progression through S and G2 even when DNA is damaged, thereby enhancing cell killing [Citation7,Citation8]. This strategy is currently under clinical investigation in cancer patients. We also reported that a subset of cell lines is very sensitive to CHK1i as a single agent because it rapidly induces DNA breaks in early S phase [Citation9]. The DNA breaks, and the associated cytotoxicity, resulting from single-agent CHK1i was prevented by incubation with low concentrations of CVT-313 that are selective for CDK2 inhibition [Citation10]. Given the established role for CDK2 in S phase progression, we were initially surprised that CVT-313 failed to prevent CHK1i-mediated abrogation of DNA damage-induced S phase arrest [Citation6]. However, we discovered that a 20-fold higher concentration of CVT-313 did prevent CHK1i-mediated S phase progression [Citation6,Citation8]. These results are consistent with different CDK2 activity thresholds being required for S phase progression versus single-agent CHK1i sensitivity as discussed above. We refer to these different states as CDK2-low and CDK2-high in this paper and propose that CDK2-low involves complex with cyclin E while CDK2-high involves complex with cyclin A.
Cyclin-dependent kinases are activated by a family of CDC25 phosphatases, which, in turn are inhibited by CHK1 [Citation11]. The inactive form of CDK2 is phosphorylated on tyrosine 15, while its dephosphorylation and activation is mediated by CDC25A. CDC25A is primarily regulated by its protein level through phosphorylation by CHK1, in concert with additional kinases [Citation12]. Both constitutive and DNA damage-activated CHK1 degrade CDC25A and thereby prevent CDK2 activation and cell cycle progression [Citation13,Citation14]. We previously reported that CHK1i induces CDC25A levels preferentially in cells sensitive to CHK1i as a single agent, suggesting that only the sensitive cells activate CDK2 in S phase in response to CHK1i [Citation9]. However, we report here that inhibition of CDK2-high also enhances the level of CDC25A. Similarly, the suppression of cyclin A elevates CDC25A. These results suggest that CDK2/cyclin A activity regulates a negative feedback loop that prevents inappropriate activation of CDK2.
Materials and methods
Cell culture
Cell lines were obtained from the Developmental Therapeutics Program of the National Cancer Institute as part of the NCI60 cell-line panel. AsPC-1 and U2OS cells were obtained from the American Type Culture Collection. Cells were expanded and stored at low passage number and experimental cultures replaced approximately every 3 months. Cells were maintained in RPMI1640 media (Corning/Mediatech) plus 10% fetal bovine serum (Hyclone), and 1% antibiotic/antimycotic (Gibco). Cell lines were confirmed negative for mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza). SMART pool siRNA for cyclin E (CCNE1) and cyclin A2 (CCNA2) were obtained from Dharmacon.
Plasmids containing mutant and wildtype CDC25A constructs were kindly provided by Dr Helen Piwnica-Worms, MD Anderson Cancer Institute [Citation12]. Each mutation was confirmed by DNA sequencing. Plasmids were transfected into cells using lipofectamine (4 µl/2 ml in 6 well plates). The cells were selected initially with 1 mg/ml G418, then subsequently maintained in 0.5 mg/ml G418.
Chemicals
MK-8776 and AZD1775 (previously MK-1775) were provided by Merck. CVT-313 and Ro3306 were purchased from Sigma. All solutions were stored at 10 mM in DMSO. SN38 was provided by Pfizer, stored in DMSO at 100 µM, and used at 25 nM (10 ng/ml).
Cell synchronization and analysis
SW620 cells were synchronized in six well plates using a double thymidine block using a schedule of 16 h 2 mM thymidine, 9 h release in media with 24 µM deoxycytidine, 16 h thymidine, and finally release into fresh media with deoxycytidine. After an additional 6 h, when cells were in G2, 100 ng/ml nocodazole was added to prevent exit from mitosis. Cells were trypsinized, fixed in 70% ethanol overnight, rehydrated and stained with Alexa 647-conjugated anti-phospho-histone H3 (ser10) (D2C8; Cell Signaling Technology) and propidium iodide. Cells were analyzed on a Becton Dickinson Gallios flow cytometer. Data was analyzed using FlowLogic.
Western blotting
Cells were rinsed in PBS, lysed in Laemmli lysis buffer, and boiled for 5 min. Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Western blotting was performed with the following primary antibodies: Cell Signaling Technologies: p-Y15-CDK1/2 (9111S and 4539S), cyclin A2 (4656S), PARP (9532S), WEE1 (13084S); EMD-Millipore: CDK2 (05–596); Thermo Fisher Scientific: CDC25A (ms-638); GeneTex: Cyclin A2 (GTX103042); Santa Cruz Biotechnology: vinculin (sc-073541), CDC25A (sc-7389), cyclin B (sc-245). Sigma: actin (A3854). Actin, vinculin and PARP were used as loading controls.
In initial studies, membranes were developed using chemiluminescence and X-ray film (). Subsequent experiments used the following fluorescent secondary antibodies from Cell Signaling Technologies: mouse IgG-DyLight 800 (5257), rabbit IgG – DyLight 800 (5151), mouse IgG-DyLight 680 (5470). Fluorescent images were obtained on a Licor Odyssey scanner and processed using Image Studio Lite. Quantitation of pCDK1/2 bands was undertaken using GelBandFitter that quantifies closely spaced bands [Citation15].
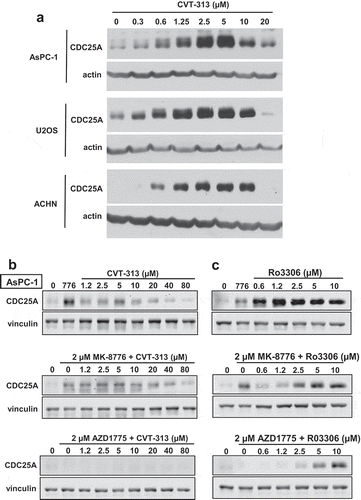
Figure 1. Changes in the level of CDC25A upon activation or inhibition of CDK1/2. (a). AsPC-1, U2OS and ACHN cells were incubated with 0–20 µM CVT-313 for 6 h, and cell lysates analyzed by western blotting for CDC25A using chemiluminescent detection. (b). AsPC-1 cells were incubated with 0–80 µM CVT-313 for 6 h alone or in combination with 2 µM MK-8776 or 2 µM AZDZ1775 as indicated. Cell lysates were analyzed by western blotting for CDC25A, with vinculin as a loading control. Fluorescent secondary antibodies were used and fluorescent images captured. (c) Cells were incubated as in B but with the addition of 0–10 µM Ro3306 rather than CVT-313
Results
CDK2 elicits feedback inhibition of CDC25A
The inhibitory phospho-tyrosine on CDK2 is removed by CDC25A, which in turn is repressed by CHK1. DNA damage activates CHK1, which phosphorylates CDC25A leading to its degradation, and thereby preventing activation of CDK2 and cell cycle progression. Consequently, incubation with CHK1i reactivates CDC25A and CDK2 inducing S and G2 phase progression even when the DNA is damaged. As we investigated the relationship between CDC25A and CDK2, we discovered that the CDK2 inhibitor CVT-313 (CDK2i) caused marked accumulation of CDC25A even in the absence of CHK1i, and in multiple cell lines ()). However, this effect was very dependent on the concentration of CVT-313, with low concentrations causing accumulation of CDC25A, whereas concentrations above 10 µM had little if any effect. We hypothesized that CDK2 acts through a feedback loop to limit the accumulation of CDC25A, but the decrease in CDC25A at higher concentrations of CVT-313 remained unresolved.
Given our more recent recognition of the importance of CDK2 activity thresholds in substrate phosphorylation [Citation5,Citation6,Citation8], we have now revisited this observation. We note that the level of induction of CDC25A by low concentrations of CVT-313 is comparable to that induced by incubation with the CHK1i MK-8776 () top). When combined with MK-8776, low concentrations of CVT-313 induced little further increase in CDC25A, but high concentrations of CVT-313 again prevented accumulation of CDC25A () middle). We surmise that high-level activity of CDK2 (CDK2-high; inhibited by low CVT-313) is responsible for the feedback loop that limits the accumulation of CDC25A, and thereby prevents over-activation of CDK2.
At the higher concentrations of CVT-313 that prevent accumulation of CDC25A, CVT-313 is no longer selective for CDK2 but also inhibits CDK1 [Citation10]. This is consistent with its ability to inhibit mitosis only at these higher concentrations [Citation5,Citation6]. Consequently, the suppression of CDC25A at high concentrations of CVT-313 could be due to inhibition of either CDK1 or CDK2-low (or both).
CHK1i preferentially activates CDK2 rather than CDK1, at least in S phase cells [Citation5,Citation6]. We confirmed this conclusion here ()). Antibodies cannot discriminate the phospho-tyrosine on CDK1 from CDK2 as it is located in a conserved amino acid sequence [Citation16]. However, we have noted that the two phosphoproteins can be resolved on western blots, particularly when using fluorescent secondary antibodies that provide greater dynamic range than chemiluminescence methods [Citation5,Citation6]. Immunoprecipitation has confirmed that the upper band is pCDK1 while the lower band is pCDK2. Phospho-CDK2 may often be overlooked as it is present at much lower levels than phospho-CDK1. In ), phospho-CDK2 was present at 17% of the level of phospho-CDK1, and only CDK2 was dephosphorylated upon incubation with MK-8776.
Figure 2. CDK2 is selectively activated by MK-8776, but both CDK1 and CDK2 are activated by AZD1775. (a) AsPC-1 cells were incubated with 0–10 µM AZD1775 or MK8776 for 6 h, and cell lysates were analyzed by western blotting. Fluorescent secondary antibodies were used. The first blot was probed for pCDK1/2; the top band of the doublet is pY-CDK1 and the lower, much weaker band is pY-CDK2. Fluorescent signals were quantified for each band as shown in the graphs. (b) MDA-MB-231 cells were arrested in S phase by 24 h incubation with SN38, then 1 µM MK-8776 was added to abrogate arrest and drive cells into G2 over the following 6 h (30 h time point). Ro3306 (0–10 µM) was added concurrent with MK-8776 and cell cycle phase determined by flow cytometry
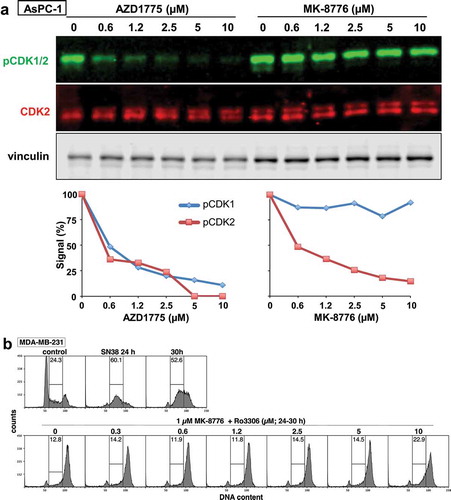
To investigate the potential involvement of CDK1 on regulation of CDC25A, we incubated cells with the WEE1 inhibitor AZD1775 which rapidly dephosphorylated both CDK1 and CDK2 ()). AZD1775 alone suppressed basal CDC25A and prevented the CVT-313-mediated accumulation of CDC25A (), bottom). This suggests that CDK1 may complement CDK2 in degrading CDC25A. This issue was investigated further by incubating cells with Ro3306 which is commonly used as a selective inhibitor of CDK1 [Citation17]. The addition of Ro3306 alone caused even greater accumulation of CDC25A than CVT-313 ()). However, these concentrations of Ro3306 are also reported to inhibit CDK2 [Citation16], although we now realize this inhibition is only of CDK2-high activity, so this observation is consistent with both CDK1 and CDK2-high contributing to the degradation of CDC25A. The only remaining CDK2 activity in Ro3306-treated cells should be CDK2-low, and this was confirmed by showing that these concentrations of Ro3306 did not prevent CHK1i-mediated S phase progression of damaged cells ()). This contrasts with high concentrations of CVT-313 that do prevent CHK1i-mediated S phase progression [Citation6]. Importantly, Ro3306 rescued CDC25A from degradation induced by AZD1775 consistent with CDK1 and CDK2-high being involved (), bottom). As discussed below, the degradation of CDC25A is also mediated by cyclin A which binds both kinases.
The biphasic response of CDC25A to CVT-313 requires further explanation, particularly the decrease at the high concentrations. At these high concentrations, there should be no CDK1 or CDK2 activity. In contrast, when cells are incubated with 10 µM Ro3306, CDK2-low is still active. This suggests that the decrease in CDC25A at high concentrations of CVT-313 is the result of inhibition of CDK2-low, thereby implying that active CDK2-low stabilizes CDC25A. The CDK2-low activity is required for replicon initiation and occurs early in S phase (likely associated with cyclin E), and is consistent with the report of a feed-forward pathway that activates cyclin E/CDK2 to drive cells into S phase [Citation18].
Degradation of CDC25A depends on cyclin A
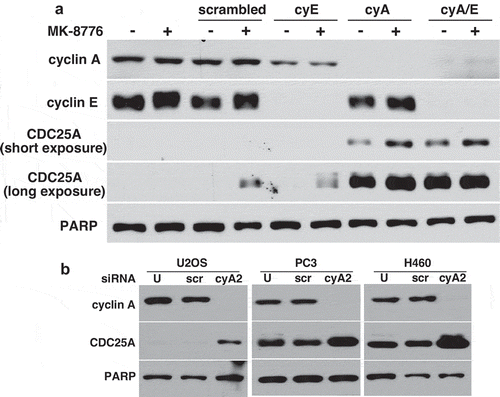
While the activation of cyclin E/CDK2 provides a feed-forward loop to drive G1 to S phase progression, the effect on CDC25A levels was not reported [Citation18]. Similarly, cyclin B/CDK1 provides a feed-forward loop to accelerate mitosis, and this is associated with stabilization of CDC25A [Citation19,Citation20]. Here, we determined the potential role of cyclins A and E in the stability of CDC25A as both form complexes with CDK2. Suppression of cyclin A2 by siRNA resulted in dramatic accumulation of CDC25A (the cells do not express cyclin A1) while suppression of cyclin E or a control siRNA had no effect ()). Concurrent suppression of cyclins A and E also resulted in dramatic accumulation of CDC25A. As cyclin A partners with both CDK2 and CDK1, it appears that cyclin A is the critical mediator of CDC25A suppression. The ability of cyclin A2 siRNA to enhance CDC25A was confirmed in three additional cell lines ()).
Figure 3. Suppression of cyclin A, but not cyclin E, results in accumulation of CDC25A. (a) AsPC-1 cells were transfected with scrambled siRNA, or siRNA targeting cyclin E, cyclin A2 or both. Cell lysates were analyzed by western blotting. (b) U2OS, PC3 and H460 cells were transfected with scrambled siRNA or siRNA targeting cyclin A2, and cell lysates analyzed by western blotting. U = untransfected. PARP was used as a loading control
Stabilization of CDC25A occurs in mitosis
We also addressed the potential stabilization of CDC25A in mitosis that is associated with CDK1/cyclin B [Citation19,Citation20]. To perform these experiments, we selected a cell line, SW620, that readily undergoes arrest and synchronized release of all the cells after a double thymidine block, whereas we find that synchronization of most cell lines results in incomplete release. First, we confirmed that SW620 cells also accumulate CDC25A at low concentrations of CVT-313 and repress it at higher concentrations ()). Upon release from the double thymidine block, SW620 cells progress rapidly through S phase, and then through mitosis after about 8 h. However, as mitosis is rapid, there was still a limited number of cells in mitosis at any specific time. To induce accumulation of cells in mitosis, we added nocodazole at 6 h, a time when most of the cells had entered G2 but had not yet reached M. By 8 h, 24% of the cells exhibited pHH3, a marker of mitosis, and this increased to 75% by 11 h ()). As cells progressed through S phase, there was a clear increase in pCDK1 (the upper band), though little if any change in pCDK2 (the lower band) ()). The onset of mitosis was associated with a rapid dephosphorylation of pCDK1 as expected. This dephosphorylation can also be seen in the blot for total CDK1 as the upper band is the phosphorylated form.
Figure 4. Cell cycle-dependent regulation of CDK1/2, cyclins A and B, CDC25A and WEE1. (a) SW620 cells were incubated with 0–80 µM CVT-313 for 6 h and analyzed by western blotting for the level of CDC25A. (b) SW620 cells were synchronized by a double thymidine block, then released for 0–12 h. Nocodazole was added at 6 h to capture cells in mitosis. Cells were stained with propidium iodide for DNA and anti-pHH3 to determine the % of cells in mitosis. Cells were analyzed by flow cytometry. (c) SW620 cells from the synchrony in B were analyzed by western blotting for the indicated antigens, followed by fluorescent secondary antibodies. Multicolor images were generated; CDK2 migrates at the lower band of the doublet for pCDK1/2. * = nonspecific band. (d) AsPC-1 cells were incubated with MK-8776 for 0–6 h and probed for expression of WEE1
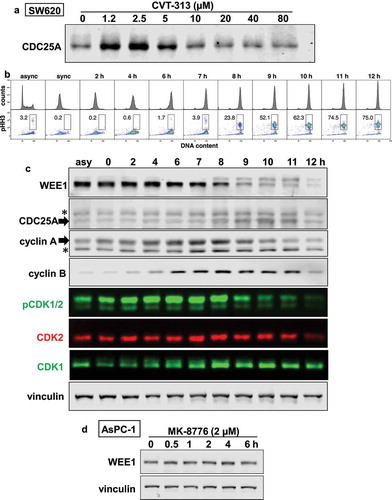
Cyclin A was expressed in the synchronized cells and increased slightly through S phase, but began to decrease around 9 h as cells entered mitosis ()). In contrast, very little cyclin B was present until cells reached G2 and this remained high in M. As expected, CDC25A also accumulated once the cells reached mitosis.
We also investigated the expression of WEE1 and observed a marked upward mobility shift as cells entered mitosis consistent with its phosphorylation. There are contradictory reports on the regulation of WEE1 at mitosis, albeit both mechanisms rely on CDK-mediated inhibition. Phosphorylation at T239-WEE1 by CDK2/cyclin A results in nuclear export of WEE1 and hence prevents its ability to phosphorylate CDK1, facilitating mitosis [Citation21]. Alternately, CDK-mediated phosphorylation on S123-WEE1 at the onset of mitosis results in ubiquitination and degradation of WEE1 (neither the CDK nor cyclin was defined) [Citation22]. The electrophoretic band shift we observed as cells enter mitosis is more consistent with the first pathway, but as both cyclin A and B are expressed at that time, does not resolve which cyclin is involved. As the band shift does not occur while cells are progressing through S phase, it suggests it is not dependent on CDK2, at least when complexed with cyclin E ()).
To further assess whether CDK2/cyclin A may contribute to the phosphorylation of WEE1, we used AsPC-1 cells that selectively activate this pathway upon inhibition of CHK1 as shown in . No band shift of WEE1 was observed ()) supporting the conclusion that the band shift is not mediated by CDK2/cyclin A, but is consistent with phosphorylation by CDK1/cyclin B. Overall, there is a clear correlation between phosphorylation of WEE1, increased CDC25A and entry into mitosis, which appears consistent with a role for CDK1/cyclin B in stabilizing CDC25A.
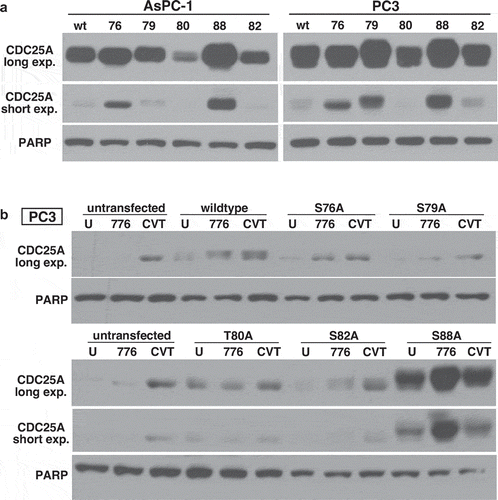
Degradation of CDC25A is induced by phosphorylation on S88
AsPC-1 and PC3 cells were transfected with plasmids encoding GST-tagged forms of CDC25A that were mutated from serine to alanine at potential phosphorylation sites. Initial transient transfections (32 h) demonstrated very little expression of most of the mutant constructs, a relatively low expression of the S79A mutant, in PC3 cells, but a very high level of the S88A mutant ()). In an attempt to obtain cells with stable expression of each mutant, we selected the cells on G418. Clones isolated from AsPC-1 cells did not express any of the various CDC25A constructs, but several PC3 transfectants did exhibit the introduced gene ()). The GST-tagged protein exhibited a slightly slower electrophoretic mobility which was evident in cells expressing the wildtype construct, albeit at a level similar to the endogenous protein. A low level of the transfected S82A mutant was also observed, although only after addition of MK-8776. The S88A construct was again expressed at much higher levels, and was further increased upon addition of MK-8776. In contrast, this construct did not change upon addition of 2.5 µM CVT-313, which is consistent with S88 being a consensus site for CDK2 phosphorylation. Furthermore, none of the other phosphorylation sites in this degron possess the CDK consensus sequence. It seems unlikely that the S88A mutant is an active phosphatase because that would lead to constitutively active CDK2 that cells are unlikely to tolerate. From these observations, we conclude that CDK2/cyclin A-mediated phosphorylation of S88 is responsible for degradation of CDC25A.
Figure 5. Serine 88 of CDC25A is required for its degradation. (a) AsPC-1 and PC3 cells were transiently transfected with wildtype CDC25A or derivatives mutated to alanine at each of the indicated amino acids. (b) Stable transfectants for each CDC25A mutant were obtained in PC3 cells, and the cells were incubated with either 2 µM MK-8776 or 2.5 µM CVT-313 for 6 h. U - = untreated. PARP was used as a loading control
Discussion
It is recognized that phosphorylation of CDC25A can lead to both its degradation and its stabilization, and it now appears that both phenomena can result from CDK1/2 phosphorylation on different serines. This is in addition to the previously reported phosphorylation on S76-CDC25A by CHK1 that initiates further phosphorylation on S82 in a “DSG” degron. There was one early report suggesting that CDK2 can mediate degradation of CDC25A [Citation23], albeit that has been overlooked in subsequent studies. The current report appears to be the first identification of the critical role of CDK2 in phosphorylating S88-CDC25A leading to its degradation. This phosphorylation appears to act as a means to limit the over-activation of CDK2 while cells are in S phase. Inappropriate regulation of CDK2 activity in S phase underlies the reason why cell lines can be very sensitive to inhibition of CHK1 [Citation8,Citation9].
These observations pose the question as to how CDK1 and CDK2 discriminate which residue of CDC25A to phosphorylate, and we conclude it depends on the different cyclin partners. Our results suggest that CDK2, when complexed with cyclin A, leads to degradation of CDC25A through phosphorylation on S88. We also propose that cyclin A in complex with CDK1 can phosphorylate S88 and degrade CDC25A. In contrast, cells in mitosis activate cyclin B/CDK1 and this stabilizes CDC25A through phosphorylation on S18 and S116 [Citation19]. During mitosis, the cyclin A/CDK1 complex decreases, so CDC25A degradation does not occur. This can be contrasted to a WEE1 inhibitor that activates all CDK1/2-cyclin complexes throughout the cell cycle resulting in CDC25A degradation. Hence, we surmise that phosphorylation on S88 is dominant over phosphorylation on S18 and S116 resulting in degradation, and even if all three phosphorylations occur simultaneously, CDC25A will be degraded.
Multiple phosphorylation events are reported around the DSG degron at S82-CDC25A. CHK1 or GSK3 mediate phosphorylation on S76, which primes CDC25A for phosphorylation on S79 and S82, the latter site in the DSG degron required for binding of the ubiquitin ligase, β-TrCP. The phosphorylation on S82 has been variously reported to involve CK1 or NEK11 [Citation12,Citation24]. Decrease of either CK1 or NEK11 reduced degradation of CDC25A. NEK11 has also been reported to phosphorylate S88-CDC25A [Citation24]. Considering that S88 is in a consensus sequence for CDK2 phosphorylation it seems more likely that CDK2 is the predominant kinase involved at that site, and perhaps this primes for phosphorylation of the other sites in the degron.
This line of investigation began with the observation that the CDK inhibitor CVT-313 elicits a biphasic effect, increasing CDC25A at low concentrations but decreasing it at high concentrations. The decrease in CDC25A at high concentrations of CVT-313 appears to occur when CDK2-low is inhibited, which is likely the form that occurs when complexed with cyclin E. This is consistent with the report of a feed-forward mechanism whereby CDK2 stabilizes CDC25A at G1/S phase [Citation18]. Consequently, we propose that, in early S phase, low levels of CDK2 activity stabilize CDC25A to continue activating CDK2 and ensure replicon initiation. As CDK2 activity rises, it complexes with cyclin A and phosphorylates alternate sites on CDC25A resulting in its degradation. Finally, when cells enter mitosis, cyclin B/CDK1 can also stabilize CDC25A, although the consequence of this remains ambivalent as there are reports that this both activates CDC25A and alternately inhibits it through binding to 14-3-3 and sequestration in the cytosol [Citation19,Citation20,Citation25].
The ability to prevent activation of CDK2/cyclin A in early S phase is of particular importance when cells are incubated with CHK1i. This feed-back inhibition protects cells from over activating CDK2/cyclin A which would otherwise elicit cytotoxicity in CHK1i sensitive cells. One interesting observation from our prior study was the large range of levels of CDC25A across a panel of cell lines, and this was independent of their sensitivity to CHK1i [Citation9]. Sensitivity to CHK1i did appear to correlate with the ability to induce CDC25A levels. The failure of CHK1i to induce CDC25A in resistant cells might therefore be the result of a more rigorous control of CDC25A by the CDK2/cyclin A feedback loop. Consequently, our observations raise the possibility that the feedback loop might be dysfunctional in sensitive cells. For example, increased dephosphorylation, decreased ubiquitination, or increased deubiquitination of CDC25A could all lead to over-activation of CDK2. These possibilities require further investigation as they might explain the variable sensitivity of cells, and provide an avenue to predict which tumors might be most response to CHK1i therapy.
Acknowledgments
This work was supported by grant CA117874 from the National Cancer Institute, and a Cancer Center Support Grant to the Norris Cotton Cancer Center (CA23108), The NCCC Immunology and Flow Cytometry Shared Resource facilitated the acquisition of flow cytometry data.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature Rev Cancer. 2009;9:153–166.
- Spencer SL, Cappell SD, Tsai FC, et al. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell. 2013;155:369–383.
- Coudreuse D, Nurse P. Driving the cell cycle with a minimal CDK control network. Nature. 2010;468:1074–1079.
- Swaffer MP, Jones AW, Flynn HR, et al. CDK substrate phosphorylation and ordering the cell cycle. Cell. 2016;167:1750–1761.
- Warren N, Eastman A. Inhibition of checkpoint kinase 1 following gemcitabine-mediated S phase arrest results in CDC7- and CDK2-dependent replication catastrophe. J Biol Chem. 2019;294:1763–1778.
- Warren NJH, Donahue K, Eastman A. Differential sensitivity to CDK2 inhibition discriminates the molecular mechanisms of CHK1 inhibitors as monotherapy or in combination with the topoisomerase I inhibitor SN38. ACS Pharmacol Transl Sci. 2019;2:168–182.
- Thompson R, Eastman A. The cancer chemotherapeutic potential of Chk1 inhibitors: how mechanistic studies impact clinical trial design. Br J Clin Pharmacol. 2013;76:358–369.
- Warren NJH, Eastman A. Comparison of the different mechanisms of cytotoxicity induced by checkpoint kinase 1 inhibitors when used as single agents or in combination with DNA damage. Oncogene. 2020;39:1389–1401.
- Sakurikar N, Thompson R, Montano R, et al. A subset of cancer cell lines is acutely sensitive to the Chk1 inhibitor MK8776 as monotherapy due to CDK2 activation in S phase. Oncotarget. 2016;7:1380–1394.
- Brooks EE, Gray NS, Joly A, et al. CVT-313, a specific and potent inhibitor of CDK2 that prevents neointimal proliferation. J Biol Chem. 1997;272:29207–29211.
- Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nature Rev Cancer. 2007;7:495–507.
- Honaker Y, Piwnica-Worms H. Casein kinase 1 functions as both penultimate and ultimate kinase in regulating Cdc25A destruction. Oncogene. 2010;29:3324–3334.
- Mailand N, Falck J, Lukas C, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429.
- Goto H, Natsume T, Kanemaki MT, et al. Chk1-mediated CDC25A degradation is a critical mechanism for normal cell cycle progression. J Cell Sci. 2019;132:jcs223123.
- Mitov MI, Greaser ML, Campbell KS. GelBandFitter - a computer program for analysis of closely spaced electrophoretic and immunoblotted bands. Electrophoresis. 2009;30:848–851.
- Sakurikar N, Eastman A. Critical reanalysis of the methods that discriminate CDK2 from CDK1. Cell Cycle. 2016;15:1184–1188.
- Vassilev LT, Tovar C, Chen S, et al. Selective small-molecule inhibitor reveals critical mitotic functions of CDK1. Proc Natl Acad Sci USA. 2006;103:10660–10665.
- Hoffmann I, Draetta G, Karsenti E. Activation of phosphatase activity of human cdc25A by a cdk2-cyclin E dependent phosphorylation at the G1/S transition. EMBO J. 1994;13:4302–4310.
- Mailand N, Podtelejnikov V, Groth A, et al. Regulation of G2/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002;21:5911–5920.
- Mazzolini L, Broban A, Froment C, et al. Phosphorylation of CDC25A on ser283 in late S/G2 by CDK/cyclin complexes accelerates mitotic entry. Cell Cycle. 2016;15:2742–2752.
- Li C, Andrake M, Dunbrack R, et al. A bifunctional regulatory element in human somatic Wee1 mediates cyclin A/Cdk2 binding and Crm1-dependent nuclear export. Mol Cell Biol. 2010;30:116–130.
- Watanabe N, Arai H, Iwaski JI, et al. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci USA. 2005;102:11663–11668.
- Ducruet AP, Lazo JS. Regulation of Cdc25A half-life in interphase by cyclin-dependent kinase 2 activity. J Biol Chem. 2003;278:31838–31842.
- Melixetian M, Klein DK, Sorensen CS, et al. NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint. Nature Cell Biol. 2009;11:1247–1253.
- Chen MS, Ryan CE, Piwnica-Worms H. Chk1 kinase negatively regulates mitotic function of Cdc25A through 14-3-3 binding. Mol Cell Biol. 2003;23:7488–7497.