
ABSTRACT
Melanoma is the deadliest form of skin cancer. While clinical developments have significantly improved patient prognosis, effective treatment is often obstructed by limited response rates, intrinsic or acquired resistance to therapy, and adverse events. Melanoma initiation and progression are associated with transcriptional reprogramming of melanocytes to a cell state that resembles the lineage from which the cells are specified during development, that is the neural crest. Convergence to a neural crest cell (NCC)-like state revealed the therapeutic potential of targeting developmental pathways for the treatment of melanoma. Neural crest cells have a unique sensitivity to metabolic dysregulation, especially nucleotide depletion. Mutations in the pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase (DHODH) particularly affect neural crest-derived tissues and cause Miller syndrome, a genetic disorder characterized by craniofacial malformations in patients. The developmental susceptibility of the neural crest to nucleotide deficiency is conserved in melanoma and provides a metabolic vulnerability that can be exploited for therapeutic purposes. We review the current knowledge on nucleotide stress responses in neural crest and melanoma and discuss how the recent scientific advances that have improved our understanding of transcriptional regulation during nucleotide depletion can impact melanoma treatment.
The cellular processes required for melanocyte development during embryogenesis and those involved in melanoma initiation and dissemination share a unique resemblance. This relationship has allowed us to gain an understanding of tumor behavior and plasticity by studying developmental programs. In this article, we outline the similarities between the transcriptional pathways involved in lineage specification and malignant transformation. We also review the unique sensitivity of these processes to disruptions in nucleotide metabolism and bring together recent work on the regulation of gene expression in neural crest and melanoma during nucleotide depletion. Finally, we discuss how in-depth knowledge of nucleotide stress pathways can inform the development of new treatment strategies for melanoma.
Melanocyte specification during development
Neural crest cells (NCCs) are a transient, migratory group of cells that arise from the embryonic ectoderm during vertebrate development [Citation1]. These multipotent progenitor cells give rise to a plethora of cell types. This includes the pigment-producing cells of the skin known as melanocytes. Melanocytes are present in the epidermis and hair follicles of the human skin and serve an important role in the protection against ultraviolet light-induced DNA damage. Melanin is one of the most potent free radicals that protects us from mutagenic reactive oxygen species that can otherwise induce structural DNA damage [Citation2]. On any given day, we generate millions of new epidermal skin cells and hairs. Both are pigmented by the transfer of melanin from melanocytes. Therefore, the demand for melanocytes is high and a tight regulation of melanocyte stem cells and their differentiation is required to ensure proper pigmentation throughout life.
Commitment of NCCs to melanocyte fate occurs via gradual lineage restriction. First, multipotent NCCs become glial-melanocyte lineage restricted cells [Citation3]. These bipotent progenitors become further restricted to the melanocyte lineage as committed, unpigmented melanocyte precursors termed melanoblasts. Melanoblasts can subsequently terminally differentiate into melanized melanocytes [Citation4]. Melanocyte fate commitment is instructed by inductive, extrinsic signals that converge on transcription. The transcription factor (TF) microphthalmia-associated transcription factor (MITF) has a central function in the specification of melanocytes. The TF is known as the master regulator of melanocyte identity and one of the earliest markers of NCC commitment to the melanocyte lineage. MITF acts as a transcriptional activator on several pigment-related genes, efficiently activating expression of genes involved in melanin synthesis such as tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1), dopachrome tautomerase (DCT/TYRP2), and melanocortin 1 receptor (MC1R) [Citation5–8]. MITF also promotes survival and proliferation of the melanocyte lineage. The TF does so via control of the inhibitor of apoptosis BCL2, regulation of cyclin-dependent kinase 2 (CDK2), and the CDK inhibitors p16 (INK4a) and p21 (CIP/WAF) [Citation9]. The importance of MITF in melanocyte development from the neural crest is exemplified by the phenotypes associated with loss of MITF function, which results in complete absence of melanocytes [Citation10,Citation11].
An important signal on which the gradual restriction toward the melanocyte lineage depends is Wnt. Wnt induces translocation of β-catenin to the nucleus, where it interacts with LEF1 at the MITF promoter to activate transcription of the master regulatory gene. By promoting melanocyte fate over glial lineage commitment, Wnt is a crucial player in the specification of melanoblasts [Citation12]. Other known neural crest specifiers, such as SOX10 and PAX3, also act as important regulators of MITF [Citation13,Citation14]. Like LEF1, SOX10 and PAX3 bind directly to their respective motifs present within the promoter of MITF and hereby active expression of the master regulator. Thus, MITF is a critical driver of pigment cell development and survival. The activation of the master regulator MITF is a key step in melanocyte development.
Developmental pathways in malignant transformation
There is increasing evidence that the transcriptional programs that govern melanocyte specification during development also contribute to tumor formation and progression. Dysregulation of the transcriptional programs that regulate NCC lineage specification and maintenance of melanocyte fate can, due to genetic mutations, transform normal melanocytes into neoplastic derivatives [Citation15]. Transformed melanocytes give rise to melanoma, a particularly aggressive and highly metastatic cancer with poor prognosis. Melanomas are enriched for embryonic neural crest specifiers such as MITF, SOX10, and PAX3 [Citation16]. While these genes are required for melanocyte specification during development, they are normally downregulated after terminal differentiation of neural crest progenitors. Reactivation of their expression in melanoma therefore implies that malignant cells have undergone transcriptional reprogramming and adopted an NCC-like fate. The re-emergence of an embryonic progenitor cell state correlates with melanoma initiation in vivo[Citation17] . Activating mutations in either the BRAF or NRAS oncogenes are present in nearly all human melanomas [Citation18]. These hallmark mutations of melanoma interact with the transcriptional programs that control NCC fate. In the presence of aberrant BRAF activity, MITF displays oncogenic activity [Citation19]. The dual expression of aberrant BRAF and MITF transforms human melanocytes into malignancies. SOX10 is a critical player in the maintenance of melanocytic neoplasms in vivo by controlling cell survival and proliferation genes. The TF cooperates with mutant NRAS, promoting NRAS-induced hyperproliferation and tumor formation [Citation16]. The lineage regulators required for proper specification of melanocytes during development thus play a role in the initiation, growth, and dissemination of melanoma. Thorough understanding of the developmental pathways and environmental signals that control lineage commitment during embryogenesis and cell fate conversion during malignant transformation aids novel therapeutic opportunities for melanoma.
Understanding melanocyte biology and melanoma development using zebrafish
A high degree of conservation in development between vertebrate species makes zebrafish (Danio rerio) a valuable model organism to study melanocyte biology [Citation20]. Similar to mice and humans, zebrafish melanocytes originate from migratory NCCs and differentiate into specialized melanin-producing pigment cells [Citation21]. Because their pigmentation patterns are clearly visible during embryogenesis and adulthood, zebrafish were central to the identification of conserved genes with previously unknown roles in the development and function of melanocytes [Citation22]. The ability to express human oncogenes, such as mutant BRAF or NRAS, in a melanocyte-specific manner furthermore instructed the generation of zebrafish melanoma models. These models revealed that zebrafish melanomas undergo fate conversion during tumor initiation, with malignant cells adopting a transcriptional signature that is similar to embryonic NCCs [Citation16].
A feature unique to the zebrafish model is the opportunity to perform large-scale chemical screens. Due to their permeability during development, phenotype-driven drug screens can be performed simply by adding chemicals to the water. This approach enables us to dissect molecular mechanisms and uncover new therapeutics that activate or inhibit specific developmental trajectories. With safety and toxicology profiles already known, chemical screens conducted with FDA-approved drugs hold significant value as repurposing of drugs for new indications provides a faster route to the clinic. These characteristics make the zebrafish an attractive model organism for studies on melanocyte biology and for the discovery of drugs that can improve patient outcomes.
Nucleotide stress inhibits melanocyte fate and melanoma
One of the first drugs identified in a zebrafish chemical screen that holds potential as melanoma therapy is leflunomide, an anti-inflammatory agent currently approved as a treatment for rheumatoid arthritis. Based on the observations that melanocytes adopt an embryonic, multipotent NCC-like fate during tumor initiation, White et al. proposed that suppressors of NCC development have implications for melanoma [Citation16]. To identify drugs that act as inhibitors of neural crest specification, the authors performed a chemical screen in zebrafish embryos. A specific class of compounds, inhibitors of the enzyme dihydroorotate dehydrogenase (DHODH), led to complete abrogation of the neural crest lineage in zebrafish. The DHODH inhibitor leflunomide, as well as its active derivate A77-1726, were identified as potent suppressors of the transcriptional programs that regulate NCC fate. Both drugs effectively block the expression of genes that are required for neural crest cell development, including mitfa and sox10. Correspondingly, also neural crest-derived lineages were affected by DHODH inhibitors. Zebrafish were devoid of pigmented melanocytes during development and in adulthood when exposed to leflunomide. Given the close relationship between melanocyte development and melanoma, the effects of leflunomide on melanoma wereassessed. Leflunomide effectively impedes tumor cell proliferation in vitro and in vivo when used alone or in combination with a selective oncogenic BRAF inhibitor [Citation23]. The contribution of NCC specifiers in the formation and dissemination of melanoma revealed the potential of targeting these developmental pathways as therapeutic strategy for melanoma.
DHODH catalyzes the fourth and rate limiting step of de novo pyrimidine (i.e. cytosine, thymidine, and uracil) synthesis. Consequently, DHODH inhibition by leflunomide leads to a depletion of nucleotide pools. Previous studies demonstrated that low nucleotide levels impede effective transcriptional elongation in vitro [Citation24]. Indeed, loss of neural crest and melanocytes following leflunomide treatment is the result of elongation defects at genes that govern neural crest specification and melanoma growth [Citation16].
Apart from direct effects on transcription, DHODH inhibition is known to alter mRNA translation and ribosome biogenesis. As one of the most energetically demanding cellular processes, ribosynthesis encompasses a substantial demand for nucleotides. The rRNA of ribosomes is where the majority of nucleotides reside [Citation25]. Depletion of the pyrimidine nucleotide pool is therefore expected to impair ribosome biosynthesis. Leflunomide-induced inhibition of DHODH decreased transcript and protein levels of crucial ribosomal components and reduced ribogenesis. The subsequent ribosomal stress leads to cell cycle arrest in tumor cells [Citation26–28].
Although DHODH inhibition would be expected to lead to ubiquitous effects in vivo, different cell lineages have varying sensitivities to DHODH inhibition depending on their ability to engage alternative mechanisms for nucleotide production such as salvage pathways. Mutations in DHODH cause a human craniofacial disorder known as Miller syndrome [Citation29,Citation30]. These craniofacial malformations are likely the result of defective NCC development since the cell lineage is prominently involved in vertebrate craniofacial development. NCCs contribute much to the cartilage, bone, and connective tissue that make up the developing head. This illustrates the unique susceptibility of NCCs to impaired nucleotide synthesis and the direct impact that loss of DHODH function has on neural crest-derived tissues. Patients undergoing cancer treatments that target nucleotide metabolism often develop resistance. Insights on the mechanism of transcriptional abrogation in response to nucleotide depletion can therefore contribute to more effective melanoma therapies.
Regulation of transcription elongation during nucleotide stress
Modulation of gene expression in response to stresses such as nucleotide depletion is essential for adaptation and survival of cells in altered environmental conditions. Changes in transcription are regulated at several levels. The ability to modify transcription initiation, elongation, and termination enables a rapid and coordinated response to stress.
Altered nucleotide abundance can impede transcriptional elongation. Transcription elongation is highly regulated following recruitment of RNA polymerase II (Pol II) to the transcriptional start site (TSS) and establishment of promoter-proximal pausing [Citation31,Citation32]. Promoter escape after phosphorylation of serine 5 (Ser5) in the Pol II carboxy-terminal domain (CTD) is insufficient for effective transcription of genes. Rather, transcription is paused after the first 20–40 nucleotides. At the promoter proximal pause site, Pol II is halted by negative elongation factor (NELF) and DRB-sensitivity inducing factor (DSIF). NELF and DSIF impair the mobility of the Pol II complex. Productive transcription elongation commences only upon phosphorylation of Ser2 of the Pol II CTD by the positive transcription elongation factor b (P-TEFb). Ser2 phosphorylation creates binding sites for additional elongation factors and mRNA processing proteins. This relieves transcriptional pausing and stimulates productive elongation [Citation33]. Ser2 phosphorylation also leads to dissociation of NELF and transforms DSIF into a positive elongation factor that associates with Pol II during elongation [Citation34].
Inhibition of effective elongation at genes required for growth and proliferation is accomplished through induction of HEXIM, a transcription elongation regulator that forms an inhibitory complex with P-TEFb. HEXIM is an RNA binding protein that engages 7SK small nuclear RNA (snRNA). As part of the 7SK small nuclear ribonucleoprotein (snRNP) complex, HEXIM negatively regulates elongation by sequestering and inactivating P-TEFb hereby mediating Pol II pause-release [Citation35]. HEXIM is sensitive to nucleotide starvation. Low nucleotide levels, such as those induced by DHODH inhibition, lead to upregulation of HEXIM at both the transcript and protein level [Citation36]. This response is specific and reversed when nucleotide levels are restored. HEXIM adaptation to altered nucleotide pools is mediated by the stress-responsive transcription factor SP1, a member of the SP/KLF family of TFs. Following activation, SP1 engages its motif located within the HEXIM promoter to mediate induction of gene expression under stress conditions.
Other TFs affected by signals of nucleotide depletion include p53, c-Myc and E2F1 [Citation37–39]. Changes in nucleotide availability and nucleolar stress alter the transcriptional activities of these factors and directly impact transcription of the corresponding target genes, among which are regulators of cell proliferation and apoptosis [Citation40,Citation41].
Increased abundance of HEXIM leads to higher engagement of the protein with P-TEFb, which sequesters the complex away from promoters and reduces productive elongation. HEXIM enforces transcriptional pausing at genes required for NCC development and melanocyte specification when nucleotides are depleted [Citation36]. This block in effective elongation leads to loss of the neural crest and melanocyte lineage. As a negative regulator of elongation, deficiency of HEXIM rescued expression of lineage regulators such as MITF and subsequently melanocyte specification during nucleotide stress conditions in vivo. Similarly, also depletion of other members of the 7SK snRNP complex rescued gene expression and melanocyte fate in vivo even when nucleotide levels are low. HEXIM, but not other members of the 7SK complex, is downregulated in melanoma. Decreased HEXIM levels are the result of epigenetic silencing by promoter hypermethylation. Loss of HEXIM accelerates melanoma, while overexpression suppresses tumor onset in vivo. Therefore, HEXIM is a tumor suppressor in melanoma.
Stress signals can also lead to the dissociation of P-TEFb from HEXIM and the 7SK snRNP complex. This increases the levels of active P-TEFb and results in stress-induced transcriptional activation. Many stress-responsive genes are already bound by Pol II in the absence of a stressor and exist in a transcriptionally paused state [Citation42,Citation43]. Upon stress, such loci recruit factors that mediate the regulated release of paused Pol II. Signal-responsive TFs can facilitate the pause-release via direct interaction with P-TEFb or through recruitment of cofactors that bind P-TEFb and promote its activity [Citation44,Citation45]. The rapid switch to productive elongation at target genes bypasses the need for assembly of the pre-initiation complex. While nucleotide stress inhibits the effective elongation of genes required for growth and proliferation, transcriptional induction of pro-apoptotic genes is observed in concert [Citation46]. The upregulation of genes involved in cell death is regulated by stress-responsive TFs such as SP1, among others [Citation47].
HEXIM not only binds 7SK RNA but engages with a variety of RNAs present in the cell. Its RNA binding pattern changes when nucleotide pools are limited. While binding to 7SK RNA remains proportionally unaffected, a change in the mRNAs bound by HEXIM is observed. HEXIM specifically engagesanti-proliferative and pro-apoptotic transcripts, including those of stress-activated genes, and stabilizes these mRNAs to promote cell death and halt cell growth [Citation36].
Regulation of the transition to effective elongation allows for a finely coordinated transcriptional response to stress. The ability of HEXIM1 to respond to stress and shift transcription toward a growth suppressive state, makes the RNA binding protein an attractive target for melanoma treatment.
Nucleotide stress is sensed by the RNA helicase DDX21
All organisms have the ability to sense the abundance of nutrients that are required for energy and macromolecule synthesis [Citation48]. A key metabolic sensor is the protein kinase mammalian target of rapamycin complex 1 (mTORC1), which integrates nutrient-related signals to control biosynthetic processes, including nucleic acid synthesis [Citation49]. mTORC1 directly regulates the activity of various nucleotide synthesis enzymes. This includes the multifunctional enzyme carbamoyl phosphate synthetase 2, aspartate transcarbamylase and dihydroorotase (CAD), which catalyzes the first three steps of the de novo pyrimidine synthesis [Citation50,Citation51]. Despite insights on sensors of nucleotide pools, the mechanism through which signals of low nucleotide levels are converted into altered transcription elongation rates during stress remains elusive.
Recently, a study aimed at understanding how a reduction in pyrimidine levels is perceived in neural crest cells and melanoma was performed. The RNA helicase DDX21 was identified as a novel mediator of the nucleotide stress response. DDX21 is a DEAD box nucleolar protein with an RNA helicase domain to which it owes its 5′ to 3′ RNA unwinding activity. The protein also contains a domain that enables the introduction of secondary structures to single-stranded RNA. DDX21 exerts important functions in rRNA production and processing in the nucleolus [Citation52,Citation53]. The nucleolus is the site of rRNA biosynthesis. Here, Pol I-mediated transcription of rDNA, processing and maturation of rRNA, and ribosome assembly occur. Loss of DDX21 results in an inhibition of rRNA production. The RNA helicase also holds a role outside the nucleolus, where it functions in the control of gene expression. DDX21 acts as a regulator of Pol II-mediated transcription in the nucleoplasm [Citation54,Citation55]. At gene promoters, DDX21 stimulates transcription via direct interactions with TFs and binding to the 7SK snRNP complex, rather than engagement with nascent mRNA transcripts at promoters. DDX21 facilitates the release of P-TEFb from the 7SK snRNP and enables commencement of effective transcription elongation. This function of DDX21 is dependent on its helicase activity. Thus, DDX21 has multiple primary functions. The RNA helicase exerts roles in rRNA metabolism in the nucleolus, while also functioning as a transcriptional regulator in the nucleoplasm () [Citation55–57].
Figure 1. RNA helicase DDX21 exerts a multitude of functions at various locations within the cell
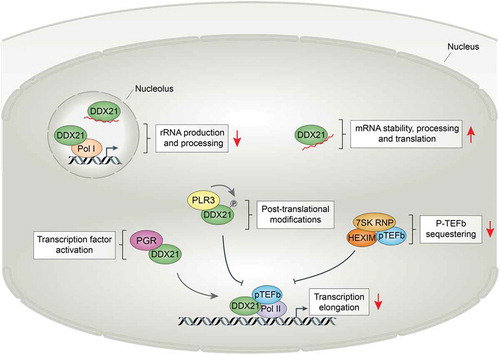
In homeostatic conditions, DDX21 binds Pol II-regulated promoters in the nucleoplasm and facilitates effective transcription elongation. However, the chromatin association of DDX21 is highly sensitive to stress. Various cellular stress conditions alter its association with Pol II-regulated genes and promote translocation of DDX21 from the nucleolus to the nucleoplasm [Citation55]. DHODH-induced nucleotide depletion leads to dissociation of DDX21 from promoters and downregulation of the corresponding genes. Loss of DDX21 from chromatin may enforce transcriptional pausing when nucleotide pools are low in order to prevent high transcriptional activity at times of metabolic stress. Instead, reduced nucleotide levels direct DDX21 to alternative functions in the nucleoplasm. DHODH inhibition leads to engagement between DDX21 and mRNAs in the nucleoplasm. Enhanced binding of DDX21 to mRNAs of genes involved in nucleotide metabolisms indicates that DDX21 aims to minimize stress defects by stabilization of transcripts that are important for adaptation. While DDX21 binding to mRNA is suggested to affect transcript stability, functions of DDX21 in mRNA splicing, processing, or nuclear export also remain a possibility. Further research will be required to decipher the exact impact of DDX21 binding on mRNAs and assess if this effect is specific to nucleotide depletion or part of a general response to stress.
The shift to the alternative, mRNA-related function of DDX21 is concomitant with its dissociation from rRNA and translocation from the nucleolus, as well as disengagement of the RNA helicase from chromatin and subsequent defective transcription elongation. A role for DDX21 in both basic cellular functions and stress adaptation is in line with dual functions described for other stress-related proteins [Citation58]. Stress mechanisms are shaped by positive natural selection to enhance an organism’s ability to cope with, and adapt to, a changing environment. The increased repertoire of extrinsic signals that can be recognized by cells and the diversification of intracellular response pathways played a fundamental role in the evolutionary success of multicellular organisms [Citation59]. A stress-related role of the already multifunctional DDX21 protein may allow cells to regulate various cellular processes at once. Affecting several intracellular systems with a single protein can facilitate a quick and coordinated adaptive response. One remaining question is whether DDX21 exerts such function only during nucleotide stress conditions or whether other types of stress also evoke this function.
Studies on neural crest and melanoma also revealed that a reduction in DDX21 expression confers resistance to transcriptional defects resulting from nucleotide depletion [Citation44]. Decreased levels of the RNA helicase during nucleotide stress might redirect residual DDX21 toward its primary functions. The transcriptional role of DDX21 on chromatin and functions in rRNA metabolisms in the nucleolus may be prioritized over mRNA binding when both nucleotide levels and DDX21 levels are low. This restores effective elongation and rescues cell fate in vivo. As a sensor of nucleotide levels, DDX21 inhibits transcription when nucleotide levels are low and shifts its binding from chromatin and rRNA to mRNA. The RNA helicase seems particularly important for the neural crest lineage. Dissociation of DDX21 from chromatin impairs craniofacial development and results in defects similar to those observed in Treacher Collins syndrome [Citation56]. Since the affected craniofacial structures are developmental derivates of NCCs, these observations suggest selective sensitivity of NCCs and derivative cell lineages not only to nucleotide depletion but also to loss of DDX21 from promoters. DDX21 is a protein with a multitude of functions in normal homeostasis and during stress that are highly interconnected and coregulated.
Extrinsic signals influence DDX21 function in neural crest and melanoma
Cell fate and function is the result of an integrated response to intracellular and extracellular signals that converge on gene expression. As a multifunctional protein responding to a variety of stressors, DDX21 function is affected by nucleotide stress conditions. The receptiveness of DDX21 to environmental signals can be leveraged to direct cells toward, or divert cells from, a particular behavior or fate.
It was recently described that progesterone and progesterone receptor (PGR) signaling affect nucleotide stress responses in the neural crest through interaction with DDX21 [Citation44]. Transcriptional defects due to nucleotide depletion can be overcome by alterations of progesterone signaling in multipotent, migratory NCCs. Changes in progesterone and PGR levels strongly suppress nucleotide stress-induced neural crest defects by bypassing the transcription elongation block, hereby rescuing the neural crest lineage and melanocyte fate.
Steroid hormones (e.g. estrogen, androgen, and progesterone) signal by binding to intracellular nuclear receptors. Hormone binding causes conformational changes of the receptor, translocation to the nucleus, and interaction with their corresponding binding elements in the DNA to regulate the transcription of target genes [Citation60]. Progesterone acts as an agonist of PGR which, upon activation, binds to progesterone response elements (PREs) near hormone response genes. Here, PGR facilitates transcription initiation and elongation through recruitment of the transcription machinery and p-TEFb, respectively [Citation61–63]. PGR also affects the expression of genes that lack defined PREs via interactions with other TFs [Citation64–66]. Additionally, PGR can exert effects through nongenomic mechanisms by altering the production of second messenger molecules and by activating signal transduction pathways in manners that are independent of its transcriptional function [Citation67,Citation68].
PGR was identified as a complex interaction partner of DDX21. Although the implications of the interaction are currently unknown, DDX21 expectedly has particular functions when associated with PGR. Modification of progesterone signaling, and more specifically a decrease in PGR transcriptional activity, ameliorates the effects of nucleotide depletion and restores NCC fate [Citation44]. Downregulation of PGR could lead to reduced TF activity and hereby reduce the transcriptional demand in NCCs. This alleviates the effects of low nucleotide availability. DDX21 acts as a mediator between nuclear hormone signaling and nucleotide stress signaling. In normal conditions, DDX21 exerts its primary functions in rRNA metabolism in the nucleolus and in Pol II-mediated transcription in the nucleoplasm [Citation55–57]. During nucleotide stress, DDX21 enforces polymerase pausing and shifts is functions toward a mRNA regulatory role. However, alterations in progesterone signaling can drive DDX21 activity back toward its primary functions, even in suboptimal conditions when nucleotide levels are low. Changes in transcriptional demand, due to reduced PGR activity, redirect DDX21 to chromatin. Loss of PGR activity could provide a second stress signal to DDX21 that overrules, or counteracts, nucleotide stress signals. In an effort to compensate for the lost PGR activity, DDX21 shifts back toward its primary roles. As a result, effective transcription is reinstated and NCC fate is restored. In conditions of competition between nucleotide stress signals and altered progesterone signaling, DDX21 activity is skewed toward the regulation of transcription.
The exact signals that direct DDX21 toward certain functions in specific cellular contexts still remain to be elucidated. DDX21 is subject to a variety of post-translational modifications and can exist in various protein complexes. Several studies have implicated the post-translational status of DDX21 as a major determinant of its activity and function [Citation54,Citation69,Citation70]. DDX21 is a phosphorylation substrate of a number of kinases that respond to environmental signals. This includes members from the mitogen-activated protein kinase (MAPK) and c-Jun protein kinase (JNK) family. While the functional impact of DDX21 phosphorylation is unknown, p38-MAPK and JNK-mediated phosphorylation promote nucleolar localization of DDX21 [Citation70,Citation71]. Furthermore, acetylation of DDX21 by the histone acetyltransferase CBP inhibits its helicase activity, while deacetylation by the deacetylase SIRT7 augments helicase activity [Citation72–74]. Additionally, DDX21 is subject to PARP-1 mediated ADP-ribosylation, which promotes the nucleolar localization of DDX21 and rDNA transcription [Citation75]. We speculate that both protein modifications and complex interactions impact DDX21 activity and localization and that these mechanisms act in a corroborative manner in certain cellular contexts. The ability of DDX21 to adapt its function based on the extrinsic signals that are present allows for a finely tuned cellular stress response.
DDX21 regulates transcriptional melanocyte stem cells
DDX21 not only holds an important function in the stress response of neural crest and melanoma cells but is also required for melanocyte differentiation during regeneration. Its role in regeneration is dependent on the interaction with phosphatase of liver regeneration 3 (PLR3) in the nucleoplasm. The phosphatase inhibits premature expansion of melanoblast progenitors and differentiation of melanocyte stem cells (MSCs) [Citation76]. PLR3 restricts differentiation by impairing productive transcription elongation of melanosome and pigment granule-related genes, hereby acting as a negative regulator of melanocytes. Suppression of effective transcription is achieved through interaction of PLR3 with, and regulation of, DDX21 on chromatin. PLR3 affects both DDX21 density and distribution specifically on genes associated with melanocyte fate. Enhanced PLR3 activity reduces promoter binding of DDX21 at its target genes [Citation76]. Simultaneously, accumulation of nascent mRNA and a reduction of Pol II activity at promoters is observed. PLR3 phosphatase activity is concurrent with reduced phosphorylation of DDX21 at a residue that is critical to melanocyte regeneration. Thus, the changes in DDX21 binding are likely dependent on the post-translational status of the RNA helicase. Transcriptional pausing enforced by PLR3 through regulation of DDX21 leads to reduced expression of melanocyte genes and prevents premature differentiation of MSCs. Modification of the post-translational status of DDX21 can provide the means to manipulate stem cell fate.
Therapeutic potential of targeting DDX21 in melanoma
Treatment options for melanoma have greatly improved over the last decade, in part, due to the development and approval of targeted therapies that provide a better prognosis for patients. However, challenges such as therapeutic resistance remain an issue. In an effort to delay or prevent tumor resistance and provoke a more durable response in patients, combinatorial strategies are currently under clinical investigation.
The DHODH inhibitor leflunomide has advanced into clinical trials for the treatment of melanoma. In pre-clinical melanoma models, leflunomide exhibits synergistic activities with the MEK inhibitor selumetinib. Combinatorial use of leflunomide with selumetinib resulted in greater apoptosis of melanoma cells than individual drug treatments alone. In xenograft mouse models, the combinatorial administration of leflunomide and selumetinib decreased both tumor volume and growth, while melanomas continued to grow when only one of the drugs was administered [Citation77]. Perhaps surprising was the premature termination of a clinical trial that investigated a combinational approach of leflunomide and vemurafenib (a mutant BRAF inhibitor) in patients with metastatic melanoma due to adverse events [Citation78]. This clinical trial revealed the potential toxicity associated with leflunomide and the complexity of combinatorial treatments. Additionally, patients undergoing chemotherapy regimen that target nucleotide metabolism often develop resistance. This raises a realistic concern for the viability of leflunomide-mediated DHODH inhibition as melanoma therapy. Leflunomide is a weak DHODH inhibitors. The novel, more potent, and selective DHODH inhibitor BAY 2402234 is currently being evaluated in a phase I clinical trial for myeloid malignancies [Citation79]. DHODH inhibitors with favorable physicochemical and pharmacokinetic properties, such as BAY 2402234, may overcome the toxicity issues that are associated with leflunomide.
Our increased understanding on the role of DDX21 in melanocyte fate and melanoma leads us to propose modulation of DDX21 activity and levels as a therapeutic avenue that warrants further investigation. Due the multitude of functions that DDX21 carries at different locations within the cell, it will be important to ensure specificity toward chromatin-bound DDX21. Affecting the post-translational status of the RNA helicase could potentially be employed as a strategy to redirect its localization and function. Targeting regulators of DDX21, such as PLR3 or PGR, may also hold therapeutic value. However, given the important role of progesterone and PGR in the endocrine system, directing DDX21 activity through hormone pathways requires additional investigation. Modifying DDX21 function could provide a novel opportunity to delay disease progression in melanoma patients.
Conclusion
Each cell type has a different tolerance to certain cellular stresses. Finding such dependencies could lead to the development of new therapies for genetic diseases and cancer. The neural crest is uniquely sensitive to low nucleotide levels. This is illustrated by patients with Miller syndrome, a disease characterized by developmental defects of the face and limbs consequent to mutations in the pyrimidine synthesis enzyme DHODH [Citation30].
When nucleotides are depleted in NCCs, transcription is slowed to conserve nucleotides until homeostasis returns. Two key players that regulate the adaption to nucleotide stress are HEXIM and DDX21. Induction of HEXIM by stress-responsive TFs leads to increased P-TEFb sequestering away from promoters and enforces transcriptional pausing of key lineage genes. Concomitantly, DDX21 dissociates from chromatin which represses effective transcription elongation and instead engages mRNA transcripts of genes required for adaptation in the nucleoplasm.
The ability of stress sensing proteins to control transcription based on the amount of nucleotide precursors available, coupled with their receptiveness to extrinsic signals, enables cells to finely tune gene expression to the cellular environment. By preventing cell growth or expansion under undesired circumstances, resources are conserved until cellular conditions become more favorable.
The programs that control neural crest development, MSC regeneration, and melanoma transformation are highly similar and uniquely sensitive to an imbalance in nucleotide levels. This metabolic vulnerability provides an opportunity that can be leveraged in anti-cancer therapies. Advancing our understanding of stress response processes in melanoma could greatly enhance our ability to effectively target cancer cells and hereby improve patient outcomes.
Acknowledgments
This work was supported by the following grants from L.I.Z: Cancer Biology R01 CA103846, NIH Melanoma PPG, P01CA63222, Melanoma Research Alliance, Starr Cancer Consortium grant. A.S. was supported by a Boehringer Ingelheim Fonds PhD Fellowship.
Disclosure statement
L.I.Z. is the founder and stockholder of Fate, Inc., Scholar Rock, Camp4 therapeutics and a scientific advisor for Stemgent.
Additional information
Funding
References
- Erickson CA, Reedy MV. Neural crest development: the interplay between morphogenesis and cell differentiation. Curr Top Dev Biol. 1998;40:177–209.
- Meredith P, Sarna T. The physical and chemical properties of eumelanin. Pigment Cell Res. 2006;19(6):572–594.
- Dupin E, Glavieux C, Vaigot P, et al. Endothelin 3 induces the reversion of melanocytes to glia through a neural crest-derived glial-melanocytic progenitor. Proc Nat Acad Sci. 2000;97(14):7882–7887.
- White RM, Zon LI. Melanocytes in development, regeneration, and cancer. Cell Stem Cell. 2008;3(3):242–252.
- Yasumoto K, Yokoyama K, Shibata K, et al. Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol. 1994;14(12):8058–8070.
- Aoki H, Moro O. Involvement of microphthalmia-associated transcription factor (MITF) in expression of human melanocortin-1 receptor (MC1R). Life Sci. 2002;71(18):2171–2179.
- Du J, Fisher DE. Identification of Aim-1 as the underwhite mouse mutant and its transcriptional regulation by MITF. J Biol Chem. 2002;277(1):402–406.
- Yasumoto K, Yokoyama K, Takahashi K, et al. Functional analysis of microphthalmia-associated transcription factor in pigment cell-specific transcription of the human tyrosinase family genes. J Biol Chem. 1997;272(1):503–509.
- McGill GG, Horstmann M, Widlund HR, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002;109(6):707–718.
- Steingrímsson E, Moore KJ, Lamoreux ML, et al. Molecular basis of mouse microphthalmia (mi) mutations helps explain their developmental and phenotypic consequences. Nat Genet. 1994;8(3):256–263.
- Tassabehji M, Newton VE, Read AP. Waardenburg syndrome type 2 caused by mutations in the human microphthalmia (MITF) gene. Nat Genet. 1994;8(3):251–255.
- Yasumoto K, Takeda K, Saito H, et al. Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J. 2002;21(11):2703–2714.
- Potterf SB, Furumura M, Dunn KJ, et al. Transcription factor hierarchy in Waardenburg syndrome: regulation of MITF expression by SOX10 and PAX3. Hum Genet. 2000;107(1):1–6.
- Bondurand N, Pingault V, Goerich DE, et al. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet. 2000;9(13):1907–1917.
- Shain AH, Bastian BC. From melanocytes to melanomas. Nat Rev Cancer. 2016;16(6):345–358.
- Shakhova O, Zingg D, Schaefer SM, et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat Cell Biol. 2012;14(8):882–890.
- White RM, Cech J, Ratanasirintrawoot S, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518–522.
- Omholt K, Platz A, Kanter L, et al. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9(17):6483–6488.
- Wellbrock C, Rana S, Paterson H, et al. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PloS One. 2008;3(7):e2734.
- Mort RL, Jackson IJ, Patton EE. The melanocyte lineage in development and disease. Development. 2015;142(4):620–632.
- Zeng Z, Richardson J, Verduzco D, et al. Zebrafish have a competent p53-dependent nucleotide excision repair pathway to resolve ultraviolet B-induced DNA damage in the skin. Zebrafish. 2009;6(4):405–415.
- Rooijen E, van, Fazio M, Zon LI. From fish bowl to bedside: the power of zebrafish to unravel melanoma pathogenesis and discover new therapeutics. Pigment Cell Melanoma Res. 2017;30(4):402–412.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–3046.
- Wada T, Takagi T, Yamaguchi Y, et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998;12(3):343–356.
- Valvezan AJ, Turner M, Belaid A, et al. mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell. 2017;32(5):624–638.e5.
- Hubackova S, Davidova E, Boukalova S, et al. Replication and ribosomal stress induced by targeting pyrimidine synthesis and cellular checkpoints suppress p53-deficient tumors. Cell Death Dis. 2020;11(2):110.
- Fairus AKM, Choudhary B, Hosahalli S, et al. Dihydroorotate dehydrogenase (DHODH) inhibitors affect ATP depletion, endogenous ROS and mediate S-phase arrest in breast cancer cells. Biochimie. 2017;135:154–163.
- Ladds MJGW, van Leeuwen IMM, Drummond CJ, et al. A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat Commun. 2018;9(1):1107.
- Rainger J, Bengani H, Campbell L, et al. Miller (Genee-Wiedemann) syndrome represents a clinically and biochemically distinct subgroup of postaxial acrofacial dysostosis associated with partial deficiency of DHODH. Hum Mol Genet. 2012;21(18):3969–3983.
- Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42(1):30–35.
- Guo J, Price DH. RNA polymerase II transcription elongation control. Chem Rev. 2013;113:8583–8603.
- Gressel S, Schwalb B, Decker TM, et al. CDK9-dependent RNA polymerase II pausing controls transcription initiation. eLife. 2017;6:R106.
- Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13(10):720–731.
- Sims RJ, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 2004;18(20):2437–2468.
- Yik JHN, Chen R, Nishimura R, et al. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell. 2003;12(4):971–982.
- Tan JL, Fogley R, Flynn R, et al. Stress from nucleotide depletion activates the transcriptional regulator HEXIM1 to suppress melanoma. Mol Cell. 2016;62(1):34–46.
- Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11.
- Russo A, Russo G. Ribosomal proteins control or bypass p53 during nucleolar stress. Int J Mol Sci. 2017;18(1):140.
- Zhang Z, Wang H, Li M, et al. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene. 2005;24(48):7238–7247.
- Riggelen J, Van, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10(4):301–309.
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24(17):2810–2826.
- Adelman K, Kennedy MA, Nechaev S, et al. Immediate mediators of the inflammatory response are poised for gene activation through RNA polymerase II stalling. Proc Natl Acad Sci U S A. 2009;106(43):18207–18212.
- Aida M, Chen Y, Nakajima K, et al. Transcriptional pausing caused by NELF plays a dual role in regulating immediate-Early expression of the junB gene. Mol Cell Biol. 2006;26(16):6094–6104.
- Rahl PB, Lin CY, Seila AC, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141(3):432–445.
- Barboric M, Nissen RM, Kanazawa S, et al. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;8(2):327–337.
- Santoriello C, Sporrij A, Yang S, et al. RNA helicase DDX21 mediates nucleotide stress responses in neural crest and melanoma cells. Nat Cell Biol. 2020;22(4):372–379.
- Schmidt T, Körner K, Karsunky H, et al. The activity of the murine Bax promoter is regulated by Sp1/3 and E-box binding proteins but not by p53. Cell Death Differ. 1999;6(9):873–882.
- Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017;45:72–82.
- Hoxhaj G, Hughes-Hallett J, Timson RC, et al. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. 2017;21(5):1331–1346.
- Ben-Sahra I, Howell JJ, Asara JM, et al. Stimulation of de Novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323–1328.
- Robitaille AM, Christen S, Shimobayashi M, et al. Quantitative phosphoproteomics reveal mTORC1 Activates de Novo pyrimidine synthesis. Science. 2013;339(6125):1320–1323.
- Henning D, So RB, Jin R, et al. Silencing of RNA helicase II/Gualpha inhibits mammalian ribosomal RNA production. J Biol Chem. 2003;278(52):52307–52314.
- Yang H, Zhou J, Ochs RL, et al. Down-regulation of RNA helicase II/Gu results in the depletion of 18 and 28 S rRNAs in Xenopus oocyte. J Biol Chem. 2003;278(40):38847–38859.
- Westermarck J, Weiss C, Saffrich R, et al. The DEXD/H-box RNA helicase RHII/Gu is a co-factor for c-Jun-activated transcription. EMBO J. 2002;21(3):451–460.
- Calo E, Flynn RA, Martin L, et al. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature. 2015;518(7538):249–253.
- Calo E, Gu B, Bowen ME, et al. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature. 2018;554(7690):112–117.
- Xing Y-H, Yao R-W, Zhang Y, et al. SLERT regulates DDX21 rings associated with Pol I Transcription. Cell. 2017;169(4):664–678.e16.
- Kültz D. Evolution of the cellular stress proteome: from monophyletic origin to ubiquitous function. J Exp Biol. 2003;206(18):3119–3124.
- Weake VM, Workman JL. Inducible gene expression: diverse regulatory mechanisms. Nat Rev Genet. 2010;11(6):426–437.
- Glass CK, Ogawa S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat Rev Immunol. 2006;6(1):44–55.
- Jacobsen BM, Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol. 2012;357(1–2):18–29.
- Bertucci PY, Nacht AS, Alló M, et al. Progesterone receptor induces bcl-x expression through intragenic binding sites favoring RNA polymerase II elongation. Nucleic Acids Res. 2013;41(12):6072–6086.
- Kininis M, Isaacs GD, Core LJ, et al. Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol Cell Biol. 2009;29(5):1123–1133.
- Clarke CL, Graham JD. Non-overlapping progesterone receptor cistromes contribute to cell-specific transcriptional outcomes. PloS One. 2012;7(4):e35859.
- Mohammed H, Russell IA, Stark R, et al. Progesterone receptor modulates ERα action in breast cancer. Nature. 2015;523(7560):313–317.
- Bamberger AM, Bamberger CM, Gellersen B, et al. Modulation of AP-1 activity by the human progesterone receptor in endometrial adenocarcinoma cells. Proc Nat Acad Sci. 1996;93(12):6169–6174.
- Bayaa M, Booth RA, Sheng Y, et al. The classical progesterone receptor mediates Xenopus oocyte maturation through a nongenomic mechanism. Proc Nat Acad Sci. 2000;97(23):12607–12612.
- Migliaccio A, Piccolo D, Castoria G, et al. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 1998;17(7):2008–2018.
- Holmström TH, Mialon A, Kallio M, et al. c-Jun supports ribosomal RNA processing and nucleolar localization of RNA helicase DDX21. J Biol Chem. 2008;283(11):7046–7053.
- Mialon A, Thastrup J, Kallunki T, et al. Identification of nucleolar effects in JNK-deficient cells. FEBS Lett. 2008;582(20):3145–3151.
- Bora P, Gahurova L, View ORCID ProfileHauserova A, et al. DDX21 is a p38-MAPK sensitive nucleolar protein necessary for mouse preimplantation embryo development and cell-fate specification. bioRxiv. 2021;4:13.439318, 2021
- Song C, Hotz-Wagenblatt A, Voit R, et al. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 2017;31(13):1370–1381.
- Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840.
- Schölz C, Weinert BT, Wagner SA, et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells. Nat Biotechnol. 2015;33(4):415–423.
- Kim D-S, Camacho CV, Nagari A, et al. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell. 2019;75(6):1270–1285.e14.
- Johansson JA, Marie KL, Lu Y, et al. PRL3-DDX21 transcriptional control of endolysosomal genes restricts melanocyte stem cell differentiation. Dev Cell. 2020;54(3):317–332.e9.
- Hanson K, Robinson SD, Al-Yousuf K, et al. The anti-rheumatic drug, leflunomide, synergizes with MEK inhibition to suppress melanoma growth. Oncotarget. 2018;9(3):3815–3829.
- Cortés H, Reyes-Hernández OD, Alcalá-Alcalá S, et al. Repurposing of drug candidates for treatment of skin cancer. Front Oncol. 2020;10:605714.
- Christian S, Merz C, Evans L, et al. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia. 2019;33(10):2403–2415.