
ABSTRACT
Studies in Caenorhabditis elegans have revealed that even a genetically identical population of animals exposed to the same environment displays a remarkable level of variability in individual lifespan. Stochasticity factors, occurring seemingly by chance or at random, are thought to account for a large part of this variability. Recent studies in our lab using C. elegans now revealed that naturally occurring variations in the levels of reactive oxygen species experienced early in life contribute to the observed lifespan variability, and likely serve as stochasticity factors in aging. Here, we will highlight how developmental events can positively shape lifespan and stress responses via a redox-sensitive epigenetic regulator, and discuss the outstanding questions and future directions on the complex relationship between reactive oxygen species and aging.
Introduction
Historically, reactive oxygen species (ROS), particular hydrogen peroxide, and superoxide anions, have been viewed as undesirable by products of respiration, highly reactive, and hence to be avoided at all costs. Based on their ability to cause irreversible oxidative damage to cellular macromolecules, ROS were held responsible for many of the phenotypes associated with aging and disease, a hypothesis known as the free radical theory of aging [Citation1]. Although the conclusion that increased oxidative damage contributes to the physiological decline of organisms over time is likely still valid, the perspective on ROS and its role in longevity has shifted dramatically over the years [Citation2–4]. In fact, entire research fields have emerged that focus on the beneficial effects of ROS in aging and disease, and many excellent review articles have been published on this exciting topic [Citation2,Citation3,Citation5–8]. It is now generally accepted that ROS concentration, subcellular location, and length of ROS exposure are all critical factors that ultimately determine whether ROS have beneficial or detrimental effects [Citation9–11]. This article will focus on yet another facet of ROS biology, the observation that even when only experienced very transiently at the appropriate times in life, ROS can have profoundly beneficial effects later in life [Citation12–16]. We will discuss effective time windows in life, and the potential mechanisms by which transient exposure to ROS can exert persistent effects on stress response pathways, resistance to future stressors, and ultimately lifespan.
ROS-induced hormesis – a health and lifespan promoting phenomenon
Hormesis, which is defined as the beneficial consequence elicited by the exposure of organisms to mild, nontoxic stressors, has been a recognized health and lifespan-promoting phenomenon for many years [Citation17]. Several prominent hormesis studies involving ROS as the instigating stressor have been conducted in C. elegans, where oxidant levels were either transiently manipulated by adding exogenous anti- or pro-oxidants or by deleting pro- or antioxidant genes at defined times during life [Citation10,Citation18,Citation19]. For instance, disruption of the electron transport chain (ETC) via the knockdown of select ETC components substantially extended lifespan in C. elegans [Citation20,Citation21], particularly when done during development [Citation12]. Importantly, this increase in longevity was abolished by interventions that attenuated ROS production, indicating that the beneficial effects are, at least in part, mediated by accumulating ROS [Citation16,Citation22]. Similar lifespan extensions have been observed when young adult worms were cultivated on low concentrations of ROS-generating compounds, such as paraquat or juglone [Citation16,Citation23–25]. Moreover, increased ROS production also appear to participate in the observed lifespan extension in response to the transient inhibition of the insulin/insulin-like growth factor 1 [Citation26], mammalian target of rapamycin (mTOR) [Citation27], and hypoxia [Citation28,Citation29]. Importantly, the beneficial effects of low levels of ROS on lifespan seem to be conserved [Citation30,Citation31] and might apply to mammals as well [Citation15,Citation32,Citation33]. Mice heterozygous for the Mlck1 gene involved in ubiquinone biosynthesis have increased levels of ROS in the mitochondria and longer lifespan [Citation15]. Additionally, a lifespan extending role for ROS has been observed in mice supplemented with D-glucosamine [Citation32] or subjected to hypothermia [Citation34,Citation35], conditions known to increase ROS formation. While these data support a potential role for ROS-mediated hormetic effects in higher organisms, the timing requirements for these actions, to our knowledge, have not been characterized. Further studies are necessary to determine whether specific changes in ROS levels early in life are sufficient to extend mammalian lifespan as has been observed in C. elegans. The hormetic effects following ROS exposure are thought to be achieved through a cooperative effort of several biological processes. The primary mechanism involves the adaptive increase in expression of stress responsive genes, which encode proteins directly involved in ROS scavenging and detoxification, such as catalase and superoxide dismutase and which prepares cells to withstand future ROS exposure without accumulating damage (reviewed in [Citation8,Citation36]). In addition, increased expression of general stress response factors such as heat shock proteins [Citation37] and/or activation of select defense pathways such as autophagy [Citation38], the unfolded protein response [Citation39], or the cellular response to hypoxia [Citation28,Citation29] may also contribute to the increase in stress resistance following transient ROS exposure. Changes to mitochondrial metabolism may also contribute to hormesis [Citation40,Citation41], particularly under circumstances of ROS-mediated hormesis following dietary restriction [Citation8].
Naturally occurring hormesis events in c. elegans
For many years, detecting changes in the endogenous ROS levels relied heavily on indirect and often very qualitative readouts, such as western blot analysis of oxidatively damaged proteins [Citation42]. More quantitative approaches analyzed changes in the cysteine oxidation states of redox-sensitive proteins [Citation43], or the levels of reduced and oxidized glutathione as read-out for endogenous redox states [Citation44]. However, the spatiotemporal resolution of these approaches is low and limited to cell or organismal population studies, and information about subcellular differences or cell-to-cell variations in ROS levels or redox states are sparse. The real breakthrough came with the development of genetically encoded redox biosensor proteins, including the H2O2 sensor HyPer and the GSSG/GSH sensor Grx1-roGFP2 [Citation45,Citation46]. Ratiometric and fluorescent, these proteins sensitively report on changes in peroxide levels or redox states on both compartmental, cellular, and tissue-wide levels [Citation45]. Implementing these redox sensor systems in C. elegans, an isogenic model system that can be easily age-matched and synchronized, revealed that the average levels of endogenous ROS fluctuate quite significantly during the lifetime of the animals [Citation14,Citation47,Citation48]. While the average redox state of developing C. elegans larvae is highly oxidizing, it rapidly becomes reducing as the worms enter young adulthood, only to get increasingly more oxidizing as the worms age [Citation14,Citation46,Citation47]. What became even more apparent when studying these redox sensor expressing worms, however, was the large heterogeneity in redox states among individuals of a well-synchronized population [Citation14,Citation47]. This variability was particularly noticeable during the larval development. Within 2 days of hatching and despite being cultured together in the same controlled environment the entire time, about 10% of the larval population displayed significantly higher levels of endogenous ROS than the rest of the population () [Citation14,Citation47]. As the worms matured, however, this difference in redox states disappeared and all worms appeared to be similarly reduced during their reproductive phase. The physiological consequences of higher or lower than average levels of endogenous ROS during development were revealed upon sorting the larval worms according to their endogenous redox states, and conducting longitudinal analyses. These studies showed that larvae with a more oxidizing redox state showed enhanced resistance to heat and oxidative stress, a more reduced redox state later in life and, most importantly, a longer lifespan than the more reduced larvae ( [Citation14]). Treating the oxidized subpopulation with the antioxidant N-acetylcysteine for a 10 h period causes a reduction in the cellular redox state and abolishes the lifespan extension. Conversely, exposing the more reduced larvae to the oxidant paraquat for 10 h during their larval stage is sufficient to extend their lifespan [Citation14]. These results suggested that a significant percentage of the worm population experiences a naturally occurring ROS-mediated hormesis event early in life. Intriguingly, the higher than average levels of ROS during development did not alter the expression of antioxidant enzymes, excluding the idea that changes in the levels of antioxidant enzymes are responsible for the protective effects later in life [Citation14]. These findings agreed with the observation that animals with increased developmental ROS were also resistant to other forms of stresses, such as heat shock [Citation14]. The results did, however, raise the important question as to how a transient accumulation of endogenous ROS during development can exert such long-lasting and beneficial effects in worms later in life, and suggested the potential involvement of ROS-mediated epigenetic changes in this process.
Figure 1. Developmental ROS vary naturally early in life. the use of genetically encoded redox sensors has revealed that significant fluctuations in the levels of ROS naturally occur in a population of age-synchronized C. elegans, with some animals having much higher levels of ROS and being more oxidized than others. this inter-individual variation is highest at the L2 stage of development, which comprises only a small period of the overall lifespan of C. elegans.
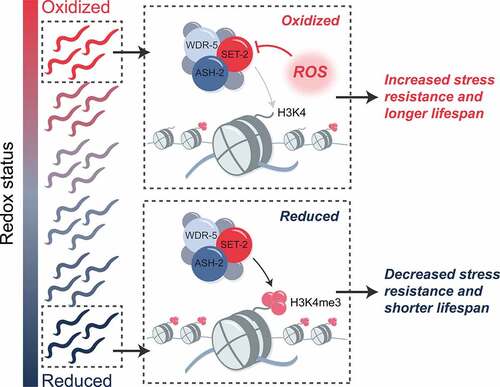
Figure 2. Developmental ROS act through H3K4me3 to influence longevity and stress resistance in C. elegans. animals that are more oxidized early in life display increased stress resistance and longer lifespan compared to animals harboring lower levels of ROS. these longevity and stress resistance benefits appear to be conferred through the ROS-mediated inactivation of the SET/MLL histone methyltransferase, which is responsible for deposition of the chromatin mark H3K4me3. as a result, oxidized animals with higher levels of developmental ROS have lower levels of H3K4me3. how increased levels of developmental ROS and changes to H3K4me3 levels in early life act to promote longevity and stress resistance later in life is currently unknown
Histone modifications, COMPASS and the role of H3K4me3 in C. elegans lifespan
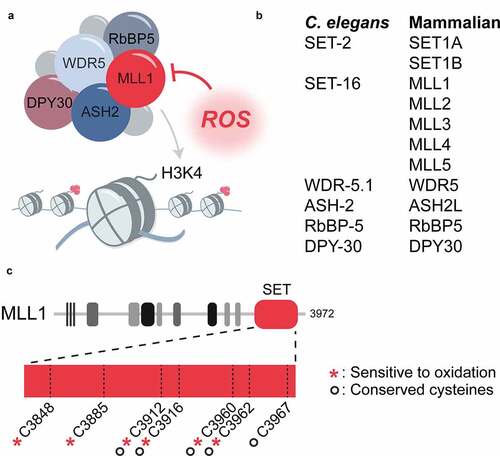
The first evidence that early-life ROS accumulation might exert its long-term effects via modifying the epigenetic landscape came from gene expression studies. The subpopulation of worms that were more oxidized during development showed gene expression pattern that were highly reminiscent of worms deficient in the absent small homeodisc protein 2 (ASH-2) [Citation14,Citation49]. This protein belongs to the core of the COMplex of Proteins ASsociated with Set1 (COMPASS), which is responsible for the trimethylation of lysine 4 of histone H3 (H3K4me3) () [Citation50]. H3K4me3 is an ubiquitous epigenetic mark that is typically associated with transcriptional activation [Citation51,Citation52]. In fact, over 80% of genes containing H3K4me3 in their promoter region are actively transcribed [Citation53]. The levels of H3K4me3 are maintained through the opposing actions of histone-lysine methyltransferases (KMT), which catalyze the sequential addition of methyl groups, and demethylases, which remove those modifications [Citation54]. One such class of H3K4me3 KMTs is the mixed-lineage leukemia (MLL-1/SET-2) family of enzymes [Citation25]. While only two members exist in C. elegans (SET-2, SET-16) [Citation49,Citation55,Citation56], six members of this family are found in mammals; SETD1A, SETD1B and MLL1–4 [Citation50] () Whereas SETD1A and B are responsible for most known H3K4me3 marks in mature cells, MLL1, and 2 are primarily responsible for H3K4me3 marks on genes expressed during development [Citation57,Citation58]. MLL3 and MLL4, on the other hand, deposit H3K4me1 marks at enhancers involved in cell type-specific gene expression [Citation59,Citation60]. Members of this family all share the Su(var)3–9, Enhancer of zeste, Trithorax (SET) domain, which catalyzes the methyltransferase reaction [Citation25]. Together with four other proteins known as WARD components, that is ASH-2, WD repeat-containing protein 5 (WDR-5), DPY-30 and RBPP-5, the MLL/SET proteins form the core COMPASS complex [Citation50] (). Additional MLL/SET specific partner proteins appear to control the extent and location of the methylation, thereby contributing to the distinct activities of the individual KMTs [Citation50].
Figure 3. Proposed mechanism for ROS-mediated inactivation of H3K4 methyltransferases. (a) the COMPASS complex responsible for the deposition of H3K4me3 consists of a SET-domain-containing histone methyltransferase of the SET-2/MLL family and several core proteins known as WARD components. the SET-domain of the histone methyltransferase is redox sensitive and inactivated by ROS. (b) H3K4me3 methyltransferases and WARD proteins in C. elegans and mammals. (c) increased levels of ROS can inactivate the COMPASS complex through oxidation of the SET domain of the histone methyltransferase (MLL1 is shown). six of the seven cysteines in the MLL1 SET domain are redox sensitive, including four of the five cysteines that are absolutely conserved between the C. elegans SET-2 and human SET1A/B and MLL1–4
First evidence that members of COMPASS and with that changes in global H3K4me3 deposition might play a role in C. elegans lifespan came from work by Greer et al., who showed that the knockdown or genetic deletion of members of COMPASS decreased global H3K4me3 levels and significantly extended worm lifespan [Citation49]. A similar lifespan extension was found in worms that overexpressed the H3K4me3 demethylase RBR-2, while increasing the global levels of H3K4me3 through the knockdown of RBR-2 shortened the lifespan in both C. elegans [Citation49] and Drosophila [Citation61]. These results led to the conclusion that the manipulation of H3K4me3 levels was sufficient to alter lifespan. In addition to extending the lifespan of organisms, reduction of H3K4me3 levels was also found to play a role in the transmission of stress resistance. Knockdown of wdr-5.1 abolished the transmission of oxidative stress resistance from exposed parental generation to subsequent generations [Citation62]. Moreover, knockdown of ash-2 was found to enhance organismal resistance to hydrogen peroxide, a heritable trait that depends on signaling from the intestine to the germline [Citation63]. However, the specific genes, whose expression is influenced by H3K4me3 and which are responsible for the inheritable stress resistance are, to our knowledge, still unknown.
MLL1/SET-2 family members are redox sensitive
Accumulation of ROS has previously shown to associate with changes in the epigenetic landscape, including histone modifications, DNA methylation, and ncRNA expression (reviewed in [Citation64,Citation65]). Yet, these alterations were thought to be primarily triggered by oxidative stress-mediated changes in key metabolites (e.g. NADH+/NAD, acetyl-CoA, alpha-ketoglutarate) or expression levels of epigenetic modifiers (reviewed in [Citation66]). In contrast, the reduction of H3K4me3 levels in oxidized larval worms or peroxide-treated HeLa cells that we observed in our study appeared to be directly due to the intrinsic redox sensitivity of members of the MLL1/SET-2 family () [Citation14]. In vitro studies confirmed that the exposure of the SET domain of mammalian MLL1 to hydrogen peroxide results in the reversible formation of intramolecular disulfide bonds and, concomitantly, to the inhibition of its methyltransferase activity [Citation14]. Under reducing conditions, the redox-sensitive cysteines form a zinc center in close vicinity to the cofactor-binding site. Given the high conservation of the cysteines in the SET domain of MLL1 with other MLL family members ()) [Citation14,Citation67] and the finding that the in vitro methyltransferase activity of purified SET1A and SET1B is equally inhibited in the presence of hydrogen peroxide [Citation14], it is seems likely that the activity of the entire family of MLL1/SET-2 enzymes is redox regulated. Deletion or knockdown of members of COMPASS eliminated any phenotypical differences previously observed in oxidized and reduced subpopulations, suggesting that the transient downregulation of H3K4m3e during development is necessary and sufficient for the improved stress resistance and longer lifespan of worms [Citation14]. Nonetheless, epigenetic marks do not work in isolation [Citation68], suggesting that transient ROS accumulation might trigger other epigenetic changes, either as a direct result of the decrease in H3K4me3 marks, or due to other epigenetic readers, writers and/or erasers, whose activity might also be redox regulated. In fact, studies in yeast revealed that low-level exposure to ROS increases H3K36me3 at select loci through the disruption of the histone demethylase Rph1p, which appears to be necessary for the hormetic adaptation of yeast to ROS [Citation69]. Whether this is due to an intrinsic redox sensitivity of Rph1p, or indirectly caused by changes in other epigenetic modifiers remains to be determined. Detailed analyses of the epigenetic changes are needed to fully characterize how transient changes in ROS levels affect the epigenetic landscape, chromatin structure and gene expression. Indeed, it would be not surprising if changes in the redox environment alter the genomic distribution of some marks at specific loci without effecting the global levels of H3K4me3.
Signaling pathways involved in ROS and H3K4me3-dependent extension of lifespan
Mitochondrial unfolded protein response (UPRmt)
Although the data demonstrate that even transient elevations in ROS levels have the capacity to extend lifespan [Citation5], the pathways involved and responsible for these effects are still largely unknown. Nevertheless, over the past years, several studies conducted in C. elegans have provided evidence that epigenetic alterations may interact with ROS-responsive signaling pathways and vice versa. As mitochondria are the primary source for the generation of intracellular ROS [Citation70], many of the adaptive responses to ROS exposure appear to involve mitochondrial stress response signaling pathways [Citation5]. One pathway with a well-documented role in mediating hormetic responses to mild oxidative stress is the mitochondrial unfolded protein response (UPRmt) [Citation71]. Transient activation of the UPRmt during development through the disruption of the ETC, which is a well-known ROS producing intervention, has been shown to cause a sustained activation of UPRmt signaling that persists into adulthood and leads to lifespan extension [Citation72]. Increased ROS, UPRmt signaling, and UPRmt-mediated longevity have also been observed upon perturbation of metabolic pathways with established roles in redox regulation, such as the pentose phosphate pathway [Citation73]. Several studies provide evidence that longevity under these conditions may be mediated by epigenetic modifiers. For example, the longevity conferred by UPRmt signaling is lost when the H3K27me3 demethylases jmjd-1.2 or jmjd-3.1 are deleted [Citation74]. Additionally, activation of UPRmt and subsequent mitochondrial stress-induced longevity were found to be dependent on the coordinated signaling between the UPRmt response factor DVE-1, the H3K9 KMT MET-2, and the chromatin remodeling factor LIN-65 [Citation75]. Moreover, H3K4me3 was found to be enriched at the promoters of mitochondrial stress response genes in worms maintained on media containing the complex III inhibitor antimycin [Citation76]. One of these stress response genes encodes for ATFS-1, the master transcriptional regulator of the UPRmt [Citation76]. While developing animals with higher levels of endogenous ROS did not have enriched expression of UPRmt genes under basal conditions, it is possible that developmental ROS drive a redistribution of H3K4me3 marks on these genes that leaves them primed for transcription in response to stress [Citation14]. Further work is necessary to determine if elevated ROS levels early in life promote longevity and stress resistance through enhanced or persistent UPRmt signaling.
Heat-shock response (HSR)
The heat-shock response (HSR), which is activated in response to a diverse set of proteostatic stresses, including elevated temperatures, oxidative stress, and viral infections, functions to maintain and restore cellular proteostasis [Citation77]. The progressive loss of the capacity to appropriately respond to such stress conditions with the upregulation of heat shock proteins and other members of the proteostasis network, is thought to be one of the hallmarks of aging and age-related pathologies [Citation78,Citation79]. Indeed, many interventions that enhance the HSR have been found to increase lifespan [Citation80–83]. Work in C. elegans revealed that the HSR peaks during development and rapidly declines as animals reach reproductive maturity [Citation80,Citation84]. This process is thought to be controlled by the H3K27me3 demethylase JMJD-3.1 [Citation84]. Induction of mild mitochondrial stress during development by disrupting the ETC appears to counteract the signaling from JMJD-3.1, allowing organisms to maintain HSR signaling throughout adulthood, and contributing to HSF-1-dependent lifespan extension [Citation85]. Gene expression studies revealed that C. elegans larvae with more oxidized redox states (similar to mutants lacking COMPASS proteins) have a substantially greater capacity to stimulate HSF-1-dependent genes (such as hsp-16.2) than their reduced counterparts [Citation14], suggesting that the lifespan extension observed in the oxidized subpopulation of C. elegans larvae might be mediated by HSF-1. It is of note that the time of HSR decline correlates with the establishment of a highly reducing redox environment [Citation14,Citation80,Citation84], making it tempting to speculate that the reduction in endogenous ROS levels at the onset of adulthood might precipitate the HSR decline. Further support for a potential role of the HSR in mediating the benefits of increased ROS levels during development, came from a study showing that the heat-shock induced expression levels of HSP-16.2 in young adult C. elegans serve as excellent predictors of lifespan and stress resistance in a genetically identical population [Citation86,Citation87]. However, the authors postulated that HSP-16.2 itself may not be solely responsible for this lifespan extending effect. Rather, the higher expression levels of HSP-16.2 may simply be a bell weather for organisms that are primed and more capable of reacting to stress conditions with a more robust expression of stress-responsive genes [Citation86,Citation87]. It would not be too surprising if the worms that were identified by their enhanced capacity to respond to heat shock treatment as young adults are in fact the grown-up versions of C. elegans larvae bestowed with increased ROS levels during development.
Other potentially relevant signaling pathways
Studies in C. elegans revealed that the lifespan extension observed in worms with reduced levels of H3K4me3 is dependent on an increase in mono-unsaturated fatty acids in the intestine [Citation88]. RNAseq experiments in developmentally oxidized worms agreed with this result and revealed that the expression of a significant number of genes involved in lipid metabolism were altered [Citation14]. These results suggest that ROS-mediated changes in the H3K4me3 landscape may extend lifespan in part through changes in lipid metabolism. However, the signaling mechanism(s) involved in this process have not been elucidated. In addition, it is likely that the DAF-16/FOXO transcription factor may also participate in ROS-mediated stress resistance and longevity. Exposure of C. elegans to mild concentrations of the ROS generator juglone administered throughout life lead to lifespan extension that is dependent in part on DAF-16 [Citation24]. Additionally, DAF-16 was found to contribute to the longevity phenotypes observed in several long-lived mitochondrial mutants [Citation89], and that activation of DAF-16 under these conditions is dependent on ROS. Overall, it seems more likely that an interlinked network of these signaling pathways act in concert to mediate the increase in stress resistance and longevity conferred by developmental ROS [Citation14].
Conclusions and outstanding questions
The discovery that naturally occurring variations in ROS serve as stochasticity factors in lifespan raises a series of intriguing questions. First, what is the cellular source of ROS during development and how are the observed variations in ROS levels generated? The effects of ROS can be very different depending on the cellular compartment in which they are generated; mitochondrially derived ROS is thought to extend lifespan, whereas ROS derived in the cytosol has been proposed to shorten lifespan [Citation9,Citation90]. Based on previous studies, it seems most likely that the mitochondria serve as the primary source of ROS, particularly as the animals are actively growing and respiring. However, analysis of the mitochondrial activity between oxidized and reduced larval subpopulation did not reveal significant differences [Citation14], in contrast to other experimental models of hormetic mitochondrial ROS production [Citation12,Citation21]. The implementation of compartment- or tissue-specific redox biosensors might help to address and identify the source(s) of ROS variability early in life. Indeed, tissue-specific effects of ROS on longevity has been observed in neurons [Citation91,Citation92], suggesting that elevated ROS in specific tissues is sufficient to affect longevity mechanisms in the organism as a whole. Another important question concerns H3K4me3; how does the distribution of H3K4me3 across the genome change in animals exposed to mild levels of ROS change, and how do these changes manifest later in life as an increase in longevity? Identification of specific genomic loci that are differentially marked by H3K4me3 in animals with higher levels of developmental ROS could lead to the discovery of novel genes responsible for enhanced stress resistance and longevity. On the basis of recent findings, ROS-mediated changes to the H3K4me3 landscape may constitute the early life events that grants a small proportion of a population of individuals with enhanced stress resistance, though how a decrease in levels of H3K4me3, an activating mark, could lead to the enhanced transcription of selective stress response genes remains an open question.
The developmental stage of life appears to be a critical window in which transient levels of ROS can set in motion events that persist into adulthood and influence lifespan and stress resistance. Reports that H3K4me3 marks that are set during development are predominantly fixed and do not change as animals age [Citation93] may provide the mechanism by which ROS-mediated changes in H3K4me3 levels early in life persist to extend lifespan in adulthood. However, several studies challenge the idea that H3K4me3 alone is indicative of transcriptional activation [Citation94–96], and that the epigenetic regulation of genes is a more complex orchestration between various activating and repressing marks and the machineries that deposit them. Additional research is clearly necessary to fully determine the impact of developmental ROS on the epigenetic landscape, and how changes to an H3K4me3 locus can affect epigenetic marks and modifiers in the same vicinity. Finally, while the subpopulation of worms with higher levels of developmental ROS may have survival advantages over the rest of the population in the face of stress, it remains to be seen whether there are trade-offs for the stress resistance and longevity benefits gained in these animals. Given that changes to H3K4me3 can elicit effects that persist not only through the life of an organism but into subsequent generations [Citation97], it is tempting to speculate that these transient changes in developmental ROS have the capacity to profoundly shape the chromatin landscape and fitness of organisms for generations to come.
Acknowledgments
Work that contributed to this perspective was supported by a Breakthrough in Gerontology award from the American Federation of Aging Research and NIH Grant GM122506 to U.J. and a BrightFocus ADR Fellowship (A2019250F) to B.J.O.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300.
- Gems D, Doonan R. Antioxidant defense and aging in C. elegans : is the oxidative damage theory of aging wrong?. Cell Cycle. 2009;8(11):1681–1687.
- Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21(10):569–576.
- Gladyshev VN. The free radical theory of aging is dead. long live the damage theory!. Antioxid Redox Signal. 2014;20(4):727–731.
- Yun J, Finkel T. Mitohormesis. Cell Metab. 2014;19(5):757–766.
- Reczek CR, Chandel NS. The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol. 2017;1(1):79–98.
- Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013;60:1–4.
- Ristow M, Schmeisser K. Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS) dose–response. Dose-Response. 2014;12(2):1.
- Van Raamsdonk JM. Levels and location are crucial in determining the effect of ROS on lifespan. Worm. 2015;4(4):e1094607.
- Wang Y, Hekimi S. Mitochondrial dysfunction and longevity in animals: untangling the knot. Science. 2015;350(6265):1204–1207.
- Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24(10):R453–R462.
- Dillin A, Hsu AL, Arantes-Oliveira N, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298(5602):2398–2401.
- Obata F, Fons CO, Gould AP. Early-life exposure to low-dose oxidants can increase longevity via microbiome remodelling in Drosophila. Nat Commun. 2018;9(1):975.
- Bazopoulou D, Knoefler D, Zheng Y, et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nat. 2019;576(7786):301–305.
- Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1± mice. J Biol Chem. 2008;283(38):26217–26227.
- Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8(12):e1000556.
- Mattson MP. Hormesis defined. Ageing Res Rev. 2008;7(1):1–7.
- Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet. 2009;5(2):e1000361.
- Bar‐Ziv R, Bolas T, Dillin A. Systemic effects of mitochondrial stress. EMBO Rep. 2020;21(6). DOI:10.15252/embr.202050094
- Lee SS, Lee RYN, Fraser AG, et al. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33(1):40–48.
- Feng J, Bussière F, Hekimi S. Mitochondrial electron transport is a key determinant of life span in caenorhabditis elegans. Dev Cell. 2001;1(5):633–644.
- Owusu-Ansah E, Song W, Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155(3):699–712.
- Schmeisser S, Schmeisser K, Weimer S, et al. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell. 2013;12(3):508–517.
- Heidler T, Hartwig K, Daniel H, et al. Caenorhabditis elegans lifespan extension caused by treatment with an orally active ROS-generator is dependent on DAF-16 and SIR-2.1. Biogerontology. 2010;11(2):183–195.
- Cypser JR, Johnson TE. Multiple stressors in Caenorhabditis elegans induce stress hormesis and extended longevity. J Gerontol Ser A Biol Sci Med Sci. 2002;57(3):B109–B114.
- Zarse K, Schmeisser S, Groth M, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15(4):451–465.
- Pan Y, Schroeder EA, Ocampo A, et al. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011;13(6):668–678.
- Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20(23):2131–2136.
- Xie M, Roy R. Increased levels of hydrogen peroxide induce a HIF-1-dependent modification of lipid metabolism in AMPK compromised C. elegans dauer larvae. Cell Metab. 2012;16(3):322–335.
- Copeland JM, Cho J, Lo T Jr, et al. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009;19(19):1591–1598.
- Scialo F, Sriram A, Fernandez-Ayala D, et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016;23(4):725–734.
- Weimer S, Priebs J, Kuhlow D, et al. D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nat Commun. 2014;5(1):3563.
- Liu X, Jiang N, Hughes B, et al. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19(20):2424–2434.
- Conti B, Sanchez-Alavez M, Winsky-Sommerer R, et al. Transgenic mice with a reduced core body temperature have an increased life span. Science. 2006;314(5800):825–828.
- Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med. 2011;51(2):327–336.
- Ludovico P, Burhans WC. Reactive oxygen species, ageing and the hormesis police. FEMS Yeast Res. 2014;14(1):33–39.
- Hartwig K, Heidler T, Moch J, et al. Feeding a ROS-generator to Caenorhabditis elegans leads to increased expression of small heat shock protein HSP-16.2 and hormesis. Genes Nutr. 2009. DOI:10.1007/s12263-009-0113-x
- Hansen M, Chandra A, Mitic LL, et al. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4(2):e24.
- Yi HS, Chang JY, Shong M. The mitochondrial unfolded protein response and mitohormesis: a perspective on metabolic diseases. J Mol Endocrinol. 2018;61(3):R91–R105.
- Shi SY, Lu SY, Sivasubramaniyam T, et al. DJ-1 links muscle ROS production with metabolic reprogramming and systemic energy homeostasis in mice. Nat Commun. 2015;6(1). DOI:10.1038/ncomms8415
- Acín-Pérez R, Carrascoso I, Baixauli F, et al. ROS-triggered phosphorylation of complex II by Fgr kinase regulates cellular adaptation to fuel use. Cell Metab. 2014;19(6):1020–1033.
- Adachi H, Fujiwara Y, Ishii N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. J Gerontol Ser A Biol Sci Med Sci. 1998;53A(4):B240–B244.
- Bazopoulou D, Knoefler D, Zheng Y, et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature. 2019;576(7786):301–305.
- Owen JB, and Butterfield DA. Measurement of Oxidized/Reduced glutathione ratio in: protein Misfolding and cellular stress in disease and aging. Methods Mol Biol. 2010:648;269–277 .
- Gutscher M, Pauleau AL, Marty L, et al. Real-time imaging of the intracellular glutathione redox potential. Nat Methods. 2008;5(6):553–559.
- Back P, De Vos WH, Depuydt GG, et al. Exploring real-time in vivo redox biology of developing and aging caenorhabditis elegans. Free Radic Biol Med. 2012;52(5):850–859.
- Knoefler D, Thamsen M, Koniczek M, et al. Quantitative in vivo redox sensors uncover oxidative stress as an early event in life. Mol Cell. 2012;47(5):767–776.
- Kirstein J, Morito D, Kakihana T, et al. Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 2015;34(18):2334–2349.
- Greer EL, Maures TJ, Hauswirth AG, et al. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466(7304):383–387.
- Meeks JJ, Shilatifard A. Multiple roles for the mll/compass family in the epigenetic regulation of gene expression and in cancer. Annu Rev Cancer Biol. 2017;1(1):425–446.
- Liang G, Lin JC, Wei V, et al. Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc Natl Acad Sci U S A. 2004;101(19):7357–7362.
- Schneider R, Bannister AJ, Myers FA, et al. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6(1):73–77.
- Zhao XD, Han X, Chew JL, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1(3):286–298.
- Hyun K, Jeon J, Park K, et al. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017. DOI:10.1038/emm.2017.11
- Simonet T, Dulermo R, Schott S, et al. Antagonistic functions of SET-2/SET1 and HPL/HP1 proteins in C. elegans development. Dev Biol. 2007;312(1):367–383.
- Fisher K, Southall SM, Wilson JR, et al. Methylation and demethylation activities of a C. elegans MLL-like complex attenuate RAS signalling. Dev. Biol. 2010;341(1):142–153.
- Hu D, Garruss AS, Gao X, et al. The Mll2 branch of the COMPASS family regulates bivalent promoters in mouse embryonic stem cells. Nat Struct Mol Biol. 2013;20(9):1093–1097.
- Rickels R, Hu D, Collings CK, et al. An evolutionary conserved epigenetic mark of polycomb response elements implemented by Trx/MLL/COMPASS. Mol Cell. 2016;63(2):318–328.
- Hu D, Gao X, Morgan MA, et al. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013;33(23):4745–4754.
- Herz HM, Mohan M, Garruss AS, et al. Enhancer-associated H3K4 monomethylation by trithorax-related, the drosophila homolog of mammalian MLL3/MLL4. Genes Dev. 2012;26(23):2604–2620.
- Li L, Greer C, Eisenman RN, et al. Essential functions of the histone demethylase lid. PLoS Genet. 2010;6(11):e1001221.
- Kishimoto S, Uno M, Okabe E, et al. Environmental stresses induce transgenerationally inheritable survival advantages via germline-to-soma communication in Caenorhabditis elegans. Nat Commun. 2017;8(1):14031.
- Nono M, Kishimoto S, Sato-Carlton A, et al. Intestine-to-germline transmission of epigenetic information intergenerationally ensures systemic stress resistance in C. elegans. Cell Rep. 2020;30(10):e4.
- Niu Y, Desmarais TL, Tong Z, et al. Oxidative stress alters global histone modification and DNA methylation. Free Radic Biol Med. 2015;82:22–28.
- Kreuz S, Fischle W. Oxidative stress signaling to chromatin in health and disease. Epigenomics. 2016;8(6):843–862.
- Kietzmann T, Petry A, Shvetsova A, et al. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br J Pharmacol. 2017;174(12):1533–1554.
- Dillon SC, Zhang X, Trievel RC, et al. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 2005;6(8):227.
- Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications - writers that read. EMBO Rep. 2015;16(11):1467–1481.
- Schroeder EA, Raimundo N, Shadel GS. Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 2013;17(6):954–964.
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–421.
- Qureshi MA, Haynes CM, Pellegrino MW. The mitochondrial unfolded protein response: signaling from the powerhouse. J Biol Chem. 2017;292(33):13500–13506.
- Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91.
- Bennett CF, Kwon JJ, Chen C, et al. Transaldolase inhibition impairs mitochondrial respiration and induces a starvation-like longevity response in Caenorhabditis elegans. PLoS Genet. 2017;13(3):e1006695.
- Merkwirth C, Jovaisaite V, Durieux J, et al. Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 2016;165(5):1209–1223.
- Tian Y, Garcia G, Bian Q, et al. Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell. 2016;165(5):1197–1208.
- Ma C, Niu R, Huang T, et al. N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat Cell Biol. 2019;21(3):319–327.
- Åkerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11(8):545–555.
- López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013. DOI:10.1016/j.cell.2013.05.039
- Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20:421–435.
- Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106(35):14914–14919.
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300(5622):1142–1145.
- Morley JF, Morimoto RI. Regulation of longevity in caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15(2):657–664.
- Steinkraus KA, Smith ED, Davis C, et al. Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell. 2008;7(3):394–404.
- Labbadia J, Morimoto RI. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell. 2015;59(4):639–650.
- Labbadia J, Brielmann RM, Neto MF, et al. Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep. 2017;21(6):1481–1494.
- Rea SL, Wu D, Cypser JR, et al. A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat Genet. 2005;37(8):894–898.
- Mendenhall AR, Tedesco PM, Taylor LD, et al. Expression of a single-copy hsp-16.2 reporter predicts life span. J Gerontol Ser A Biol Sci Med Sci. 2012;67(7):726–733.
- Han S, Schroeder EA, Silva-García CG, et al. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature. 2017;544(7649):185–190.
- Senchuk MM, Dues DJ, Schaar CE, et al. Activation of DAF-16/FOXO by reactive oxygen species contributes to longevity in long-lived mitochondrial mutants in Caenorhabditis elegans. PLoS Genet. 2018;14(3):e1007268.
- Schaar CE, Dues DJ, Spielbauer KK, et al. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015;11(2):e1004972.
- Schmeisser S, Priebe S, Groth M, et al. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol Metab. 2013;2(2):92–102.
- Kreis P, Gallrein C, Rojas-Puente E, et al. ATM phosphorylation of the actin-binding protein drebrin controls oxidation stress-resistance in mammalian neurons and C. elegans. Nat Commun. 2019;10(1). DOI:10.1038/s41467-019-08420-w
- Pu M, Wang M, Wang W, et al. Unique patterns of trimethylation of histone H3 lysine 4 are prone to changes during aging in Caenorhabditis elegans somatic cells. PLoS Genet. 2018;14(6):e1007466.
- Douillet D, Sze CC, Ryan C, et al. Uncoupling histone H3K4 trimethylation from developmental gene expression via an equilibrium of COMPASS, Polycomb and DNA methylation. Nat Genet. 2020;52:615–625.
- Howe FS, Fischl H, Murray SC, et al. Is H3K4me3 instructive for transcription activation?. Bioessays. 2017;39(1):1–12.
- Margaritis T, Oreal V, Brabers N, et al. Two distinct repressive mechanisms for histone 3 lysine 4 methylation through promoting 3ʹ-end antisense transcription. PLoS Genet. 2012;8(9):e1002952.
- Greer EL, Maures TJ, Ucar D, et al. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature. 2011;479(7373):365–371.