
ABSTRACT
N6-methyladenosine (m6A) modification, the most abundant internal methylation of eukaryotic RNA transcripts, is critically implicated in RNA processing. There is extensive evidence indicating that long non-coding RNAs (lncRNAs) serve as key regulators of oncogenesis and tumor progression in humans. Through prior study has assessed that LIFR-AS1 plays a key role in various kinds of malignant tumors. However, the exact role of m6A induced LIFR-AS1 in pancreatic cancer (PC) and its potential molecular mechanisms remain largely unknown. In this study, we determined that PC cell lines and tumors exhibit increased LIFR-AS1 expression that correlates with larger tumor size, lymph node metastasis, and more advanced TNM stage. Functionally, loss-of-function studies indicated that LIFR-AS1 knockdown decreased the proliferation, migration, and invasion of PC cells in vitro. Mechanistically, we found that METTL3 induced m6A hyper-methylation on the 3′ UTR of LIFR-AS1 to enhance its mRNA stability and LIFR-AS1 could directly interact with miR-150-5p, thereby indirectly up-regulating VEGFA expressions within cells. Through rescue experiments, we were able to confirm that the unfavorable impact of LIFR-AS1 knockdown on VEGFA /PI3K/Akt Signaling could be reversed via the inhibition of miR-150-5p expression. Together, these findings indicate that a noval m6A-LIFR-AS1 axis promotes PC progression at least in part via regulation of the miR-150-5p/VEGFA axis, indicating that this regulatory axis may be a viable clinical target for the treatment of PC.
Introduction
Pancreatic cancer (PC), one of the most frequent cancer types in the world, is a deadliest solid malignancy with poor prognosis [Citation1–3]. In spite of recent improvements in diagnosis and surgical treatment, the estimated 5-year survival rate for pancreatic cancer continues to be poor [Citation4,Citation5]. As the cause of PC is complex and multifactorial, deeply investigating the cancer-associated molecular events could be the key to understanding the progression of this deadly disease and thus the cornerstone of developing a potential therapy strategy [Citation6,Citation7].
The N6-methyladenosine (m6A) modification is the most abundant internal methylation of RNA transcripts in eukaryotes [Citation8]. More and more evidences show that m6A modification has a significant and comprehensive impact on a variety of biological regulation processes [Citation9]. In recent years, many studies have described the key role of the m6A mechanism in different human cancers [Citation10]. METTL3 has been identified as a carcinogenic driver of PC [Citation11]. However, the exact function and potential regulatory mechanism of METTL3 in PC are still unclear.
Long noncoding RNAs (lncRNAs) have recently been shown to play diverse regulatory roles in a range of cancers [Citation12–14]. Leukemia inhibitory factor receptor antisense RNA 1 (LIFR-AS1), a novel cancer-related lncRNA, is transcribed from the LIFR gene located on human chromosome 5p13.1 in an antisense manner [Citation15]. Recent studies indicated that LIFR-AS played a key role in several solid tumors, such as breast cancer [Citation16] and colorectal cancer [Citation17]. However, the exact role of LIFR-AS1 in PC and its potential molecular mechanisms remain largely unknown, and as such the present study was designed to explore the physiological relevance and functional role of this lncRNA in PC in an effort to provide novel therapeutic insights.
Material and methods
Tissue collection and ethics statement
A total of 139 pair patients’ tissues analyzed in this study underwent resection of the primary pancreatic cancer at Changhai Hospital, Shanghai, China. All collected PC tissue samples were immediately snap frozen in liquid nitrogen and stored at −80°C until required. Our study was approved by the Research Ethics Committee of Changhai Hospital (No. CHKY2014-03-019), and written informed consent was obtained from all patients. The clinical characteristics of the pancreatic cancer patients are summarized in .
Table 1. The relationship between LIFR-AS1 expression and PC patient clinic-pathological features.
Cell culture
Five pancreatic cancer cell lines (AsPC-1, BxPC-3, Mia-capa2, HPAC, and PANC-1) and adherent cell line of pancreatic normal ductal epithelium (HPDE) were obtained from the American Type Culture Collection (Manassas, VA). All of the cell lines were grown and maintained in RPMI 1640 or DMEM Medium (Invitrogen) and supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 mg/ml streptomycin (Invitrogen, Shanghai, China) at 37°C with 5% CO2.
Cell transfection
Small interfering RNA (siRNA) constructs specific for targeting METTL3 (si-METTL3), LIFR-AS1 (si-LIFR-AS1) and non-targeting control (si-NC) constructs were from Guangzhou RIBOBIO Co., Ltd (Guangzhou, China). miR-150-5p agomir/antagomir (agomir-150-5p and antagomir-150-5p) and negative control agomir/antagomir (agomir-NC and antagomir-NC) constructs were from GenePharma Co., Ltd (Shanghai, China), as were VEGFA overexpression pcDNA3.1-VEGFA (pc-VEGFA) and empty pcDNA3.1 control vector. For transfection experiments, cells were plated for 24 h in 6-well plates, after which Lipofectamine 2000 (Invitrogen, USA) was used for cellular transfection with appropriate constructs.
qRT-PCR
TRIzol (Invitrogen) was used to isolate sample RNA, after which a NanoDrop instrument (NanoDrop Technologies, USA) was used to evaluate sample quantity and quality. A Mir-X™ miRNA First-Strand Synthesis Kit (TaKaRa Biotechnology, Dalian, China) was used to quantify miR-150-5p expression together with a Mir-X™ miRNA qRT-PCR TB Green® Kit (TaKaRa). Levels of LIFR-AS1 and VEGFA expression were measured with a PrimeScript™ RT Reagent Kit (TaKaRa) together with a SYBR-Green PCR Master Mix (TaKaRa). U6 and Actin were used as normalization controls for miRNA and mRNA expression levels, respectively, with the 2−ΔΔCq method being employed to assess relative gene expression. Primers used for reverse transcription-quantitative PCR were listed in Supplement Table S1.
CCK-8 assay
The proliferation of cells was measured via CCK-8 assay. Briefly, cells were rested for 24 h post-transfection, after which they were added to 96-well plates (2 × 103/well). At appropriate time points, 10 μL of CCK-8 solution (Shanghai Haling Biotechnology, Co., Ltd., Shanghai, China) was added to each well and the plates were then incubated for 2 h at 37°C. A microplate reader (Bio-Rad, CA, USA) was then employed for assessing absorbance at 450 nm in each well.
Transwell assays
At 48 h post-transfection, cells used for migration or invasion assays. Briefly, cells (5 × 104 in 200 µL of serum-free DMEM) were put in the upper portion of a Matrigel (BD Biosciences, NJ, USA)-coated Transwell assay chamber (8-µm pore size; Corning, NY, USA). Next, the lower chamber was filled with 600 µL of media with 10% FBS, followed by a 24 h incubation at 37°C. A cotton swab was then used to carefully remove noninvasive cells, with the remaining cells being subjected to fixation using methanol followed by crystal violet staining. The migration of these cells was quantified via the same approach, omitting only the Matrigel pre-coating of the Transwell inserts. A microscope (Olympus Corporation, Tokyo, Japan) was then used to quantify cells in five random fields of view in order to measure cellular migration and invasion.
RNA immunoprecipitation (RIP) assay
Interactions between mIR-150-5p and LIFR-AS1 were measured with a Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, MA, USA). Following lysis, cellular lysates were combined overnight at 4°C with human AGO2 antibody- or control IgG-conjugated magnetic beads (Millipore). Beads were then collected and all precipitated RNA was isolated for analysis via qRT-PCR.
Luciferase reporter assay
GenePharma synthesized LIFR-AS1 fragments containing wild-type (WT) or mutated (MUT) versions of the putative miR-150-5p binding sequence. These fragments were inserted into the pmirGLO Dual-luciferase Vector (Promega, WI, USA), yielding WT- and MUT-LIFR-AS1 reporter plasmid constructs. A comparable approach was used to prepare VEGFA reporter plasmids. For transfection, cells were plated overnight in 24-well plates until 60–70% confluent, at which time they were co-transfected using appropriate reporter plasmids and agomir-150-5p or agomir-NC. After 48 h, cells were collected and a Dual-Luciferase Reporter Assay System (Promega) was used based on provided direcitons, with Renilla luciferase activity being employed for normalization purposes.
Western blotting
RIPA buffer (Beyotime Institute of Biotechnology, Shanghai, China) was used to isolate total protein from cell samples, after which protein levels were quantified via BCA assay kit (Beyotime Institute of Biotechnology). Equal protein amounts from each sample were then separated via SDS-PAGE prior to transfer onto PVDF membranes (Millipore). Blots were blocked for 2 h with 5% nonfat milk, after which they were probed overnight at 4°C using primary antibodies. Blots were then washed and probed using a secondary HRP-conjugated antibody (1:5,000; ab205718; Abcam), and an Immobilon Western Chemiluminescent HRP Substrate kit (EMD Millipore) was used to quantify protein expression levels. Primaryantibodies were as follows: rabbit polyclonal anti-VEGFA (1:1000, ab46154, Abcam, UK), anti-VEGFR2 (1:1000, ab39256, Abcam, UK), anti-phospho (Y1175)-VEGFR2 (1:1000, ab194806, Abcam, UK), anti-Akt (#9272, Cell Signaling Technology, USA), anti-phospho (Ser473)-Akt (1:2000, #4060, Cell Signaling Technology, USA), anti-mTOR (1:1000, ab2732, Abcam, UK), anti-phospho (S2448)-mTOR (1:1000, ab84400, Abcam, UK) anda mouse anti-human actin (1:5000, ab128915 Abcam, UK).
m6A-qPCR
m6A-IP enrichment followed by RT-qPCR to quantify the changes in m6A methylation of the target gene was performed using Magna MeRIP m6A Kit (Millipore, MA) following the manufacturer’s instructions. Primers used for m6A-qPCR were listed in Supplement Table S2.
RNA stability assays
PC cells were seeded in 12-well plates overnight, and then treated with actinomycin D (5 μg/mL, HY-17559, MedChem Express) at the 0, 3, 6 h. Total RNA was then isolated by TRIzol and analyzed by qPCR. The mRNA expression for each group at the indicated time was calculated and normalized by Actin.
The cytoplasmic and nuclear fractionation assay
The cytoplasmic and nuclear fractionation assay was carried out by the use of PARIS™ Kit (Invitrogen, USA) following the supplier’s suggestion. GAPDH and U6 were used as cytoplasmic or nuclear fractionation indicators.
Statistical analysis
Data are means ± SD from triplicate experiments. The relationship between PC patient clinicopathological characteristics and LIFR-AS1 levels was assessed via a chi-squared test. Other data were compared via Student’s t-tests or one-way ANOVAs with Tukey’s post hoc test as appropriate. Survival of PC patients was assessed via the Kaplan–Meier approach, with log-rank tests used to compare these survival rates. Spearman’s correlation analyses were used to examine the relationship between the expressions of specific genes of interest in PC patient samples. P < 0.05 was the significance threshold.
Results
PC samples exhibit LIFR-AS1 increased
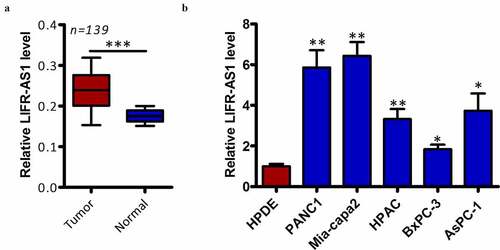
We began by quantifying LIFR-AS1 expression levels in 139 pairs of clinical PC tumor and normal para-cancerous tissues via qRT-PCR. This analysis indicated that LIFR-AS1 expression is significantly higher in PC tumors relative to levels in normal surrounding tissue (, P < 0.05). We then extended this analysis in vitro, confirming that LIFR-AS1 levels were also higher in PC cell lines (AsPC-1, BxPC-3, Mia-capa2, HPAC, and PANC-1) relative to the control HPDE cell line (, P < 0.05). We next explore the clinical relevance of this lncRNA by dividing PC patients into two groups based on the median LIFR-AS1 levels in this cohort, yielding LIFR-AS1-low and LIFR-AS1-high patient groups. When clinical findings were compared between these groups, we determined that higher LIFR-AS1 expression was associated with larger tumor size (P = 0.031), lymph node metastasis (P = 0.011), and more advanced TNM stage (P = 0.001) (). Together, these findings highlight a potential role for LIFR-AS1 in the onset or progression of PC.
Figure 1. PC samples exhibit the upregulation of LIFR-AS1.
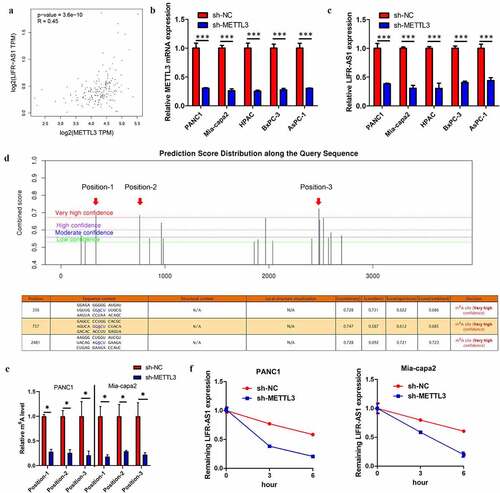
METTL3 induced LIFR-AS1 m6A to enhance its mRNA stability
Based on GEPIA database analysis, the LIFR-AS1 expression was positive with METTL3 in PC (), indicating that LIFR-AS1 might be regulated by METTL3 in an m6A pattern. qRT-PCR indicated that knockdown of METTL3 leads to a decrease LIFR-AS1 in PC cell (). To identify the specific m6A methylation loci of LIFR-AS1, we apply the SRAMP website(http://www.cuilab.cn/sramp/) indicating that three positions existed abundance of m6A methylation loci (). Following m6A-qPCR indicated that these positions did exist abundance of m6A methylation loci and METTL3-induced LIFR-AS1 m6A hyper-methylation in PC cell lines (Supplement Figures S1 and S2E). Previous study reported that m6A modified could enhance the mRNA stability [Citation9]. Indeed, our results confirm that silencing of METTL3 reduces mRNA of LIFR-AS1 stability in PANC1 and Mia-capa2 cells (). Above results indicated that METTL3-induced LIFR-AS1 m6A hypermethylation to enhance its mRNA stability in PC.
Figure 2. METTL3 induced LIFR-AS1 m6A to enhance ItsmRNA stability in PC.
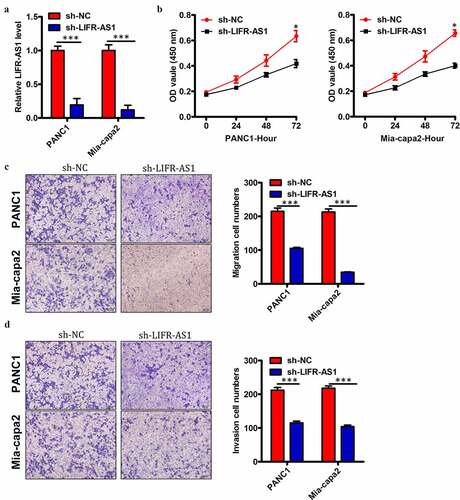
LIFR-AS1 knockdown suppressed PC malignancy in vitro
We elected to use these two PC cell lines for mechanistic analyses of the functional role of this lncRNA. To that end, we transfected these two cell lines (LIFR-AS1 levels were highest in PANC1 and Mia-capa2 cells of the 5 tested PC cell line) with si-LIFR-AS1 in order to knock down LIFR-AS1 levels therein, using a si-NC construct as a control (). We found that LIFR-AS1 knockdown impaired the proliferation of both tested PC cell lines in a CCK-8 assay relative to si-NC transfected cells (). Knockdown of LIFR-AS1 also markedly impaired both the migration () and invasion () of these cells relative to si-NC-transfected controls. This thus suggested that LIFR-AS1 functions in a pro-oncogenic manner in PC.
Figure 3. Reduced levels of LIFR-AS1 impair the proliferation, invasion, and survival of PC cellsin vitro.
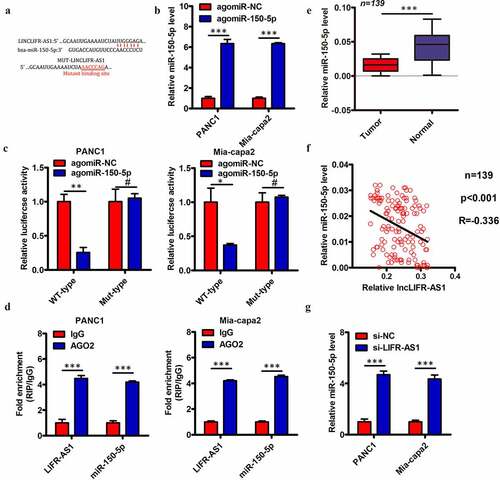
LIFR-AS1 binds to and sequesters miR-150-5p in PC cells
Several prior studies have found that lncRNAs can serve as competing endogenous RNAs (ceRNAs) capable for binding and sequestering specific target miRNAs. Subcellular fractionation assay indicated that LIFR-AS1 was predominantly located in the cytoplasm of PC cells (Supplement Figure S2). We therefore utilized the starBase 3.0 algorithms to identify putative miRNA targets of LIFR-AS1, revealing miR-150-5p () to have a sequence complementary to that found within LIFR-AS1. We therefore selected this miRNA for further validation experiments, given that it has previously been reported to suppress PC progression [Citation18]. We began by using a luciferase reporter assay to demonstrate the direct interaction between miR-150-5p and LIFR-AS1 in PC cells. Briefly, we transfected PANC1 and Mia-capa2 cells with luciferase reporter constructs containing WT or MUT versions of the predicted miR-150-5p binding site together with either agomir-150-5p or agomir-NC. We confirmed that agomir-150-5p transfection significantly increased expression of this miRNA in these target cell lines (). We additionally found that agomir-150-5p transfection was sufficient to reduce WT but not MUT LIFR-AS1 luciferase reporter activity in both PANC1 and Mia-capa2 cells (). We additionally confirmed that endogenous miR-150-5p and LIFR-AS1 were able to interact with one another via a RIP assay, revealing significant enrichment for LIFR-AS1 and miR-150-5p in Ago2-containing immunoprecipitates relative to those prepared with a control IgG (). We additionally quantified the expression of miR-150-5p in the 139 previously analyzed PC and paracancerous control tissue samples, revealing lower miR-150-5p levels in tumors relative to control samples (). We further found that LIFR-AS1 and miR-150-5p expression were negatively correlated in these PC tissues (; Spearman r = −0.336, P < 0.001). We then knocked down LIFR-AS1 expression in PANC1 and Mia-capa2 cells and measured miR-150-5p levels, revealing that the knockdown of this lncRNA led to significant upregulation of miR-150-5p in these same cells (). Together, these findings indicate that LIFR-AS1 can function as a ceRNA that binds to and sequesters miR-150-5p in PC cells.
Figure 4. LIFR-AS1 interacts with and sequesters miR-150-5p within PC cells.
LIFR-AS1 sequesters miR-150-5p and thereby upregulates VEGFA in PC cells
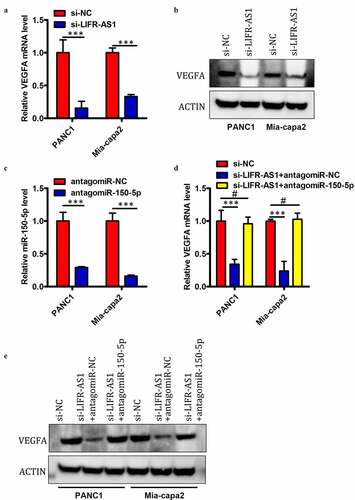
Previous research indicates that miR-150-5p can target and downregulate VEGFA [Citation18]. Given that we identified miR-150-5p as a LIFR-AS1 target in PC cells, we next sought to assess whether this lncRNA is able to regulate VEGFA expression in these same cells. We found that siRNA-mediated knockdown of LIFR-AS1 was associated with a significant drop in VEGFA mRNA () and protein levels () in PANC1 and Mia-capa2 cells. We then assessed whether this relationship between LIFR-AS1 and VEGFA expression was attributable to the ceRNA-mediated sequestration of miR-150-5p by co-transfecting PC cells with si-LIFR-AS1 plus antagomir-150-5p or antagomir-NC. After confirming that antagomir-150-5p significantly decreases miR-150-5p expression in both tested cell lines (), we determined that antagomir-150-5p transfection was also able to reverse the observed LIFR-AS1-mediated reductions in VEGFA mRNA () and protein levels () in PANC1 and Mia-capa2 cells. These findings thus indicated that LIFR-AS1 can positively regulate VEGFA expression in PC cells via serving as a ceRNA for miR-150-5p.
Figure 5. LIFR-AS1 serves as a ceRNA that sequesters miR-150-5p and thus alters the expression of VEGFA in PC cells.
LIFR-AS1 targets miR-150-5p to activate VEGFA/Akt/mTOR signaling
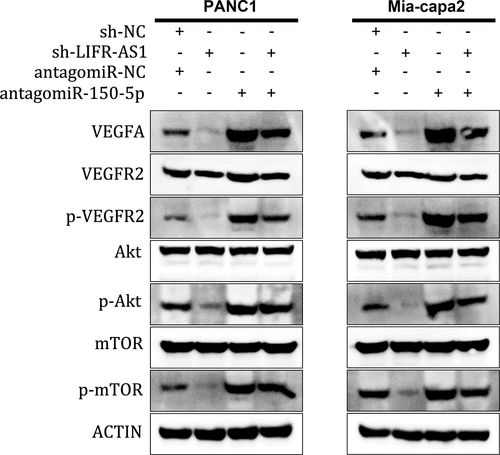
To further explore whether LIFR-AS1 enhanced PC development through VEGFA-mediated Akt/mTOR signaling pathway, we found that knockdown LIFR-AS1 significantly decreased the level of VEGFA, and then significantly suppressed the phosphorylation of VEGFR2 (p-VEGFR2), Akt (p-Akt) and mTOR (p-mTOR) in PANC1 and Mia-capa2 cells, while miR-150-5p inhibitor could at least partially increase the expression of p-VEGFR2, p-Akt and p-mTOR (). These results revealed that LIFR-AS1 exerted its oncogene function via the miR-150-5p /VEGFA /Akt/mTOR signaling pathway.
Figure 6. LIFR-AS1 targets miR-150-5p to activate VEGFA/Akt/mTOR Signaling.
Discussion
Previous reports have demonstrated that lncRNA expression patterns become dysregulated in PC, contributing to the development and progression of this deadly disease [Citation19]. As such, there is clear value in the further study of PC-related lncRNAs as such research may offer novel insights into the management of this disease. Prior studies have assessed that LIFR-AS1 play key role in various kinds of malignant tumors [Citation20–23]. However, the exact role of LIFR-AS1 in PC and its potential molecular mechanisms remain largely unknown. In this study, we therefore explored the oncogenic role of LIFR-AS1 in PC. We began by demonstrating that this lncRNA is highlighted upregulated in PC tumor samples, and that higher LIFR-AS1 expression is positively correlated with tumor size, lymph node metastasis, and advanced TNM stage in PC patients. The existing findings show that LIFR-AS1 can be a biomarker for PC.
m6A modification is implicated in diverse biological processes including tumorigenesis. m6A modification regulates RNA fate and such functions as mRNA stability, nuclear processing, transport, localization, translation, primary microRNA processing, and RNA–protein interactions [Citation9]. METTL3 is known to be involved in all stages in the life cycle of RNA. Recently studies have reported that METTL3 played a key role in PC. In our study indicated that METTL3-induced LIFR-AS1 3ʹUTR m6A hyper-methylation to enhance LIFR-AS1 mRNA stability in PC. These suggested that METLL3 could exhibit its oncogene roles in PC by regulating LIFR-AS1 mRNA levels via the m6A modification.
Through mechanistic experiments, we also determined that LIFR-AS1 knockdown was sufficient to impair PC cell proliferation, migration, and invasion. There are multiple different mechanisms whereby lncRNAs can impact tumor progression, but the ceRNA mechanism is thought to be the most prominent and best understood of these mechanisms [Citation24]. Previous studies have assessed that LIFR-AS1 may target miR-29a in colorectal cancer resistance to photodynamic therapy, and we wonder if there exist novel target genes. In an effort to establish the novel mechanistic basis whereby LIFR-AS promoted PC malignancy, we employed predictive bioinformatics analyses to identify miR-150-5p as a potential binding target for this lncRNA. We then confirmed the direct interaction of LIFR-AS1 and miR-150-5p via luciferase reporter and RIP assays. In further support of a regulatory relationship between these molecules, we found miR-150-5p to be expressed at low levels in PC tissues and to be negatively correlated with LIFR-AS1 levels. The downregulation of LIFR-AS1 was also sufficient to increase the expression of miR-150-5p, in turn leading to the downregulation of VEGFA. Together, these results thus support a model wherein LIFR-AS1 is a ceRNA for mIR-150-5p, thereby indirectly regulating the expression of VEGFA in PC cells.
Akt and mTOR are both downstream targets of VEGFA. Previous studies have reported that the Akt/mTOR signaling pathway played a critical role in various biological processes of cancers, such as proliferation, metastasis, survival and angiogenesis [Citation25–27]. We further examined whether LIFR-AS enhanced tumor initiation and progression by activating the VEGFA/VEGFR2/Akt/mTOR signaling pathway, and our data showed that knockdown LIFR-AS could significantly reduce the expression of VEGFA and reduce the activity of VEGFR2 and the downstream Akt/mTOR signaling pathway. Together, these results highlight VEGFA as a promising therapeutic target in cancer. Our results indicate that the LIFR-AS1/miR-150-5p/VEGFA axis can regulate Akt/mTOR signaling and PC cell malignancy in vitro. Specifically, we found that LIFR-AS1 can serve as a ceRNA that binds and sequesters miR-150-5p within PC cells, thereby reducing the expression of this miRNA while enhancing VEGFA expression. This regulatory axis may thus offer novel insights into the development or progression of PC, while also highlighting potential targets for future therapeutic interventions aimed at treating patients with this disease.
In summary, our study demonstrated the pivotal roles of the m6A-LIFR-AS1 axis mediated VEGFA/VEGFR2/Akt/mTOR signaling pathway on the tumor genesis and progression of PC. This research not only provides a new insight into the mechanism of PC progression but also highlights LIFR-AS1 as a potential diagnostic biomarker and therapeutic target of PC.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Supplemental Material
Download MS Word (17.4 KB)Supplemental Material
Download MS Word (364.6 KB)Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Chen JQ, Tao YP, Hong YG, Li HF, Huang ZP, Xu XF, Zheng H, Hu LK. M6A-mediated up-regulation of LncRNA LIFR-AS1 enhances the progression of pancreatic cancer via miRNA-150-5p/ VEGFA/Akt signaling. Cell Cycle. 2021 Dec;20(23):2507–2518.
- Lin QJ, Yang F, Jin C, et al. Current status and progress of pancreatic cancer in China. World J Gastroenterol. 2015;21:7988–8003.
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
- Heestand GM, Kurzrock R. Molecular landscape of pancreatic cancer: implications for current clinical trials. Oncotarget. 2015;6:4553–4561.
- Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63:318–348.
- Oldfield LE, Connor AA, Gallinger S. Molecular events in the natural history of pancreatic cancer. Trends Cancer. 2017;3:336–346.
- Cicenas J, Kvederaviciute K, Meskinyte I, et al. Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers (Basel). 2017;9:42.
- Ma Z, and Ji J. N6-methyladenosine (m6A) RNA modification in cancer stem cells. Stem Cells. 2020 Sep 27.
- Gan H, Hong L, Yang F, et al. Progress in epigenetic modification of mRNA and the function of m6A modification. Sheng Wu Gong Cheng Xue Bao. 2019;35:775–783.
- Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613.
- Xia T, Wu X, Cao M, et al. The RNA m6A methyltransferase METTL3 promotes pancreatic cancer cell proliferation and invasion. Pathol Res Pract. 2019;215:152666.
- Chi Y, Wang J, Wang J, et al. Long non-coding RNA in the pathogenesis of cancers. Cells. 2019;8:1015.
- Lv Y, Huang S. Role of non-coding RNA in pancreatic cancer. Oncol Lett. 2019;18:3963–3973.
- Penna GC, Vaisman F, Vaisman M, et al. Molecular markers involved in tumorigenesis of thyroid carcinoma: focus on aggressive histotypes. Cytogenet Genome Res. 2016;150:194–207.
- Frey HA, Stout MJ, Pearson LN, et al. Genetic variation associated with preterm birth in African-American women. Am J Obstet Gynecol. 2016;215:235.e1–8.
- Wang L, Li J, Zhao H, et al. Identifying the crosstalk of dysfunctional pathways mediated by lncRNAs in breast cancer subtypes. Mol Biosyst. 2016;12:711–720.
- Liu K, Yao H, Wen Y, et al. Functional role of a long non-coding RNA LIFR-AS1/miR-29a/TNFAIP3 axis in colorectal cancer resistance to pohotodynamic therapy. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2871–2880.
- Chen X, Xu X, Pan B, et al. miR-150-5p suppresses tumor progression by targeting VEGFA in colorectal cancer. Aging (Albany NY). 2018;10:3421–3437.
- Tang XJ, Wang W, Hann SS. Interactions among lncRNAs, miRNAs and mRNA in colorectal cancer. Biochimie. 2019;163:58–72.
- Xu F, Li H, Hu C. LIFR-AS1 modulates Sufu to inhibit cell proliferation and migration by miR-197-3p in breast cancer. Biosci Rep. 2019;39. DOI:10.1042/BSR20180551
- Ding H, Cui L, and Wang C. Long noncoding RNA LIFR-AS1 suppresses proliferation, migration and invasion and promotes apoptosis through modulating miR-4262/NF-κB pathway in glioma. Neurol Res. 2020;43(3): 210–219.
- Wang HF, Lv JQ, Li HH, et al. High long non-coding LIFR-AS1 expression correlates with poor survival in gastric carcinoma. Eur Rev Med Pharmacol Sci. 2020;24:5378–5384.
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917.
- Yin C, Mou Q, Pan X, et al. MiR-577 suppresses epithelial-mesenchymal transition and metastasis of breast cancer by targeting Rab25. Thorac Cancer. 2018;9:472–479.
- Trinh XB, Tjalma WAA, Vermeulen PB, et al. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br J Cancer. 2009;100:971–978.
- Rodrigues DA, Guerra FS, Sagrillo FS, et al. Design, synthesis, and pharmacological evaluation of first-in-class multitarget N-Acylhydrazone derivatives as selective HDAC6/8 and PI3Kalpha inhibitors. ChemMedChem. 2020;15:539–551.
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318.