
ABSTRACT
Cyclin-dependent kinase 4 (CDK4) is a master integrator that couples mitogenic/oncogenic signaling with the cell division cycle. It is deregulated in most cancers and inhibitors of CDK4 have become standard of care drugs for metastatic estrogen-receptor positive breast cancers and are being evaluated in a variety of other cancers. We previously characterized the T-loop phosphorylation at T172 of CDK4 as the highly regulated step that determines the activity of cyclin D-CDK4 complexes. Moreover we demonstrated that the highly variable detection of T172-phosphorylated CDK4 signals the presence or absence of the active CDK4 targeted by the CDK4/6 inhibitory drugs, which predicts the tumor cell sensitivity to these drugs including palbociclib. To date, the phosphorylation of CDK4 has been very poorly studied because only few biochemical techniques and reagents are available for it. In addition, the available ones including 2D-IEF separation of CDK4 modified forms are considered too tedious. The present report describes the generation, selection and characterization of the first monoclonal antibodies that specifically recognize the active CDK4 phosphorylated on its T172 residue. One key to this success was the immunization with a long phosphopeptide corresponding to the complete activation segment of CDK4. These monoclonal antibodies specifically recognize T172-phosphorylated CDK4 in a variety of assays, including western blotting, immunoprecipitation and, as a capture antibody, a sensitive ELISA from cell lysates. The specific immunoprecipitation of T172-phosphorylated CDK4 allowed to clarify the involvement of phosphorylations of co-immunoprecipitated p21 and p27, showing a privileged interaction of T172-phosphorylated CDK4 with S130-phosphorylated p21 and S10-phosphorylated p27.
Abbreviations: 2D: two-dimensional; CAK: CDK-activating kinase; CDK: cyclin-dependent kinase; HAT: Hypoxanthine-Aminopterin-Thymidine; FBS: fetal bovine serum; IP: immunoprecipitation; ID: immunodetection; mAb: monoclonal antibody; PAGE: polyacrylamide gel electrophoresis; PBS: phosphate buffer saline; pRb: retinoblastoma susceptibility protein; SDS: sodium dodecyl sulfate; DTT: dithiotreitol; TET: tetracyclin repressor; Avi: Avi tag; TEV: tobacco etch virus cleavage site; EGFP: enhanced green fluorescent protein; BirA: bifunctional protein biotin ligase BirA; IRES: internal ribosome entry site; HIS: poly-HIS purification tag; DELFIA: dissociation-enhanced lanthanide fluorescent immunoassay; 3-MBPP1: 1-(1,1-dimethylethyl)-3[(3-methylphenyl) methyl]-1H-pyrazolo[3,4-d] pyrimidin-4-amine; BSA: bovine serum albumin; ECL: Enhanced chemiluminescence
Introduction
Multiple mitogenic and growth-inhibitory signals must be integrated to determine if it is appropriate for a cell to divide. Much of this regulation is directed by the kinases that mediate the earliest transition in the G1 phase of the cell cycle. CDK4 (and/or CDK6) activity is generally the point at which this integration occurs [Citation1–5]. CDK4 activity requires its binding to cyclins D (CCND1-3 genes). INK4 CDK4 inhibitors such as p16 (CDKN2A-D genes) compete for this binding. As commonly considered, mitogenic/oncogenic signaling cascades activate CDK4/6 by inducing at least one cyclin D. However, the CDK4 activation is much more complex [Citation3,Citation6,Citation7], involving distinct regulations of (i) the assembly of cyclin D-CDK4 complexes [Citation8,Citation9] that are generally stabilized by binding to p21 or p27 [Citation10–13], (ii) their nuclear import [Citation14,Citation15] and (iii) the activating T-loop phosphorylation of CDK4 at Thr172 [Citation16–23]. So far, this required activating T172-phosphorylation of CDK4 has been much less studied due to lack of easy detection tools. By separating the modified forms of CDK4 by two-dimensional (2D) gel electrophoresis, we showed that the activating T172-phosphorylation of CDK4 bound to cyclins D is exquisitely regulated and that it is the central rate-limiting event (integrating all previous steps) in CDK4 activation [Citation18–21,Citation24,Citation25]. This event thus determines pRb phosphorylation and cell cycle commitment in pRb-proficient cells [Citation7,Citation21,Citation23]. The mechanisms and pathways implicated in the various regulations of CDK4 T172-phosphorylation are still poorly defined. The phosphorylated T-loop threonine is uniquely followed by a proline in CDK4 [Citation26] and we and others identified several proline-directed CDK4-activating kinases [Citation21,Citation23,Citation27]. Phosphorylations of p21 and p27 were found to be critical for CDK4 T172-phosphorylation and activation [Citation21,Citation22,Citation27,Citation28]. Finally, the T172-phosphorylation of CDK4 is labile [Citation21,Citation23] but CDK4 T172-phosphatases remain enigmatic.
Because CDK4 is downstream from most of the oncogenic deregulations of mitogenic signaling pathways, it has been proposed to represent the therapeutic target for which there should be the fewest bypass mechanisms [Citation29]. Cell cycle deregulation in cancer is often linked to addiction to CDK4 activity [Citation30–32]. The CDK4/6 inhibitors including PD0332991/palbociclib, LEE-011/ribociclib and LY2835219/abemaciclib have now become standard of care combined with endocrine therapy for advanced estrogen receptor positive breast cancers [Citation33–35]. These drugs are tested in a large number of phase II/III clinical trials against various cancers while diverse combination therapies involving CDK4/6 inhibitors are being envisaged [Citation36,Citation37]. In breast cancer tumor samples and cell lines, we demonstrated that the highly variable detection of T172-phosphorylated CDK4 signals the presence or absence of the active CDK4 targeted by the CDK4/6 inhibitory drugs, which was associated with the sensitivity or insensitivity of tumor cells to palbociclib [Citation38]. CDK4 T172-phosphorylation might thus be the best biomarker of potential tumor sensitivity to CDK4/6 inhibitory drugs.
The corresponding T-loop phosphorylations of CDK1 and CDK2 are easily studied thanks to wide collection of polyclonal and monoclonal phosphospecific antibodies. By contrast, reproducible or high-quality antibodies were hardly developed to detect CDK4 T172 phosphorylation. Here, we describe the generation of the first T172-phosphospecific CDK4 monoclonal antibodies (mAb). Keys to this achievement were the immunization with a long phosphopeptide corresponding to the complete activation segment of CDK4 [Citation39] and the development of screening and characterization tools based on phosphorylated and non-phosphorylated cyclin D/CDK4 fusions coupled to a biotinylatable tag [Citation40]. We characterized these antibodies for use in immunoblotting, (co)immunoprecipitation and ELISA. The specific immunoprecipitation of T172-phosphorylated CDK4 allowed to clarify the involvement of co-immunoprecipitated p21 and p27 phosphorylated forms.
Material and methods
Generation of anti-PT172 CDK4 mAbs
Mouse mAbs specific for human CDK4 phosphorylated on T172 were obtained by immunizing mice with either a short (EGT578) or a long KLH fusion peptide (EGT577) (). Mice immunization and hybridoma production were performed by Eurogentec (Seraing, Belgium) mAb production customer service. Briefly, for each peptide four BALB/c mice were immunized by four injections on days 0, 14, 28 and 42. Sera of immunized mice collected on days 35 and 49 after immunization were tested by indirect ELISA for reactivity against the immunogen. The best responding mice (M1 and M4, EGT577) were selected for the fusion of splenocytes with Sp2/OAg 14 myeloma cells. After selection of hybridoma cells by treatment with Hypoxanthine-Aminopterin-Thymidine (HAT), IgG-positive hybridoma supernatants were screened by indirect ELISA for reactivity against the phosphorylated antigen and the non-phosphorylated one. Hybridoma supernatants giving positive results in these assays were further screened using a dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) that we developed using an immobilized cyclin D3/CDK4 proteic fusion expressed in MCF7 cells vs the same proteic fusion containing the T172A mutation (see below). They were then tested by western blotting from one and two-dimension electrophoresis gels. Hybridomas of interest were subcloned by limiting dilution and the obtained subclones were tested similarly as described above for the parental clones. Two of them, NB8-AD9 and LA2-AD4, were selected for this study and were purified on Protein A-Sepharose. Both mAbs were determined to be IgG1.
Table 1. Phospho-peptide sequences EGT578 is a typical 13 aa-long immunizing peptide, while EGT577 is 28 aa-long that corresponds to the full activation segment (T-loop) of CDK4
Cell culture and transfection
T98G, HCT116K7AS [Citation41] and CHO cells were cultured as described [Citation20,Citation21]. After starvation without FBS for 3 days, T98G and HCT116 K7AS cells were growth stimulated by 10% FBS. 3-MBPP1 5 μM (Calbiochem) was added 1 hour before FBS to allow CDK7 inhibition in HCT116 K7AS. CHO cells were transfected for 48 h using JetPEI (Polyplus Transfection) with 8 µg of pcDNA3 vector encoding HA-tagged CDK4 WT or HA-tagged CDK4 T172A, and Xpress-cyclin D3 [Citation20]. MCF7 cells were cultured as described [Citation42] in the presence of 5% FBS and 6 ng/ml insulin. C2C12, BT-20, MDAMB-231, SKBR3, BT20, MDAMB-361, MDAMB-468 were cultured in DMEM containing 10% FBS; MDAMB-134 VI in DMEM 10% FBS supplemented with 1% non-essential aminoacids. BT-474, ZR75.1, HCC 1569, T47D, HCC 1954, HCC 1806, HCC 1500, HCC 38, BT-549, HCC 1937, HCC 70 were maintained in RPMI media containing 10% FBS. MDAMB-436 cells were grown in DMEM:Ham’s F12 medium (1:1 mixture) with 10% FBS [Citation38].
Cells expressing cyclin D3/CDK4 fusion gene
MCF7 genome was modified as described [Citation40] by three consecutive lentiviral transductions. Cell populations were selected by FACS sorting or antibiotic (neomycin and puromycin) selection. Briefly, 10,000 trypsinized MCF7 cells were resuspended in 200 µl of thawed lentiviral supernatant previously prepared by calcium phosphate transfection of HEK293T cells as described [Citation21]. Suspended cells were plated (200 µl/well) and incubated 72 h into 96-well plates. Viral supernatant was discarded; cells were washed thrice with PBS, trypsinized and stepwize expanded twice before being selected. Cells were first transduced with a lentiviral construct driving the constitutive expression of the rTTA3 doxycyclin-reponsive transcription regulator and the DsRed fluorescent protein under the control of an IRES sequence as detailed in [Citation40]. Red fluorescent cells were then transduced with a lentiviral construct driving the constitutive expression of the humanized BirA biotin ligase of E. Coli and the neomycin phosphotransferase under the control of an IRES sequence as detailed in [Citation40]. Neomycin-resistant DsRed-positive cells were finally transduced with a lentiviral construct driving the expression of WT or T172A-mutated cyclin D3/CDK4 fusion proteins described in [Citation40,Citation43], fused to EGFP and tagged with the BirA-biotinylable AviTag as well as a puromycin resistance gene detailed in [Citation40] (). Transduced MCF7 cells were cultured as described except that expression of cyclin D3/CDK4 fusions and their biotinylation were induced for the last 24 h before lysis by adding doxycycline (1 µg/ml) and biotin (0.1 mM) ()).
Figure 1. Screening of anti-phosphoT172 CDK4 mAbs using immobilized cyclin D3/CDK4 proteic fusions.
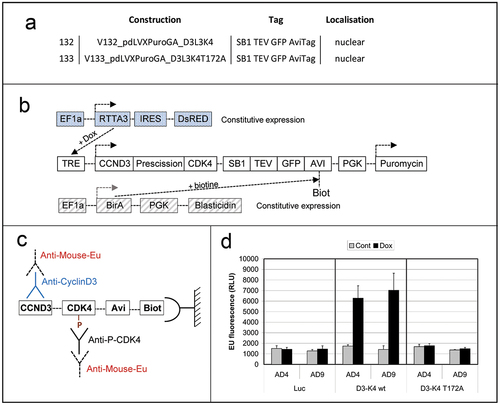
Dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA)
25 µg (proteic mass) of lysates in 100 µl of NP40 buffer [Citation18] obtained from MCF7 cells transduced as above were loaded in Multiscreen Filter plates (MSBVN1210, Merck) and incubated for 3 h at 4°C under slight agitation in the presence of 3 μl streptavidin-coated sepharose beads prewashed twice with NP40 buffer. Then, biotinylated cyclin D3/CDK4 reporter bound to streptavidin-coated beads was washed in filter plates three times with NP40 buffer and once with 1x DELFIA Wash buffer (Perkin Elmer 000063700) before overnight incubation in 50 µl of assayed immune sera or hybridoma culture supernatants, or DCS-22 monoclonal cyclin D3 antibody (Neomarkers/ThermoFisher), diluted in DELFIA assay buffer (Perkin Elmer 1244–111) supplemented with 0.1% BSA. Beads in plates were next washed four times with DELFIA wash buffer before incubation under mild shaking for 2 h at room temperature with 50 μl DELFIA Assay buffer supplemented with 0.1% BSA and 300 ng/ml Eu-N1-labeled anti mouse IgG (AD0124, Perkin Elmer). Plates were next washed 4 times with DELFIA wash solution before incubation for 10 min at room temperature with DELFIA enhancement solution (Perkin Elmer 1244–105). The solution containing the released Eu-cryptates was collected by centrifugation in a reading plate (Yellow 96 well plate low fluorescence, Perkin Elmer, AAAND-0001) and fluorescence measured after a delay of 100 μsec for 400 μsec on a TECAN Infinite Pro 2000 fluorimeter equipped with a 340 nm excitation filter and a 610 nm emission filter.
Immunoblot analyses
Equal amounts of whole cell extract protein were separated according to molecular mass and immunodetected using the following antibodies: monoclonal mouse antibodies against cyclin D3 (DCS-22) (Neomarkers/ThermoFisher); cyclin D1 (DCS-6), p16 (DCS-50), CDK4 (DCS-31) (Santa Cruz Biotechnology); p21, p27, CDK4 antibodies (respectively 12D1, D69C12, D9G3E, Cell Signaling Technology); or rabbit polyclonal (H22) CDK4 and anti-phospho-p27 (S10) antibodies (Santa Cruz Biotechnology). The purified NB8-AD9 mAb was diluted at 1.9 µg/ml for immunoblot detections. Secondary antibodies were coupled to horseradish peroxidase (Cell Signaling Technology). The proteins were detected using Western Lightning Plus ECL (Perkin Elmer), and viewed in Fusion FX gel documentation system using the Solo7S camera (Vilber Lourmat, France).
Immunoprecipitation
Co-immunoprecipitations were performed as described [Citation18,Citation20] using the following antibodies: cyclin D1 rabbit mAb (EPR2241, Abcam), cyclin D3 mouse mAb (DCS-28, Neomarkers/ThermoFisher), a mixture of the K25020 anti-p27 mAb from BD-PharMingen and the C-19 p27 polyclonal antibody (Santa Cruz Biotechnology). Immunoprecipitation of T172-phosphorylated CDK4 was performed with 3.5 µg of purified LA2-AD4 mAb and 1 mg (proteic mass) of cell lysate.
In vitro phosphorylation of cyclin D3-CDK4/6 complexes by CAK
Cyclin D3 complexes containing CDK4 and CDK6 from HCT116 K7AS cells FBS-stimulated for 16 h were immunoprecipitated using the DCS-28 cyclin D3 mAb (Neomarkers/ThermoFisher) and used as substrate for phosphorylation of CDK4 and CDK6 by CAK as described [Citation21]. The immunoprecipitates were washed three times with NP-40 lysis buffer and then three times with CAK buffer (80 mM β-glycerophosphate [pH 7.3], 15 mM MgCl2, 20 mM EGTA, and 5 mM dithiothreitol) [Citation44]. The beads were resuspended in 50 μl of CAK buffer containing protease and phosphatase inhibitors with or without 1 μg of recombinant CDK7-cyclin H-MAT1 complex (Upstate, Charlottesville, Virginia). After 1 mM ATP addition, the suspensions were incubated at 30°C for 1 h. After three washes in the appropriate buffer, the immunoprecipitated proteins were prepared for 2D gel electrophoresis analysis of CDK4 and CDK6 (C21, Santa Cruz Biotechnology).
Two-dimensional (2D)-gel electrophoresis
As described [Citation18], immunoprecipitated protein complexes were denatured in a buffer containing 7 M urea and 2 M thiourea. Alternatively, lysates were prepared directly by scraping cells in this urea buffer. Proteins were separated by isoelectric focusing on immobilized linear gradient (pH 5 to 8 [11 cm] or 3 to 10 [18 cm], Bio-Rad and GE healthcare respectively) strips, separated by SDS-PAGE and immunoblotted with rabbit monoclonal antibodies against CDK4 (D9G3E), p21 (12D1), or p27 (D69C12) (Cell Signaling Technology), rabbit polyclonal anti-p27 (C15) and anti-phospho-p27 (S10) antibody (Santa Cruz Biotechnology), and monoclonal anti-phosphotyrosine antibody (4G10, Upstate). Fc fragment specific secondary antibodies coupled to horseradish peroxidase (AB_2337937 and AB_2338506, Jackson Immunoresearch) were used for detection of p27 after co-immunoprecipitation. Chemiluminescence images of blots were captured with a Vilber-Lourmat Solo7S camera and quantified using the Bio1D software (Vilber-Lourmat).
Indirect immunofluorescence
Cells were fixed in 4% paraformaldehyde for 10 min at 4°C, then methanol for 10 min at −20°C, and permeabilized with 0.1% Triton X-100 before immunofluorescent detection. Anti-HA probe (F7, Santa Cruz Biotechnology) or anti-phospho-CDK4 (T172) monoclonal (NB8-AD9) antibodies were followed by a biotinylated anti-mouse immunoglobulin antibody (GE Healthcare) and Alexa Fluor 488-conjugated streptavidin (Jackson Immunoresearch).
ELISA
Microtitration plates (96-well, Maxisorp, Thermo scientific) were first coated with the LA2-AD4 mAb (0.75 μg/ml in carbonate buffer, pH 9.5) overnight at 4°C. After draining, the plates were blocked with PBS-0.05% Tween20 (PBS-T) plus 5% skim milk powder (PMT) for 2 h at 37°C. Wells were then washed three times with PBS-T, followed by the addition of cell lysates diluted in lysis buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 1% Na Deoxycholate, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 1 mM sodium orthovanadate, 1 mM β-glycerophosphate, 1 mM DTT, protease inhibitors (Pefabloc, leupeptin), and 10% glycerol) for an overnight incubation at 4°C under gentle agitation. After three washes with PBS-T, anti-CDK4 rabbit biotinylated-mAb (D9G3E, Cell Signaling Technology) was added (1:200 dilution in PMT) supplemented with human IgG (50 μg/ml, Jackson Immunoresearch) and horse serum (1/20, Gibco), and incubated for 1.5 h at room temperature under 500 rpm agitation. After four PBS-T washes, wells were incubated with streptavidin-Poly-HRP80 (1:3500, SDT (Stereospecific Detection Technologies) Germany) for 45 min at room temperature at 500 rpm. After four PBS-T washes, the reaction was developed for 10 min in the dark with Pierce Ultra TMB under agitation, then stopped by the addition of 2 M sulfuric acid. Absorbance was read on a microplate reader (iMark™ Microplate Absorbance Reader) at 450 nm and 655 nm. Lysis buffer was used as a blank. In parallel, a CDK4 sandwich ELISA was also developed based on the same methodology except that CDK4 DCS-35 mAb (1 μg/ml in PBS, Santa Cruz Biotechnology) was used to coat wells, and that in some experiments recombinant CDK4 protein (RayBiotech) was used instead of lysates.
Results
Screening of sera
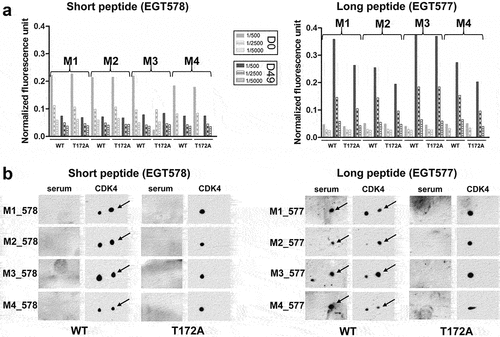
Sera obtained at day 0 (pre-immune), or at days 35 and 49 after peptide injection from the 8 mice were assayed in standard peptide ELISA with the phosphorylated versus non-phosphorylated CDK4 peptides used for immunization (Eurogentec). Day 35 and 49 sera were all reactive (), but only the sera from the 4 mice immunized with the long phospho-peptide (EGT577) did show a stronger reactivity against the phosphorylated peptide compared to the non-phosphorylated one. We went on with the screening using the DELFIA assay (characterized in ) that we developed using an immobilized cyclin D3/CDK4 proteic fusion expressed in MCF7 cells versus the same proteic fusion containing the T172A mutation (as schematized in ). This assay allowed us to show that the sera from the four mice immunized with the short peptide (EGT578) were all negative, while the sera from the four mice immunized with the long peptide (EGT577) all showed positivity against the cyclin D3/CDK4 fusion with partial phospho-specificity in the case of M1, M2 and M4 mice ()). Phospho-specific immune responses against CDK4 were confirmed on 2D-gel western blots of CHO cells transfected with WT or T172A CDK4 and cyclin D3 constructs ()). Interestingly, this assay showed that the sera from the four mice immunized with the long phospho-peptide only recognized the T172-phosphorylated CDK4 form, whereas the sera from the four mice immunized with the short phospho-peptide did not detect any CDK4 form ()). The EGT577 M1 and M4 mice were chosen for the splenocyte-myeloma cell fusion.
Figure 2. Sera evaluation by CDK4 peptide ELISA.
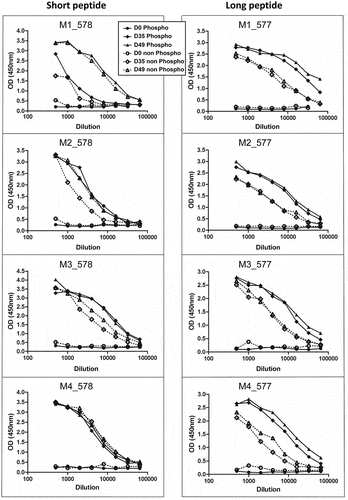
Figure 3. Evaluation of CDK4 T172-phosphospecificity of immune sera.
Hybridoma supernatants screening and selection of mAbs
Three hybridoma fusions were obtained for M1, all phospho-specific in indirect ELISA (Figure S1), and 23 hybridoma fusions for M4, with 16 displaying partial or complete phosphospecificity. Further screening of the hybridoma fusion culture supernatants with the DELFIA assay showed (Figure S2(a)) that (i) from the three M1 hybridoma fusions, only one, GB4, was positive but not phosphospecific; (ii) from the 23 M4 hybridoma, 9 were positive and more or less phospho-specific, LA2, NB8, MD8 and RF7 displaying the best responses. It should be noted that some supernatants like RF5 were only positive in the DELFIA assay.
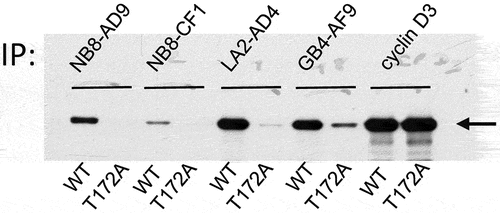
In parallel, the hydridoma fusion culture supernatants were tested on immunoblots obtained by 1D-gel separation of MCF7 cells expressing a WT (132) or T172A (133) cyclin D3/CDK4 fusion or obtained by 2D-gel separation of CHO cells transfected to express WT or T172A CDK4 and cyclin D3. GB4, JE3, LA2, LC7, MD8, NB8, RC4 and RF7 showed phospho-specificity on the cyclin D3/CDK4 fusion in 1D-gel immunoblots (Figure S2(b)) and in 2D-gel immunoblots of CDK4 transduced with cyclin D3 in CHO cells (Figure S2(c)). NB8 and LA2 were successfully cloned giving respectively two different and 14 identical clones. GB4 also yielded two apparently identical clones. NB8-AD9, NB8-CF1, LA2-AD4 and GB4-AF9 were produced ().
Figure 4. CDK4 T172-phosphospecificity of cloned mAbs.
Characterization of purified antibodies NB8-AD9 and LA2-AD4
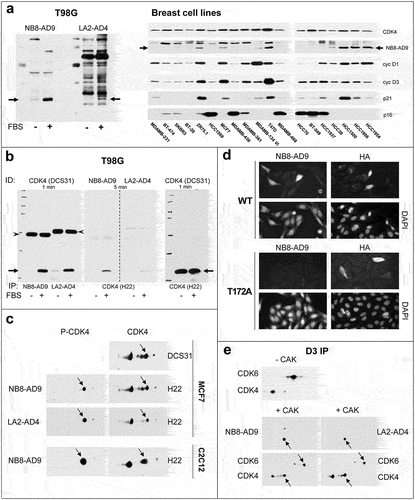
Immunoblotting experiments showed that NB8-AD9 and LA2-AD4 detect endogenous T172-phosphorylated CDK4 from human 16-h FBS-stimulated T98G cells or breast cancer cell lines () and from mouse C2C12 cells ()). As previously reported [Citation38], the T172-phosphorylation of CDK4 was undetectable using NB8-AD9 in those breast cancer cell lines that were previously documented to be unresponsive to palbociclib (HCC1569, MDAMB-436, MDAMB-468, HCC70, BT-549, HCC1937) ()). NB8-AD9 and LA2-AD4 precipitated endogenous phospho-CDK4 ()) and NB8-AD9 discriminated in immunofluorescence experiments between WT or T172A CDK4 in transfected CHO cells ()). Both NB8-AD9 and LA2-AD4 detected CDK4, but not CDK6, that were phosphorylated in vitro by CAK ()). This observation is interesting because the sequences around the phosphorylated threonine are very similar in CDK4 and CDK6, mainly differing in +1 position by the presence of a proline in CDK4 and a serine in CDK6.
Figure 5. Characterization of purified mAbs NB8-AD9 and LA2-AD4.
Several (mostly polyclonal) phospho T172-CDK4 antibodies have been commercialized recently. Although we did not intend to test all such antibodies, we compared NB8-AD9 to a few of them. (Figure S3(a,b)) shows that ThermoFisher PA5-64,482 failed to detect the WT CDK4/cyclin D3 (132) or T172A CDK4/cyclin D3 (133) fusion expressed in MCF7 cells or an overexpressed WT or T172A CDK4 protein in CHO cells, analyzed by 2D-gel immunoblotting. The antibodies commercialized by other brands (Abbexa, antibodies.com, Biorbyt, Signalway Antibody, Absci) might be identical to this antibody as they share the same datasheet pictures. 9H2L7 (ThermoFisher, #702,556), a rabbit mAb directed against a short phosphopeptide (MALTPVV) that is also present in other proteins (focadhesin, fibrocystin), which is specified for immunofluorescence or immunocytochemistry, gave only a faint signal on 2D-gel western blot (Figure S3(b)). It is finally interesting to note that technical sheets of some commercialized T172-phospho CDK4 antibodies use HeLa cells as positive control (LSBio, Abcam). However HeLa cells did not express T172-phospho CDK4 (Figure S3(c)), which we ascribe to their very high p16-INK4A (CDKN2A) expression resulting from pRb inactivation by the E7 viral oncogene of HPV.
Use of LA2-AD4 in ELISA for T172-phosphorylated CDK4
A sandwich ELISA was generated using LA2-AD4 as a capture antibody and a biotinylated rabbit mAb against CDK4 (D9G3E) as the detection antibody (). Its development was performed using lysates of CHO cells transfected with WT or T172A CDK4 and cyclin D3 constructs as positive or negative controls respectively. LA2-AD4 was preferred over NB8-AD9 because in preliminary experiments it provided higher positive/negative signal ratios in CHO cell lysates expressing WT versus T172A CDK4 or in WT CDK4 CHO lysates versus lysis buffer. Since LA2-AD4 correctly discriminated between T172-phosphorylated and non-phosphorylated CDK4, i.e. between WT or T172A CDK4 in transfected CHO cells, we used graded dilutions of these extracts of CHO cells transfected with WT CDK4 to establish a standard curve to measure the amount of T172-phosphorylated CDK4 in unknown samples ()). The absolute amount of phosphorylated CDK4 in these WT CDK4 CHO cell extracts could be determined by (i) measuring their amount of total CDK4 by comparison in a total CDK4 ELISA (using the DCS-35 CDK4 mAb as a capture antibody) with a standard curve performed by 2 fold serial dilutions (range 25 to 0.39 ng) of recombinant CDK4 ()), and (ii) measuring by 2D-gel immunoblotting the proportion of T172-phosphorylated versus total CDK4 in these WT CDK4 CHO cell extracts. The ELISA using LA2-AD4 correctly measured the variable abundance of T172-phosphorylated CDK4 from whole cell extracts of HCT116 CDK7AS/AS (expressing analog sensitive (AS) CDK7 alleles) stimulated by FBS and subjected or not subjected to CDK7 inhibition by 3-MBPP1 [Citation21,Citation23], T98G cells stimulated or not stimulated by FBS [Citation20], or breast cancer cells expressing or not expressing T172-phosphorylated CDK4 [Citation38] (). In these experiments, the highest dilution (1/640) of WT CDK4 CHO cell extracts was consistently detected and measured to contain approximatively 130 pg/ml of T172-phosphorylated CDK4 (3.9 pM = 0.39 fmoles in 100 µl). The cellular concentration of T172-phosphorylated CDK4 was calculated to be approximatively 140 pg/mg protein in stimulated HCT116 cells and 1.4 ng/mg protein in T47D cells ()).
Figure 6. T172-phosphospecific CDK4 sandwich ELISA.
Involvement of phosphorylations of p21 and p27 in CDK4 activation
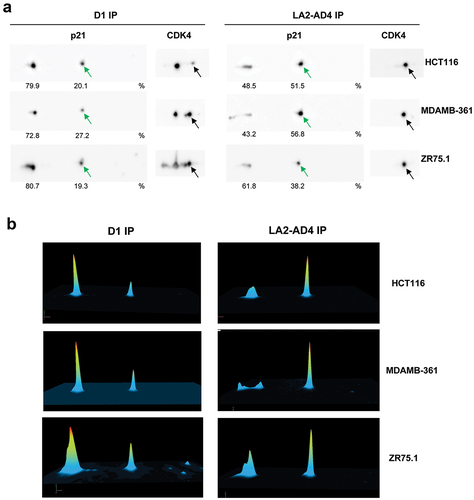
Since our phospho-CDK4 mAbs specifically immunoprecipitate phospho-T172-CDK4, we exploited LA2-AD4 to investigate which phosphorylated p21 or p27 forms are associated to T172-phosphorylated CDK4. We have previously shown that p21 is mainly phosphorylated on S130 and to a lesser extent on S98 [Citation21] and that 50% of p27 is phosphorylated on S10 [Citation20]. However, p27 is reported to be also phosphorylated on other residues including T157, T187, T198 and Y74, Y88 and Y89 [Citation45]. Previously, we fully characterized the 2D-gel electrophoresis profile of p21 using phosphatase treatment, mutagenesis of phosphorylation sites and in vitro phosphorylation of p21 by various kinases [Citation21]. We co-immunoprecipitated p21 through its association with cyclin D1 or T172-phosphorylated CDK4 (using LA2-AD4) and analyzed its phosphorylation profile as separated by 2D-gel electrophoresis (). In the three cell lines studied (HCT116, MDA-MB361 and ZR75.1), we observed a marked enrichment of the form of p21 that is phosphorylated solely on S130 in co-immunoprecipitates of T172-phosphorylated CDK4 as compared to p21 that is bound to total CDK4 in immunoprecipitated cyclin D1-CDK4 complexes ().
Figure 7. S130-phosphorylated p21 is enriched in phospho-CDK4 immunoprecipitation.
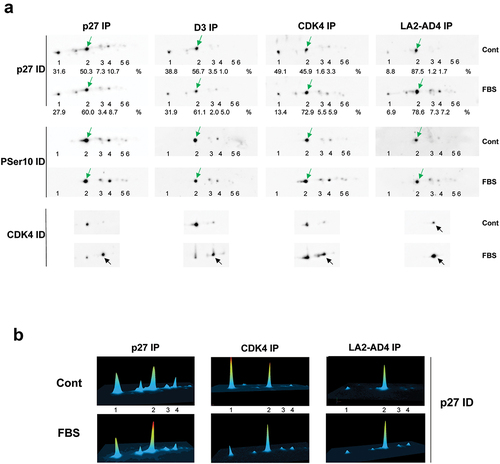
In similar experiments, we compared the 2D-gel separation of p27 bound to T172-phosphorylated CDK4 co-immunoprecipitated using LA2-AD4 to total p27 (immunoprecipitated using p27 antibody) or p27 bound to cyclin D-CDK4 complexes (co-immunoprecipitated using CDK4 or cyclin D antibodies) ( and Figure S4). As previously reported [Citation20,Citation24], the relative abundance of the T172-phosphorylated form of CDK4 increased in FBS-stimulated T98G cells, not only in complexes immunoprecipitated using CDK4 and cyclin D3 antibodies but also in CDK4 bound to p27 ()). The relative abundance of the p27 form that is phosphorylated solely on S10 (previously identified using 32P phosphate incorporation [Citation20] and a S10-phosphospecific p27 antibody) also increased in response to FBS stimulation of T98G cells () in total p27 (slightly) and more in p27 bound to CDK4 complexes. This single S10-phosphorylated form was the only enriched abundant form of p27 in LA2-AD4 immunoprecipitations (). Accordingly, anti-phosphotyrosine antibodies (4G10 or phospho-Y88-p27 antibody) failed to detect p27 in these co-immunoprecipitations (Figure S4). We also confirmed the preferential association of the single S10-phosphorylated p27 form with T172-phosphorylated CDK4 in ZR75.1 (Figure S5(a)) and MDA-MB361 (Figure S5(b)) breast cancer cells.
Figure 8. S10-phosphorylated p27 is enriched in phospho-CDK4 immunoprecipitation.
Discussion
Because the activity of CDK4 is not restricted by stoichiometrically abundant inhibitory phosphorylations [Citation7,Citation16,Citation20], at variance with Y15 and T14 phosphorylations of CDK2 and CDK1 [Citation46,Citation47], and also because the T-loop T172 phosphorylation of CDK4 depends on previous steps including binding to cyclins D [Citation3], this phosphorylation represents the rate-limiting step of CDK4 activation. As such it integrates the various mitogenic and antimitogenic pathways to commit the cell cycle at restriction point in G1 phase [Citation7,Citation21,Citation23]. The T172 phosphorylation is required for the opening of the catalytic site of CDK4 [Citation48,Citation49]. It is thus not only critical for the activity of CDK4 complexes [Citation16,Citation20], but also for the engagement of CDK4 by ATP-competitive inhibitors like palbociclib. The lack of T172-phosphorylation of CDK4 that we observed in some tumors and cancer cell lines, partially due to high p16 CDKN2A expression [Citation38], likely explains why in such models palbociclib is observed not to bind to its target [Citation50,Citation51], leading to complete insensitivity to the anti-cancer drug.
Despite these critical roles, the activating T172 phosphorylation of CDK4 has been very infrequently investigated so far. Because CDK4 is a low abundance protein, the initial two-dimensional analysis of its tryptic 32P-labeled phosphopeptides might have been a rather heroic approach to demonstrate the regulation of endogenous CDK4 by T172-phosphorylation [Citation17]. For about one decade, CDK4 T172-phosphorylation was no longer studied directly, due to lack of easy methodological tools, and it was only inferred from CDK4 activity. For years, we have circumvented this difficulty by exploiting the high-resolution power of the 2D-IEF gel electrophoresis to separate the different phospho-forms of CDK4 and other CDKs [Citation46,Citation47]. We identified the most negatively charged forms as the T172-phosphorylated CDK4 thanks to various approaches including [32P]phosphate incorporation, immunoblotting using a first trial rabbit polyclonal phospho-specific-CDK4 (T172) antibody (shared by Cell Signaling Technology, this antibody was deemed insufficient for commercialization and could not be reproduced), in vitro phosphorylation by recombinant CAK, and analysis of T172A-mutated CDK4 [Citation20,Citation26].
Here we wanted to complete this array of experimental tools in a thorough attempt to generate mAbs specific to T172-phosphorylated CDK4. Nearly all phospho-specific antibodies are now produced using as antigens chemically synthesized phosphopeptides coupled to a weakly immunogenic carrier protein such as KLH [Citation52–54] to immunize animals to prepare either polyclonal antibodies, rendered specific through extensive purification [Citation55], or monoclonal antibodies. The generation of phospho-specific mAbs, by screening large numbers of candidate hybridoma clones against phosphorylated and non-phosphorylated targets, has been considered more challenging by the rarity of phospho-specific mAb clones [Citation56], sometime estimated to be only 0.1–5% [Citation57,Citation58]. Typically, the desired phosphorylated amino acid is placed in the middle of a 10–14 amino acids long peptide in order to allow sufficient exposure of the phosphorylation, while restricting antibodies to the non-phosphorylated portions of the peptide sequence. Because antibody epitopes can comprise as few as three or four amino acid residues, it has been recommended that, when feasible, only three residues are placed C-terminal to phosphoserine/threonine, as antibodies directed against unphosphorylated epitopes are preferentially produced if there are more residues on this external side [Citation52]. On the other hand, it is recognized that the peptide’s conformation might be far from the natural state of a cellular protein. Some first phospho-specific antibodies were successfully prepared by immunization of rabbits with native purified protein. However, many phosphorylated proteins are believed to undergo rapid dephosphorylation during immunization, leading to the loss of the desired phospho-epitope. Moreover, holoproteins generally contain multiple immunogenic epitopes, decreasing the probability of clonal dominance for the desired phospho-specific epitope.
With all these possible limitations in mind, we compared mice immunizations using a short KLH-coupled 13aa phosphopeptide designed according to current prediction algorithms or a KLH-coupled 28aa phosphopeptide that corresponds to the full activation segment (T-loop) of CDK4, which we expected to better preserve the conformation and exposure of the phospho-epitope. Whereas the short phosphopeptide completely failed to generate a phospho-specific immune response against CDK4, plausibly reproducing previous failures, the long “T-loop” KLH-coupled phosphopeptide proved to be exceptionally immunogenic in the four assayed mice. Indeed, even before hybridoma cloning, the sera of the four mice only recognized the T172-phosphorylated form of CDK4 but not at all its unphosphorylated forms, at least when assayed by immunoblotting. This unique yield of phospho-specificity was confirmed (with two exceptions in the DELFIA screening using the cyclin D3/CDK4 fusion) at the splenocyte fusion and hybridoma cloning steps, representing to our knowledge the first successful generation of mAbs specific for T172-phosphorylated CDK4. Interestingly, the mAbs raised in the present study did not display any cross-reactivity against T177-phosphorylated CDK6 despite the high identity of the activation segments of CDK4 and CDK6, which differ essentially by the residue at +1 position after the phosphorylated threonine (Pro in CDK4, Ser in CDK6) [Citation26]. Given the critical influence of proline in secondary structures, this observation indicates again that maintaining the conformation of the epitope might have been important. Indeed this characteristic differs from the first (non-commercialized) polyclonal antibody produced by Cell Signaling Technology against a short CDK4 phosphopeptide, which recognizes equally the phosphorylation sites of CDK4 and CDK6 [Citation20,Citation26]. Whether our approach based on the use of the whole phosphorylated activation segment as antigen might be generalized to other protein kinases including CDK6 remain to be explored.
Our present antibodies allow the direct detection of endogenous phospho-CDK4 from human and mouse cells by a variety of techniques including immunoblotting, immunoprecipitation and immunofluorescence (the latter depending on overexpression). To achieve a maximal sensitivity of the immunoblotting detection of T172-phosphorylated CDK4 form, endogenous CDK4 complexes might be first concentrated by immunoprecipitation using antibodies directed against CDK4 (such as EPR17525 from Abcam) or cyclins D. Used as capture antibody, LA2-AD4 also allowed us to develop a robust and sensitive ELISA to measure the concentration of T172-phosphorylated CDK4 in various cellular contexts. The NB8-AD9 antibody most likely recognizes the denatured CDK4 better, as it has superior performance and specificity in immunoblotting and immunofluorescent applications, whereas LA2-AD4 displays greater reactivity in assays involving non-denatured CDK4 including the DELFIA phospho-specific detection of the cyclin D3/CDK4 proteic fusion, immunoprecipitation and specific capture of T172-phosphorylated CDK4 in ELISA. The success of these achievements should be appreciated considering the very low cellular concentration of CDK4 (likely 1 or 2 orders of magnitude lower than other signal transduction kinases such as ERK2, AKT1 or other CDKs including CDK1, based on spectral count evidence [Citation59,Citation60]) and the small relative proportion of T172-phosphorylated CDK4 that may represent not more than 5% of total CDK4 in normal proliferating cells as evaluated by 2D-gel separations [Citation20].
Importantly, the LA2-AD4 allows for the first time to co-immunoprecipitate proteins specifically bound to T172-phosphorylated CDK4. The mechanisms involved in the regulation of CDK4 phosphorylation inside its different complexes with D-type cyclins stabilized by p21 or p27 remain poorly known and debated. Various phosphorylations of p27 and p21 were claimed to facilitate the activation of CDK4 and/or its T172-phosphorylation, however a direct association of the different phospho-forms of p21 or p27 with T172-phosphorylated CDK4 could not be verified. We have previously considered that the likelihood of a specific phosphorylated residue of p21 or p27 playing a significant role in the activation or activity of CDK4 bound to p21 or p27, would depend on the relative abundance of this phosphorylation [Citation7]. Our previous 2D-IEF analyses have shown that the S130-phosphorylation is by far the most abundant one in p21, that it is highly regulated and that it is required for CDK7-dependent T172-phosphorylation of CDK4 [Citation21]. Consistent with these observations, we showed here that the p21 form phosphorylated only at S130 is selectively enriched in CDK4 complexes precipitated by LA2-AD4 in different cell lines. p21 S130 can be phosphorylated by numerous kinases including various MAP kinases [Citation27,Citation61,Citation62], CDK2 [Citation21,Citation63,Citation64] and, most efficiently when p21 is engaged in cyclin D-CDK4 complexes, by CDK4 and CDK6 [Citation21]. Therefore, the direct association of S130-phosphorylated p21 with T172-phosphorylated CDK4 fully supports our conclusions that the S130-phosphorylation of p21 plays a major role both in the initiation of the activation of p21-stabilized cyclin D-CDK4 complexes [Citation27] and in its maintenance or amplification by positive feedback mechanisms mediated by CDK4/6 or even CDK2 [Citation21].
We also preliminarily explored the association of T172-phosphorylated CDK4 with phospho-forms of p27. Phosphorylations of p27 at one of its tyrosine residues (Y88 [Citation65,Citation66], Y89 [Citation22] or Y74 [Citation28]) were reported to facilitate the activation and activating phosphorylation of p27-bound CDK complexes, including cyclin D-CDK4. However, we observed here that p27 phosphorylated solely at S10 is the most enriched form in immunoprecipitates of T172-phosphorylated CDK4 using LA2-AD4. As initially characterized by phosphopeptide mapping and phosphoamino acid analysis, phosphorylation at S10 accounts for approximately 70% of the total phosphorylation of p27 (whereas tyrosine phosphorylation was undetectable) [Citation67]. 2D-IEF separation of immunoprecipitated p27 combined with immunoblotting with S10-phosphospecific p27 antibody also demonstrated S10 as the major phosphorylation site of p27 in a variety of cell types [Citation20,Citation68]. By contrast we failed to detect a significant abundance of Y-phosphorylation in previous experiments and in the present ones (Figure S4) using the Y88-phosphospecific p27 mAb (shared by Ludger Hengst [Citation69]) or 4G10 phosphotyrosine mAb. p27 S10 has been reported as a substrate for a variety of protein kinases, including KIS [Citation70], KSHV v-cyclin-CDK6 [Citation71], DYRK1A [Citation72], PCTAIRE1 [Citation73] and JNKs [Citation74]. S10 phosphorylation is generally associated with increased stability and cytoplasmic translocation of p27 [Citation67,Citation75,Citation76]. However, other studies have shown that S10 phosphorylation of p27 is unlikely to be fully sufficient for nucleus export [Citation20,Citation68,Citation77] as the S10-phosphorylation-dependent association of p27 with CRM1 exportin is counterbalanced by the nuclear reentry of p27, mediated by the association of its strong NLS with importin α [Citation78]. Despite the roles of S10 phosphorylation in cytoplasmic relocalization of p27 and its involvement in cell migration [Citation76,Citation79], we and others previously observed the association of CDK4 with S10-phosphorylated p27 [Citation20,Citation68]. The investigation of the structural impact of S10-phosphorylation of p27 on the activation and activity of cyclin-CDK complexes has been hampered because p27 is an intrinsically unstructured protein that folds only upon encountering interactors like cyclins [Citation80] and because crystals of cyclin A-CDK2 [Citation81] and cyclin D1-CDK4 [Citation28] could only be obtained with the kinase-inhibitory domain of p27 (residues 28–96 or 25–93 respectively). These lack the N-terminal extremity and all the C-terminal part of p27, and hence its major phosphorylation sites. Like the T187 residue of p27 and the S130 residue of p21, S10 is followed by a proline in p27, indicating that it could be a substrate for different proline-directed kinases including CDKs. Further experiments should thus decipher whether the association of T172-phosphorylated CDK4 with S10-phosphorylated p27 might reflect a facilitating role of S10-phosphorylation in CDK4 activation, phosphorylation of S10 by activated CDK4 itself, or both as in the bidirectional causal relationship that we demonstrated between S130-phosphorylation of p21 and T172-phosphorylation of p21-bound CDK4 [Citation21]. The subcellular location of cyclin D-CDK4 complexes is largely determined by their interaction with p21 or p27 [Citation20,Citation82]. We previously observed that the relocation of cyclin D3-CDK4 to cytoplasm, induced by binding to a p27 truncated from its C-terminal NLS, does not impair CDK4 T172-phosphorylation [Citation20]. One hypothesis could thus be that the dynamic nucleus/cytoplasm shuttling mediated in part by S10-phosphorylation of p27 may facilitate a cooperation between the cytoplasmic CDK4-activating kinases that we postulated and nuclear CAK/CDK7 to initiate and sustain the T172-phosphorylation of CDK4 [Citation7,Citation21,Citation27].
To conclude, the use as antigen of the full activation segment rather than a short phosphopeptide allowed the successful generation of new phospho-specific mAbs to detect and measure in a variety of assays the phosphorylation of CDK4 at T172 that governs the cell cycle commitment. These unique reagents will greatly help to improve our understanding of the regulatory mechanisms of CDK4 activation in normal and cancer cells.
Supplemental Material
Download Zip (14.2 MB)Acknowledgements
We thank Prof. Jacques Dumont for continued interest and support, Olivier Sintobin for technical help in the generation of the lentiviral constructs and the generation of cell lines, Florence Lemmens for contribution to the development of the DELFIA assay, and Drs Ludger Hengst and Heidelinde Jaekel (Innsbruck Medical University) for sharing the Y88-phospho p27 mAb and purified recombinant p27 (phosphorylated or not phosphorylated by Abl). We also thank Dr Hengst for discussions regarding cyclin D/CDK4 proteic fusions.
Disclosure statement
KC, VV, SP, JMP, RC, ER and PPR report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6(4):353–367.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166.
- Bockstaele L, Coulonval K, Kooken H, et al. Regulation of CDK4. Cell Div. 2006;1(1):25.
- Asghar U, Witkiewicz AK, Turner NC, et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–146.
- Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28(33):2925–2939.
- Blain SW. Switching cyclin D-Cdk4 kinase activity on and off. Cell Cycle. 2008;7(7):892–898.
- Paternot S, Bockstaele L, Bisteau X, et al. Rb inactivation in cell cycle and cancer: the puzzle of highly regulated activating phosphorylation of CDK4 versus constitutively active CDK-activating kinase. Cell Cycle. 2010;9(4):689–699.
- Depoortere F, Van Keymeulen A, Lukas J, et al. A requirement for cyclin D3-cyclin-dependent kinase (cdk)-4 assembly in the cyclic adenosine monophosphate-dependent proliferation of thyrocytes. J Cell Biol. 1998;140(6):1427–1439.
- Cheng M, Sexl V, Sherr CJ, et al. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc Natl Acad Sci U S A. 1998;95(3):1091–1096.
- Cheng M, Olivier P, Diehl JA, et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18(6):1571–1583.
- Blain SW, Montalvo E, Massague J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem. 1997;272(41):25863–25872.
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512.
- LaBaer J, Garrett MD, Stevenson LF, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11(7):847–862.
- Reynisdottir I, Massague J. The subcellular locations of p15(Ink4b) and p27(Kip1) coordinate their inhibitory interactions with cdk4 and cdk2. Genes Dev. 1997;11(4):492–503.
- Depoortere F, Pirson I, Bartek J, et al. Transforming growth factor beta(1) selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-cdk4 with nuclear p27(kip1). Mol Biol Cell. 2000;11(3):1061–1076.
- Kato JY, Matsuoka M, Strom DK, et al. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Cell Biol. 1994;14(4):2713–2721.
- Kato JY, Matsuoka M, Polyak K, et al. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell. 1994;79(3):487–496.
- Coulonval K, Bockstaele L, Paternot S, et al. The cyclin D3-CDK4-p27kip1 holoenzyme in thyroid epithelial cells: activation by TSH, inhibition by TGFbeta, and phosphorylations of its subunits demonstrated by two-dimensional gel electrophoresis. Exp Cell Res. 2003;291(1):135–149.
- Paternot S, Coulonval K, Dumont JE, et al. Cyclic AMP-dependent phosphorylation of cyclin D3-bound CDK4 determines the passage through the cell cycle restriction point in thyroid epithelial cells. J Biol Chem. 2003;278(29):26533–26540.
- Bockstaele L, Kooken H, Libert F, et al. Regulated activating Thr172 phosphorylation of cyclin-dependent kinase 4(CDK4): its relationship with cyclins and CDK “inhibitors”. Mol Cell Biol. 2006;26(13):5070–5085.
- Bisteau X, Paternot S, Colleoni B, et al. CDK4 T172 phosphorylation is central in a CDK7-dependent bidirectional CDK4/CDK2 interplay mediated by p21 phosphorylation at the restriction point. PLoS Genet. 2013;9(5):e1003546.
- Ray A, James MK, Larochelle S, et al. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol. 2009;29(4):986–999.
- Merzel-Schachter M, Merrick KA, Larochelle S, et al. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell. 2013;50(2):250–260.
- Paternot S, Roger PP. Combined inhibition of MEK and mammalian target of rapamycin abolishes phosphorylation of cyclin-dependent kinase 4 in glioblastoma cell lines and prevents their proliferation. Cancer Res. 2009;69(11):4577–4581.
- Rocha AS, Paternot S, Coulonval K, et al. Cyclic AMP inhibits the proliferation of thyroid carcinoma cell lines through regulation of CDK4 phosphorylation. Mol Biol Cell. 2008;19(11):4814–4825.
- Bockstaele L, Bisteau X, Paternot S, et al. Differential regulation of cyclin-dependent kinase 4 (CDK4) and CDK6, evidence that CDK4 might not be activated by CDK7, and design of a CDK6 activating mutation. Mol Cell Biol. 2009;29(15):4188–4200.
- Colleoni B, Paternot S, Pita JM, et al. JNKs function as CDK4-activating kinases by phosphorylating CDK4 and p21. Oncogene. 2017;36(30):4349–4361.
- Guiley KZ, Stevenson JW, and Lou K, et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science. 2019;366(6471):eaaw2106.
- Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3(1):39–55.
- Choi YJ, Li X, Hydbring P, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22(4):438–451.
- Puyol M, Martin A, Dubus P, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18(1):63–73.
- Reddy HK, Grana X, Dhanasekaran DN, et al. Requirement of Cdk4 for v-Ha-ras-induced breast tumorigenesis and activation of the v-ras-induced senescence program by the R24C mutation. Genes Cancer. 2010;1(1):69–80.
- Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016;18(1):17.
- Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17(4):425–439.
- Hortobagyi GN, Stemmer SM, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375(18):1738–1748.
- Knudsen ES, Shapiro GI, Keyomarsi K. Selective CDK4/6 inhibitors: biologic outcomes, determinants of sensitivity, mechanisms of resistance, combinatorial approaches, and pharmacodynamic biomarkers. Am Soc Clin Oncol Educ Book. 2020;40(40):115–126.
- Alvarez-Fernandez M, Malumbres M. Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell. 2020;37(4):514–529.
- Raspe E, Coulonval K, Pita JM, et al. CDK4 phosphorylation status and a linked gene expression profile predict sensitivity to palbociclib. EMBO Mol Med. 2017;9(8):1052–1066.
- Lolli G, Johnson LN. Recognition of Cdk2 by Cdk7. Proteins. 2007;67(4):1048–1059.
- Raspe E, Roger PP, Coulonval K, et al. inventors. Immobilised cyclin-dependent kinase 4 fusion proteins and uses thereof. 2015 US Patent App. 14/425,380
- Larochelle S, Merrick KA, Terret ME, et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25(6):839–850.
- Paternot S, Colleoni B, Bisteau X, et al. The CDK4/CDK6 inhibitor PD0332991 paradoxically stabilizes activated cyclin D3-CDK4/6 complexes. Cell Cycle. 2014;13(18):2879–2888.
- Rao RN, Stamm NB, Otto K, et al. Conditional transformation of rat embryo fibroblast cells by a cyclin D1-cdk4 fusion gene. Oncogene. 1999;18(46):6343–6356.
- Matsuoka M, Kato JY, Fisher RP, et al. Activation of cyclin-dependent kinase 4 (cdk4) by mouse MO15-associated kinase. Mol Cell Biol. 1994;14(11):7265–7275.
- Bencivenga D, Caldarelli I, Stampone E, et al. p27(Kip1) and human cancers: a reappraisal of a still enigmatic protein. Cancer Lett. 2017;403:354–365.
- Coulonval K, Bockstaele L, Paternot S, et al. Phosphorylations of cyclin-dependent kinase 2 revisited using two-dimensional gel electrophoresis. J Biol Chem. 2003;278(52):52052–52060.
- Coulonval K, Kooken H, Roger PP. Coupling of T161 and T14 phosphorylations protects cyclin B-CDK1 from premature activation. Mol Biol Cell. 2011;22(1):3971–3985.
- Day PJ, Cleasby A, Tickle IJ, et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci U S A. 2009;106(11):4166–4170.
- Takaki T, Echalier A, Brown NR, et al. The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc Natl Acad Sci U S A. 2009;106(11):4171–4176.
- Green JL, Okerberg ES, Sejd J, et al. Direct CDKN2 modulation of CDK4 alters target engagement of CDK4 inhibitor drugs. Mol Cancer Ther. 2019;18(4):771–779.
- Nomanbhoy TK, Sharma G, Brown H, et al. Chemoproteomic evaluation of target engagement by the cyclin-dependent kinase 4 and 6 inhibitor palbociclib correlates with cancer cell response. Biochemistry. 2016;55(38):5434–5441.
- Taya Y, Nakajima K, Yoshizawa-Kumagaye K, et al. Generation and application of phospho-specific antibodies for p53 and pRB. Methods Mol Biol. 2003;223:17–26.
- Archuleta AJ, Stutzke CA, Nixon KM, et al. Optimized protocol to make phospho-specific antibodies that work. Methods Mol Biol. 2011;717:69–88.
- Brumbaugh K, Liao WC, Houchins JP, et al. Phosphosite-specific antibodies: a brief update on generation and applications. Methods Mol Biol. 2017;1554:1–40.
- Arur S, Schedl T. Generation and purification of highly specific antibodies for detecting post-translationally modified proteins in vivo. Nat Protoc. 2014;9(2):375–395.
- Koerber JT, Thomsen ND, Hannigan BT, et al. Nature-inspired design of motif-specific antibody scaffolds. Nat Biotechnol. 2013;31(10):916–921.
- DiGiovanna MP, Stern DF. Activation state-specific monoclonal antibody detects tyrosine phosphorylated p185neu/erbB-2 in a subset of human breast tumors overexpressing this receptor. Cancer Res. 1995;55(9):1946–1955.
- Dopfer EP, Schopf B, Louis-Dit-Sully C, et al. Analysis of novel phospho-ITAM specific antibodies in a S2 reconstitution system for TCR-CD3 signalling. Immunol Lett. 2010;130(1–2):43–50.
- Huttlin EL, Jedrychowski MP, Elias JE, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143(7):1174–1189.
- Ochoa D, Jarnuczak AF, Vieitez C, et al. The functional landscape of the human phosphoproteome. Nat Biotechnol. 2020;38(3):365–373.
- Kim GY, Mercer SE, Ewton DZ, et al. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J Biol Chem. 2002;277(33):29792–29802.
- Hwang CY, Lee C, Kwon KS. Extracellular signal-regulated kinase 2-dependent phosphorylation induces cytoplasmic localization and degradation of p21Cip1. Mol Cell Biol. 2009;29(12):3379–3389.
- Zhu H, Nie L, Maki CG. Cdk2-dependent Inhibition of p21 stability via a C-terminal cyclin-binding motif. J Biol Chem. 2005;280(32):29282–29288.
- Bornstein G, Bloom J, Sitry-Shevah D, et al. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278(28):25752–25757.
- Jakel H, Peschel I, Kunze C, et al. Regulation of p27 (Kip1) by mitogen-induced tyrosine phosphorylation. Cell Cycle. 2012;11(10):1910–1917.
- Patel P, Asbach B, Shteyn E, et al. Brk/Protein tyrosine kinase 6 phosphorylates p27KIP1, regulating the activity of cyclin D-cyclin-dependent kinase 4. Mol Cell Biol. 2015;35(9):1506–1522.
- Ishida N, Kitagawa M, Hatakeyama S, et al. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem. 2000;275(33):25146–25154.
- Bencivenga D, Tramontano A, Borgia A, et al. P27Kip1 serine 10 phosphorylation determines its metabolism and interaction with cyclin-dependent kinases. Cell Cycle. 2014;13(23):3768–3782.
- Grimmler M, Wang Y, Mund T, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell. 2007;128(2):269–280.
- Boehm M, Yoshimoto T, Crook MF, et al. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21(13):3390–3401.
- Sarek G, Jarviluoma A, Ojala PM. KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood. 2006;107(2):725–732.
- Soppa U, Schumacher J, Florencio Ortiz V, et al. The down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle. 2014;13(13):2084–2100.
- Yanagi T, Krajewska M, Matsuzawa S, et al. PCTAIRE1 phosphorylates p27 and regulates mitosis in cancer cells. Cancer Res. 2014;74(20):5795–5807.
- Mielke K. Growth-arrest-dependent expression and phosphorylation of p27kip at serine10 is mediated by the JNK pathway in C6 glioma cells. Mol Cell Neurosci. 2008;38(3):301–311.
- Ishida N, Hara T, Kamura T, et al. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277(17):14355–14358.
- Besson A, Gurian-West M, Chen X, et al. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20(1):47–64.
- Delmas C, Aragou N, Poussard S, et al. MAP kinase-dependent degradation of p27Kip1 by calpains in choroidal melanoma cells. Requirement of p27Kip1 nuclear export. J Biol Chem. 2003;278(14):12443–12451.
- Shin I, Rotty J, Wu FY, et al. Phosphorylation of p27Kip1 at Thr-157 interferes with its association with importin alpha during G1 and prevents nuclear re-entry. J Biol Chem. 2005;280(7):6055–6063.
- McAllister SS, Becker-Hapak M, Pintucci G, et al. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23(1):216–228.
- Lacy ER, Filippov I, Lewis WS, et al. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11(4):358–364.
- Russo AA, Jeffrey PD, Patten AK, et al. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382(6589):325–331.
- Reynisdottir I, Polyak K, Iavarone A, et al. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995;9(15):1831–1845.