
ABSTRACT
In the present study, we determined the effects of the Src family kinase (SFK) inhibitor, Bosutinib, and the engineered loss of the Lyn SFK on all-trans retinoic acid-induced leukemic cell differentiation. Retinoic acid (RA) is an embryonic morphogen and dietary factor that demonstrates chemotherapeutic efficacy in inducing differentiation of a non-APL AML cell model, the HL-60 human myeloblastic (FAB-M2) leukemia cell line, via activation of a novel signalsome containing an ensemble of signaling molecules that drive differentiation. Bosutinib is an inhibitor of SFKs used to treat myeloid leukemias where prominent high expression of SFKs, in particular Lyn, has been observed. Using either Bosutinib or loss of Lyn expression due to shRNA promoted RA-induced phenotypic differentiation, G0 arrest, and respiratory burst (functional differentiation) of HL-60 cells. Signaling events putatively seminal to RA-induced differentiation, the expression of Fgr, Cbl, Slp-76 and Vav, and the phosphorylation of c-Raf (pS259), Vav (p-tyr), and Slp76 (p-tyr) were not inhibited by Bosutinib or loss of Lyn. Nor was RA-induced upregulation of p-tyr phosphorylation of p47phox, a member of the NADPH complex that produces ROS, a putative phosphorylation dependent signaling regulator. Surprisingly, Bosutinib still works in the absence of Lyn to enhance RA-induced differentiation and neither compromised RA-induced expression, nor phosphorylation of signaling molecules that drive differentiation. These findings suggested there is a novel, off-target, Lyn-independent effect of Bosutinib that is of therapeutic significance to differentiation therapy.
Introduction
All-trans-retinoic acid (RA) is a well-known regulator cell differentiation and cell cycle in numerous contexts. It is used to cause differentiation and G0 cell cycle arrest of leukemia cells, specifically the acute promyelocytic (APL) subtype of acute myeloid leukemia (AML). However, it also induces differentiation and cell cycle arrest in HL-60 human myeloblastic lineage leukemia cells [Citation1]. APL is a rare subtype myeloid leukemia classified as the FAB (French American British classification) M3 subtype of AML. APL is characterized cytogenetically by a t (15;17) (q22; q12) translocation, which results in a PML-RARα, fusion protein, which is seminal to the disease [Citation2]. The treatment of acute promyelocytic leukemia (APL), has been transformed by RA differentiation therapy. APL, which was once considered very difficult to medically manage, is now one of the most treatable, with remission rates of 80–90% using a combination of RA and arsenic trioxide therapy [Citation1]. The overwhelming success of APL treatment with RA, as well as the lower toxicity of RA compared to existing chemotherapeutic agents, and the lack of secondary tumors emerging much later as a sequela, has sparked intense interest in its use in other cancers and leukemias, especially in acute myeloid leukemia (AML), where non-terminally differentiated myeloid precursors proliferate uncontrollably [Citation3,Citation4]. Cytotoxic chemotherapy for AML can reportedly induce remission in 60 to 80% of individuals under the age of 60 [Citation5]. The majority, however, relapse with treatment-resistant cancers within 2–3 years, with overall survival rates as low as 30% [Citation6,Citation7]. Unfortunately, in AML, RA therapy only promotes apoptosis or proliferation inhibition, not differentiation [Citation8–10]. Thus it is of significant interest to incorporate RA with other agents, such as other differentiation-inducing compounds or kinase inhibitors, to effect RA-induced differentiation of APL as well as non-APL AML. HL-60 cells have been found to bear fidelity to a subtype of non-APL AML that is RA responsive, and are highly studied as a model of a myeloblastic (non-APL) leukemia that does differentiate in response to RA with the goal of bringing effective differentiation therapy to non-APL AML as well as APL [Citation11].
The Src family of protein tyrosine kinases (SFKs) are group of enzymes that play a significant role in leukemia cell proliferation, survival, adhesion, and differentiation through regulating signal transduction by a diverse set of cell surface receptors in a variety of cells [Citation12,Citation13]. SFKs have been proven to effectively regulate MAPK signaling, cell proliferation, and cell transformation [Citation14,Citation15]. Abnormal expression of SFKs in many cancer types has been documented and associated with acute and chronic myeloid malignancies, as well as metastasis [Citation16]. Members of the Src family include c-Src, Fyn, c-Yes, Lck, Hck, c-Fgr, Lyn, Blk, and Yrk. Src, Fyn, and Yes are ubiquitously expressed, while Lck, Hck, Fgr, Lyn, and Blk are identified most often in hematopoietic cells [Citation17,Citation18]. Lyn, a member of the SFK family, is prominently expressed in AML cells in an active form and its hyper activation is considered a contributor to AML pathogenesis [Citation18,Citation19]. The HL-60 human AML leukemia cell line, which has cytological features of promyelocytes, has been a useful model to study-to-study leukemic cell differentiation [Citation20–22]. Lyn has been shown to be the most activated SFK in HL-60 cells, despite the presence of other SFK members such as Lck, Fyn, and Hck in the cells [Citation19]. Its prominence makes it an attractive target SFK [Citation23].
Bosutinib (SKI-606), 4-[(2,4-dichloro-5-methoxyphenyl)amino]-6-methoxy-7-[3-(4-methyl-1-piperazinyl) propoxy]-3-quinolinecarbonitrile monohydrate is a competitive inhibitor of both Src and ABL tyrosine kinases [Citation24,Citation25]. Bosutinib has been shown to bind to the kinase domain BCR-ABL1 [Citation26]. Bosutinib has also been shown to inhibit the Src family kinases Src, Lyn, and Hck, as well as the platelet-derived growth factor (PDGF) receptor and c-Kit [Citation27]. Src-transformed fibroblasts and Src overexpressing HT29 colon tumors subcutaneously transplanted into athymic nu/nu mice were successfully inhibited by bosutinib [Citation28]. Bosutinib is now in approved use for treatment of CML.
In this study, we examined the effects of RA/Bosutinib co-treatments on HL-60 cells and HL-60 Lyn KD cells to determine the effects of Bosutinib and losing Lyn expression on RA-induced differentiation and cell cycle arrest. RA induced progressively increasing expression of the CD11b differentiation marker at 48 and 72 h, which was enhanced by addition of Bosutinib to the RA treatment. Loss of Lyn in Lyn KD stable transfectants of the parental HL-60 enhanced the expression of CD11b induced by RA and RA plus Bosutinib. Likewise, cell cycle distribution analysis showed G1/G0 enrichment at 48, and 72 h in the RA-treated cells, and this response was increased by adding Bosutinib and by the loss of Lyn. RA caused an increase in functional differentiation measured by inducible reactive oxygen species (ROS) production compared to untreated cells, and adding Bosutinib to RA further increased inducible oxidative metabolism compared to RA alone. Loss of Lyn increased these effects of RA and Bosutinib. Hence Bosutinib still enhanced RA-induced differentiation despite loss of Lyn. RA-induced differentiation is putatively driven by up-regulated expression and phosphorylation of signaling molecules within the signalsome, and in particular kinases, Raf, Lyn and Fgr, and adaptor/scaffold functioning molecules, Slp-76, Cbl and Vav, have been implicated. These potentially sensitive targets were surveyed for aberrations associated with Bosutinib or Lyn KD in RA-treated cells. Neither upregulation of Raf, Lyn, Fgr, Slp-76, c-Cbl or Vav, nor phosphorylation of c-Raf (pS259), Vav (p-tyr), and Slp76 (p-tyr) was inhibited by Bosutinib. And neither upregulation of Raf, Fgr, Slp-76, c-Cbl or Vav nor phosphorylation of c-Raf (pS259), Vav (p-tyr), and Slp76 (p-tyr) was inhibited by Lyn KD. These up-regulations and phosphorylations persisted in RA-treated cells that had both Bosutinib and loss of Lyn. These findings suggest that there is a novel off-target Lyn-independent effect of Bosutinib that promotes RA-induced differentiation of these non-APL AML cells that is of therapeutic significance to differentiation therapy. Combining RA and Bosutinib therapy in AML differentiation therapy may ergo be beneficial because of an off-target effect of Bosutinib.
Materials and methods
Cell culture and treatments
Human myeloblastic leukemia cells (HL-60) were grown in a humidified environment of 5% CO2 at 37°C and maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented by 1% antibiotic/antimycotic (Sigma, St. Louis, MO) and 5% heat-inactivated bovine fetal serum (Hyclone, Logan, UT). Experimental cultures were initiated at a density of 0.2 × 106 cells/mL for lysate collection 72 hours later after treatment for all experiments. The hemocytometer and 0.2% trypan blue (Invitrogen, Carlsbad, CA) were used for the dye exclusion assay of cell growth and viability. The same RA (Sigma) treatment conditions (0 μM or 1 μM) were used for all cells and lysate obtained. At least three biological replicates of each experiment were performed. There were four treatment regimens studied: (1) untreated, (2) RA, (3) Bosutinib and (4) RA plus Bosutinib. Bosutinib (Sigma) was used from a stock of 5 mM in Dimethyl sulfoxide (DMSO; Sigma) to make the final concentrations in culture 0.25 µM Bosutinib. Lyn KD cells were HL-60 stable transfectants expressing an shRNA targeting Lyn as reported previously by Rashid, et al [Citation29].
Antibodies and reagents
Antibodies for flow cytometric analysis, PE-conjugated CD38 (clone HIT2) and APC-conjugated CD11b (clone ICRF44) conjugated with allophycocyanin (APC), were from BD Biosciences (San Jose, CA). SLP-76, Lyn, Fgr, Vav1, p-tyr, HRP mouse and rabbit antibodies were purchased from Cell Signaling Technologies (Danvers, MA). Anti-c-Cbl (clone C-15, catalog number sc-170, lot H0414) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The c-Raf antibody was from BD Biosciences (San Jose, CA). Protease and phosphatase inhibitors were purchased from Sigma (St. Louis, MO). Protein G magnetic beads used for immunoprecipitation were from Millipore (Billerica, MA).
CD38, CD11b quantification and phenotypic analysis
Immunostaining for CD38 and CD11b was performed as previously stated [Citation30] and fluorescence was detected using a Becton Dickinson LSR II flow cytometer (San Jose, CA). Flow cytometric phenotypic analysis gating for positives was set to exclude 95% of the untreated cells in HL-60 wildtype and Lyn KD cells samples.
Measurement of respiratory burst (inducible oxidative metabolism)
0.5 × 106 cells from HL-60 cultures were harvested by centrifugation and resuspended in 200 μL PBS with 10 μM 5-(and-6)-chloromethyl-2ʹ,7ʹ – containing Acetyl ester of dichlorodihydrofluorescein diacetate (H2 -DCF, Molecular Probes, Eugene, OR) and 0.4 μg/mL 12-O-tetradecanoylphorbol-13-acetate (TPA, Sigma). Samples were incubated at 37°C in a humidified atmosphere of 5% CO2 for 20 minutes. Flow cytometric analysis was performed (BD LSRII flow cytometer) using 488-nm laser excitation and 505 long-pass collected emission. The fluorescence intensity shift in response of TPA was used to evaluate the percentage of cells with inducible generation of superoxide. Gates to assess the percentage of positive cells were set to exclude 95% of the control cells that did not get TPA to induce the respiratory burst distinguishing mature cells. Samples with or without TPA of cells that have not been RA-treated and without TPA of RA-treated cells showed indistinguishable DCF fluorescence histograms [Citation31]
Cell-cycle quantification
A total of 1 × 106 cells were centrifuged and resuspended in 200 μL of cold propidium iodide (PI) hypotonic staining solution containing 50 μg/mL of propidium iodide, 1 μL/mL of triton X-100 and 1 mg/mL of sodium citrate. Cells were incubated for 1 hour at 4°C and analyzed by flow cytometry using 488-nm excitation and emission measured with a 576/26 band-pass filter (BD LSRII flow cytometer). Doublets were identified and gated out from the analysis by a PI signal width versus area map.
Western blotting and immunoprecipitation
Cell fractionation was performed with the NE-PER kit (Pierce) in accordance with the manufacturer’s instructions, with the addition of protease and phosphatase inhibitors, and all lysates were stored at −80°C before use. After lysate collection, cell debris was cleared by centrifugation at 13,000 rpm for at least 10 minutes, and protein concentrations were quantified using the BCA (Pierce) assay.
For Western blotting, 25 μg protein per lane was resolved on a 12% polyacrylamide gel. The electro- transfer was done at 400 mA for 1 hour. The membranes were blocked in milk for 1 hour before adding the primary antibody and were incubated overnight at 4°C. The images were captured on a ChemiDoc XRS Bio-Rad Molecular Imager.
For immunoprecipitation, equal amounts of lysate were pre-cleared with Pure Proteome Protein G magnetic beads (Millipore, Billerica, MA, USA) for 2 hours and then incubated overnight with beads and 1 μg primary antibody. The beads were washed, boiled for 10 minutes, and SDS/PAGE analysis resolved the dissociated proteins, followed by electro-transfer to polyvinylidene fluoride (PVDF) membranes (Millipore).
Statistical analysis
The experiments were triplicate biological replicates, and the findings are shown as mean and standard deviation (SD). For the estimation of the difference between two classes, a two-tailed paired t test was used. A p value less than 0.05 was considered significant.
Results
Bosutinib or loss of Lyn does not cripple RA-induced differentiation but enhances it
To determine how Bosutinib and/or loss of Lyn affected RA-induced differentiation, we first determined the effect of Bosutinib on RA induced differentiation in HL-60 and Lyn KD cells by comparing cells that were untreated, treated with RA or Bosutinib alone or with RA in combination with Bosutinib The Lyn KD cells were created using shRNA and resulted in essentially total loss of Lyn [Citation29]. The concentration of Bosutinib used was 0.25 µM which was established previously as the lowest dose that enhances RA-induced differentiation in HL-60 cells and has no detectably overt toxicity [Citation32,Citation33].We measured induced differentiation over a 72 h treatment period by assessing CD38 and CD11b expression, two cell surface receptors that are differentiation markers. Expression of the CD38 and CD11b cell surface differentiation markers was measured by immunofluorescence and flow cytometry. CD38 is an ectoenzyme receptor, and CD11b is the integrin receptor β subunit. Expression of these cell differentiation markers was compared for cells treated with RA (10−6 M) alone, Bosutinib (B) alone, or RA in combination with Bosutinib (RA/B) over 8, 24, 48, and 72 h treatment periods in HL-60 and Lyn KD cells cultures. For both HL-60 and Lyn KD cells, CD38 expression was nearly was 75% positive in the RA and RA/Bosutinib treated cells at 8 h and 100% in both RA and RA/Bosutinib treated cells at 24, 48, and 72 h, respectively (). Neither the SFK inhibitor, Bosutinib, nor Lyn KD affected RA-induced CD38 expression.
Figure 1. Phenotypic cell surface differentiation marker analysis of HL-60 wt and Lyn KD cells that were untreated Control or treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated at the time points: 8, 24, 48, and 72 h. (a) CD38 expression was assessed by flow cytometry following the treatment period. Gating to discriminate positive cells was set to exclude 95% of untreated controls. Percentage of cells positive for CD38 expression is shown (n = 3). Error bars indicate SEM. (b) CD11b expression was assessed by flow cytometry after 72 h treatment periods. Gating to discriminate positive cells was set to exclude 95% of untreated controls (n = 3). Percentage of cells positive for CD11b expression at 72 h is shown (n = 3). * p < .05 comparing RA-treated HL-60 wt samples to RA Lyn KD cells samples
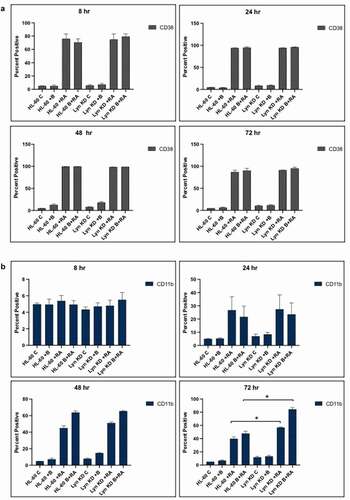
Bosutinib significantly increased RA-induced CD11b expression in HL-60 cells as well as Lyn KD cells (). The RA-induced expression of CD11b at 48 and 72 h was increased by addition of Bosutinib for both HL-60 and Lyn KD cells. Thus RA-induced expression of CD11b was increased by addition of Bosutinib even in the absence of Lyn. Interestingly, in the absence of RA and Lyn, Bosutinib induced low level expression of CD11b. The data show that even in the absence of Lyn expression, Bosutinib can improve aspects of RA-induced differentiation, suggesting an off target effect separate from Lyn kinase inhibition of Bosutinib.
Bosutinib or loss of Lyn does not cripple RA-induced G1/G0 arrest but enhances it
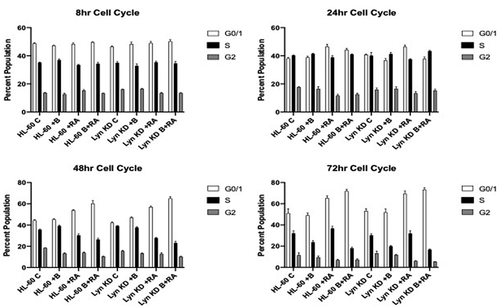
To determine how Bosutinib and/or loss of Lyn affected G1/G0 cell cycle arrest, which is typically associated with differentiation, the effect of Bosutinib on RA-induced cell cycle arrest of HL-60 and Lyn KD cells was compared. Cells were untreated, treated with RA alone, Bosutinib alone, or with RA in combination with Bosutinib. Cell cycle phase distribution was measured by flow cytometry in these HL-60 and Lyn KD cells. HL-60, and Lyn KD cells were grown in parallel untreated and RA-treated (10−6 M) cultures with and without Bosutinib. Cell cycle distribution analysis showed RA-induced G1/G0 enrichment at 48, and 72 h for both HL-60 and Lyn KD cells. Addition of Bosutinib increased the RA-induced G1/G0 enrichment for both HL-60 and Lyn KD cells (). Thus RA-induced cell cycle arrest was enhanced by Bosutinib even in the absence of Lyn, suggesting this Bosutinib effect is independent of Lyn.
Figure 2. Cell cycle distribution showing the percentages of cells in G1, S, G2/M for HL-60 wt and Lyn KD cells that were untreated Control or treated with 1 µM RA, 0.25 µM Bosutinib, or 1 µM RA plus 0.25 µM Bosutinib as indicated at the time points: 8, 24, 48, and 72 h. DNA-stained cells were analyzed using flow cytometry with propidium iodide staining. Error bars indicate SEM (n = 3). * p < .05 comparing untreated samples to RA/ treated samples
Bosutinib or loss of Lyn does not cripple, but enhances, RA-induced oxidative metabolism (ROS production)
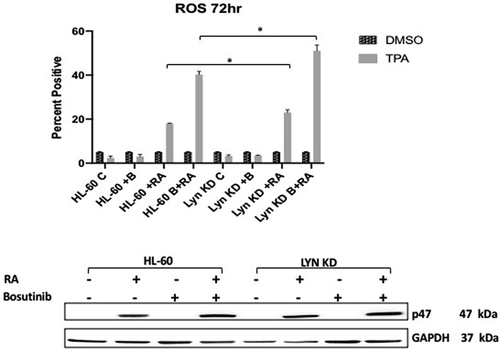
To determine how Bosutinib or loss of Lyn affected an RA-induced functional differentiation marker for mature myelo-monocytic cells, namely inducible respiratory burst, we measured induced reactive oxygen species production (ROS). HL-60 and Lyn KD cells were untreated, treated with RA alone, Bosutinib alone, or with RA in combination with Bosutinib. Inducible reactive oxygen species production (ROS) was measured by flow cytometry at 72 h. RA caused an increase in ROS compared to untreated cells that was enhanced by co-treatment with Bosutinib. The same thing happened with Lyn KD cells, indicating that this effect of Bosutinib on the RA-induced functional differentiation marker did not depend on Lyn (). Interestingly, as for CD11b, Lyn KD enhanced the RA/Bosutinib-induced response. RA-induced p47phox, a component of the NADPH oxidase machinery that generates the inducible oxidative metabolism response, largely paralleled this as expected, corroborating the ROS results.
Figure 3. Functional differentiation marker, TPA-induced respiratory burst, analysis of HL-60 wt and Lyn KD cells that were untreated Control or treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated at 72 h. (a) Respiratory burst was analyzed by measuring TPA-inducible reactive oxygen species (ROS) production by flow cytometry using the 2′,7′-dichlorofluorescein (DCF) assay. Gates were set to exclude 95% of the DMSO-treated control population (carrier control) for each culture condition (n = 3). Error bars indicate SEM. Inducible ROS production is betrayed by the shift in the TPA histogram compared to the DMSO histogram. Percentage of cells showing inducible oxidative metabolism is shown. (b) Expression of p47phox by HL-60 wt versus Lyn KD cells that were untreated or treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated. Cells were cultured for 72 h and the whole cell lysate was collected. Twenty-five microgram of lysate per lane was used. Western blots of SDS PAGE-resolved lysates were probed for p47phox, (n = 3 biological repeats). GAPDH was used as loading controls following the procedure described above
Bosutinib or loss of Lyn does not cripple RA-induced upregulation/phosphorylation of molecules in the signalsome that are putative drivers of differentiation
To determine how Bosutinib, an SFK inhibitor, or loss of the Lyn affected putative signalsome components implicated in control of RA-induced differentiation in HL-60 cells, we assessed expression and phosphorylation of signasome molecules previously found to drive RA-induced differentiation. HL-60 and Lyn KD cells were untreated, treated with RA or Bosutinib alone, or with RA in combination with Bosutinib. After a 72 h treatment period, we collected cell lysate and analyzed the proteins via Western blot and immunoprecipitation.
It is known that the SFKs contribute to myeloid cell leukemia and cancer [Citation32,Citation34]. It is also known that SFKs, Fgr and Lyn, are upregulated by Retinoic acid (RA) to propel differentiation and growth arrest of myeloblastic leukemia cells such as HL-60 [Citation35–38]. The regulatory role of these kinases is ergo complicated. To determine whether Fgr upregulation by RA was affected by Bosutinib or Lyn KD as well as whether Lyn upregulation by RA was affected by Bosutinib, their expression was measured. HL-60 and Lyn KD cells were untreated, treated with RA or Bosutinib alone, or with RA in combination with Bosutinib (). Bosutinib did not cripple RA-induced upregulation of Fgr or Lyn; indeed, co-treatment with Bosutinib actually modestly enhanced expression. Lyn KD also did not cripple RA-induced upregulation of Fgr expression. In Lyn KD cells, Lyn expression was only marginally detectable after RA treatment, and remained minimal with Bosutinib co-treatment. Hence the RA-induced expression of Fgr was not crippled by Bosutinib or by Lyn KD; nor was RA-induced Lyn expression crippled by Bosutinib.
Figure 4. Regulation of putative differentiation regulating molecules in HL-60 wt versus Lyn KD cells by RA and Bosutinib. HL-60 wt and Lyn KD cells were untreated and treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated. Cells were cultured for 72 h, and the whole cell lysate was collected. Twenty-five microgram of lysate per lane was used. Western blots of SDS PAGE-resolved lysates were probed for Lyn, Vav 1, Fgr, c-Cbl, and Slp76 (n = 3 biological repeats). GAPDH was used as the loading control following the procedure described above
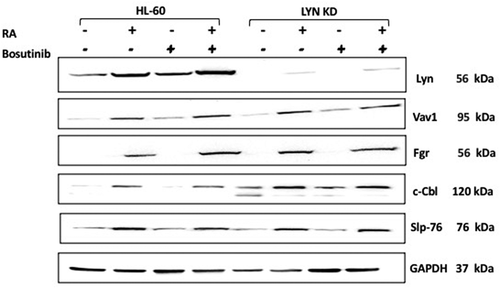
Vav, Cbl, and Slp-76 are putative promoters of myeloid differentiation that are upregulated by RA [Citation39].Vav1 is a guanine nucleotide-exchange factor (GEF); and it’s the only member of the Vav family expressed in hematopoietic cells [Citation40]. It can also function as an adaptor [Citation41]. It has been reported to be necessary for myelopoiesis [Citation41–43]. Both c-Cbl and SLP-76 are adaptor proteins that drive sustained MAPK signaling to facilitate RA induced cell differentiation [Citation39]. To determine whether Vav1, c-Cbl and SLP-76 upregulation by RA were affected by Bosutinib or Lyn KD, their expression was measured. HL-60 and Lyn KD cells were untreated, treated with RA or Bosutinib alone, or with RA in combination with Bosutinib. RA increased the expression of Vav, c-Cbl and SLP-76 in the HL-60 wt cells, and this increase was not crippled by either Bosutinib or by Lyn KD. Interestingly, in RA-treated cells, Lyn KD actually increased the expression of c-Cbl and this persisted with Bosutinib co-treatment.
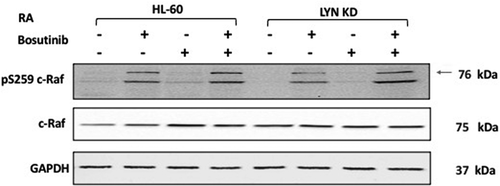
The expression and activation of the Raf kinase is known to propel RA-induced differentiation [Citation44,Citation45]. Raf is an element of the canonical Raf/Mek/Erk cascade of MAPK signaling necessary for differentiation [Citation46]; and pertinent to the present consideration, SFKs can stimulate the MAPK signaling axis. We analyzed the effect of Bosutinib and Lyn KD on RA-induced c-Raf activation. To determine whether regulation of c-Raf by RA was affected by Bosutinib or Lyn KD, phosphorylation and expression was measured in HL-60 and Lyn KD cells that were untreated, treated with RA or Bosutinib alone, or with RA in combination with Bosutinib (). We evaluated the phosphorylation of c-Raf at the S259 regulatory site by Western blot. Neither Bosutinib nor Lyn KD crippled the Retinoic acid (RA) induced phosphorylation of c-Raf. Nor was expression of c-Raf crippled.
Figure 5. Regulation of pS259 Raf and Raf in HL-60 wt versus Lyn KD cells by RA and Bosutinib. HL-60 wt and Lyn KD cells were untreated Controls or treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated. Cells were cultured for 72 h, and the whole cell lysate was collected. Twenty-five microgram of lysate per lane was used. Western blots of SDS PAGE-resolved lysates were probed for pS259 c-Raf and c-Raf, (n = 3 biological repeats). GAPDH was used as loading control following the procedure described above
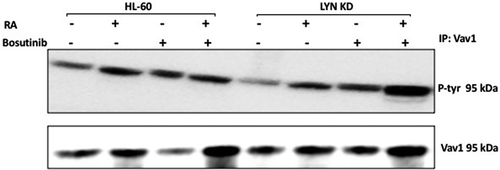
Historically identified as a GEF, Vav was found to also act as an adaptor [Citation43]. PTKs (protein tyrosine kinases) can phosphorylate Vav and activate it as an adaptor [Citation41,Citation47,Citation48]. RA causes upregulation of Vav associated with induced myeloid differentiation [Citation49]. Consistent with the need for Vav to propel myeloid differentiation, loss of Vav was found to disrupt myeloid cell differentiation in certain animal models, albeit not necessarily all [Citation49], suggesting the possibility that its phosphorylation status may determine its function. To determine if RA regulated Vav, tyrosine phosphorylation of Vav and the effect of Bosutinib or loss of Lyn thereon was measured. HL-60 and Lyn KD cells were untreated, treated with RA or Bosutinib alone, or with RA in combination with Bosutinib (). Tyrosine phosphorylation of Vav increased after treating with RA in HL-60 wt cells and in Lyn KD cells. Cells were grown in parallel untreated and treated cultures, harvested after 72 h, and lysates subject to immunoprecipitation using anti-Vav as bait and probed for p-tyr by Western blotting. RA caused tyrosine phosphorylation of Vav in wt HL-60 cells and in Lyn KD cells. This was not crippled by either Bosutinib or by Lyn KD. Interestingly, Bosutinib by itself increased tyrosine phosphorylation of Vav, and in the Lyn KD cells we also observed an increase in the tyrosine phosphorylation of Vav after Bosutinib and RA co-treatment. The results are consistent with a potential novel off-target effect of Bosutinib on Vav that is independent of Lyn.
Figure 6. Regulation of Vav1 tyrosine phosphorylation in HL-60 wt and Lyn KD cells by RA and Bosutinib. HL-60 wt and Lyn KD cells were untreated Control or treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated. Cells were cultured for 72 h. Vav1 was immunoprecipitated and immunoprecipitates were probed with anti-p-tyr for tyrosine phosphorylation by Western blotting (upper). Presence of Vav1 bait in immunoprecipitate was confirmed (lower). All blots shown are representative of three biological replicates, and the trend for changes in signal intensity levels are consistent among the repeats
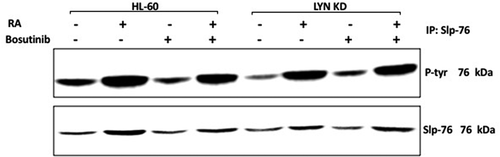
Slp-76 was identified to be one of the most highly tyrosine phosphorylated molecules induced in RA-treated HL-60 cells [Citation39]. Its over expression contributes propulsion to RA-induced differentiation in conjunction with c-FMS [Citation39]. To determine whether Slp-76 tyrosine phosphorylation was affected by Bosutinib or Lyn KD, tyrosine phosphorylated Slp-76 was measured. HL-60 and Lyn KD cells were untreated, treated with RA or Bosutinib alone, or with RA in combination with Bosutinib. After 72 h the cells were harvested and lysates subject to analysis for tyrosine phosphorylation of Slp-76 by immunoprecipitation using Slp-76 as bait and probed with anti-p-tyr Western blotting (). HL-60 wt cells showed prominent RA-induced Slp-76 tyrosine phosphorylation. Neither Bosutinib nor Lyn KD crippled this.
Figure 7. Regulation of Slp-76 tyrosine phosphorylation in HL-60 wt and Lyn KD cells by RA and Bosutinib. HL-60 wt and Lyn KD cells were untreated Controls or treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated. Cells were cultured for 72 h. Slp-76 was immunoprecipitated and immunoprecipitates were probed with anti-p-tyr for tyrosine phosphorylation by Western blotting (upper). Presence of Slp-76 bait in immunoprecipitate was confirmed (lower). All blots shown are representative of three biological replicates, and the trend for changes in signal intensity levels are consistent among the repeats
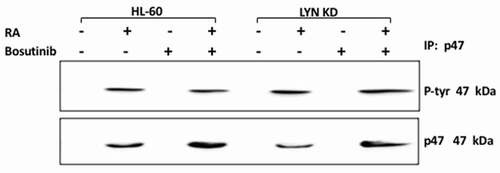
p47 phox, is a phosphoprotein that is not expressed but rapidly prominently induced in RA-treated HL-60 cells, suggesting potential regulatory significance in promoting signaling seminal to differentiation. It is historically classified as a main component of the NADPH oxidase complex, which mediates inducible oxidative metabolism which functionally characterizes mature myeloid cells. It is also thought to regulate the intracellular oxidative state which regulates signaling pathways associated with both peptide and steroid hormones – all of which are putatively involved in the present context. Evidence of RA-induced tyrosine phosphorylation would potentially point to regulatory significance for this early, rapidly induced phospho-protein in the signaling driving differentiation. HL-60 and Lyn KD cells were untreated, treated with RA or Bosutinib alone, or with RA in combination with Bosutinib, and harvested after 72 h for analysis of tyrosine phosphorylation of p47 phox (). Lysates were immunoprecipitated using anti-p47 phox as bait and probed for p-tyr by Western blotting. In HL-60 wt cells p47 phox tyrosine phosphorylation was initially undetectable until RA induced its prominent expression. Neither co-treatment with Bosutinib or Lyn KD diminished this. Hence there was no crippling of this early molecular marker of cells undergoing differentiation by either Bosutinib or Lyn KD. Bosutinib thus had no crippling effect on neither the rapid nor slower molecular signaling responses tested here.
Figure 8. Regulation of P47 phox tyrosine phosphorylation in HL-60 wt and Lyn KD cells by RA and Bosutinib. HL-60 wt and Lyn KD cells were untreated Controls or treated with 1 µM RA, or 0.25 µM Bosutinib or 1 µM RA and 0.25 µM Bosutinib as indicated. Cells were cultured for 72 h. p47 phox was immunoprecipitated and immunoprecipitates were probed with anti-p-tyr for tyrosine phosphorylation by Western blotting (upper). Presence of p47 phox bait in immunoprecipitate was confirmed (lower). All blots shown are representative of three biological replicates, and the trend for changes in signal intensity levels are consistent among the repeats
DISCUSSION
The present study probed the mechanism by which the kinase inhibitor, Bosutinib, enhanced RA-induced differentiation of a non-APL AML cell line, HL-60. It is a generally taken tenet that SFK hyper-activity contributes to myeloid leukemogenesis. We have previously established that RA-induced leukemic cell differentiation in this model is enhanced by Bosutinib co-treatment [Citation33]. Lyn is the most prominent SFK in these cells. We also found that essentially eliminating Lyn expression by shRNA KD enhanced RA-induced differentiation [Citation29]. If we accept that Bosutinib is an inhibitor of SFKs and that the primary SFK over expressed in these leukemic myeloid cells and targeted by Bosutinib is Lyn, then loss of Lyn should cripple the ability of Bosutinib to enhance RA-induced differentiation. Many kinase inhibitors have effects other than inhibiting the target kinase, hence this is a test of whether there are off-target effects of the drug that could be beneficial in an application other than cytotoxic attack, namely in differentiation therapy. The novel finding was that Bosutinib still enhanced RA-induced differentiation even if there was no Lyn to target. There is an ensemble of molecules that constitute a signalsome that propels RA-induced differentiation [Citation50]. RA-induced activation of the signalsome is betrayed by upregulation/phosphorylation of certain components thereof [Citation36,Citation51]. Surveying some of them already individually known to drive differentiation to determine if Bosutinib or loss of the Lyn kinase disrupted any of these [Citation29,Citation33], we found no induced phosphorylation crippled by the Bosutinib or loss of the Lyn kinase amongst these candidates. Interestingly Bosutinib actually apparently enhanced tyrosine phosphorylation of Vav, a signalsome component reported to be needed for sustaining normal myeloid cell differentiation [Citation52]. Hence we take this as evidence that Bosutinib used in co-treatment with RA enhances the RA-induced differentiation in an off-target effect distinct from inhibition of Lyn.
Although the Src family of kinases has been known for nearly a century, a complete conceptual model of their action as mediators of biological signals and as kinases is still actively pursued. Myelo-monocytic differentiation, which yields mature granulocytes and monocytes/macrophages, is an essential element of the host defense mechanism against invading microorganisms. That being said, in myeloid leukemias, genetic changes prevent myeloid differentiation. Once this takes place, immune responses are defeated, and the accumulation of proliferative blasts causes bone marrow crowding and the onset of symptomatic leukemia. There is the hope that molecular targeting of specific signaling pathways and proteins might be an effective method for restoring normal differentiation in myeloid leukemias. Regimens with RA could relieve the differentiation block and are a successful therapeutic strategy in acute promyelocytic leukemia. To figure out ways and reagents for triggering differentiation in other myeloid leukemias, it is essential to know the molecular pathways that drive the normal signaling pathway.
Src family kinases tend to be negative regulators of myelopoiesis, while the MEK/ERK pathway, which they can target, is an essential key regulator of both granulocytic and monocytic differentiation. This strongly indicates that SFK inhibitors may be effective in recovering or boosting myeloid differentiation when it is blocked in leukemia. The FDA approved the SFK inhibitor Bosutinib for use in imatinib-resistant CML. Bosutinib is a tyrosine kinase inhibitor that is readily available in the United States for the treatment of chronic myeloid leukemia. Bosutinib is ergo of potential interest in the treatment of differentiation-resistant AML, either alone or in combination with differentiation inducers.
In this study, we investigated how Bosutinib enhanced RA-induced differentiation of HL-60 cells in pursuit of exploring its therapeutic utility in differentiation therapy of non-APL AML. In particular, we asked if this depended on Lyn by testing if Bosutinib still enhanced RA-induced differentiation in the absence of Lyn. Combination therapy with RA has piqued the interest of many researchers as a means of improving RA-induced differentiation and loss of the transformed/cancer phenotype. We recently reported that combined RA/Bosutinib treatment enhanced several myeloid lineage differentiation markers compared to RA treatment alone: CD11b expression, G1/G0 cell cycle arrest, and inducible respiratory burst, a functional marker of mature myeloid series cells [Citation33]. In contrast, RA-induced expression of CD38, which is an early marker of RA-induced differentiation, shows no significant difference attributable to Bosutinib. We have previously reported that RA-induced CD38 expression may not be necessary for the RA-induced differentiation process [Citation11,Citation33,Citation53]. Bosutinib is an SFK inhibitor, but RA causes Lyn to be upregulated. Lyn’s function in RA-induced differentiation is thus enigmatic. In this regard, it is noteworthy that SFK inhibitors may also serve as activators in a number of situations [Citation54], suggesting that context is of significance. To gain insight into this paradox, we examined several mediators of signaling seminal to RA-induced differentiation. Since Bosutinib is a SFK inhibitor, we assessed its effects on the two prominent SFK members expressed in RA-treated HL-60 cells, Fgr and Lyn, the primary active SFKs expressed in AML cells [Citation19,Citation55]. We found that RA increased Lyn expression in HL-60 wt cells, and the expression was further enhanced by co-treatment with Bosutinib. There is no Lyn expression in Lyn KD cells, but it is minimally induced by RA without/or with co-treatment with Bosutinib. For HL-60 wt and Lyn KD cells, the expression of Fgr was induced by RA treatment, and Bosutinib co-treatment caused no significant change from that induced by RA alone. We then examined Vav1 expression. Vav1, a guanine nucleotide exchange factor, is linked to the differentiation marker, CD38, and plays a key role in the activation of the MAPK signaling cascade [Citation56]. We observed that in HL-60 wt cells, RA increased Vav1 expression, and Bosutinib co-treatment did not alter it. RA also increased Vav1 expression in Lyn KD cells, and this enhancement also was not affected by co-treatment with Bosutinib. Nor was RA-induced tyrosine phosphorylation of Vav compromised by either Bosutinib or by Lyn KD. RA-induced differentiation and G1/G0 arrest of HL-60 cells are also facilitated by the adaptor proteins c-Cbl and SLP-76, both of which immunoprecipitated with Lyn [Citation35]. Therefore we looked at the levels of these adaptor proteins which are linked to RA-induced differentiation and known to be abnormally activated in AML [Citation30,Citation35,Citation57]. RA induced the prominent upregulation of c-Cbl and SLP-76 expression in HL-60 wt cells, which was nor impaired by Bosutinib. RA-induced tyrosine phosphorylation of Slp-76 was also not compromised by either Bosutinib or by Lyn KD. These kinases and adaptors are well known regulators of classical MAPK pathway signaling of which the Raf/Mek/Erk axis is imbedded in the activated signalosome with other individual components such as Lyn, Fgr, Slp-76, and Cbl, which have all been implicated in propelling RA-induced differentiation. Raf could be considered the prototype of signalsome constituents providing propulsion for RA-induced HL-60 cell differentiation by its activation and nuclear translocation associated with targeting of RB [Citation44,Citation45,Citation58,Citation59]. Given that Bosutinib enhanced RA-induced differentiation, we asked how it affected Raf. RA enhanced c-Raf expression and prominently induced pS259 phosphorylation of Raf in HL-60 wt cells, and neither was compromised by either Bosutinib or by Lyn KD. There was ergo no obvious evidence that Bosutinib was acting as a Lyn inhibitor to enhance RA-induced differentiation, suggesting there is an off-target effect. To our knowledge, this is the first indication that Bosutinib enhances RA’s ability to induce myeloid cell differentiation in the absence of Lyn.
Acknowledgments
We are grateful to Dr. John Babish and Paracelsian for generous support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- De Vos S, Koeffler HP. Differentiation induction in leukemia and lymphoma. Nutrition Oncol. 2006:491
- Grignani F, Fagioli M, Alcalay M, et al. Acute promyelocytic leukemia: from genetics to treatment. 1994.
- Bunaciu RP, Yen A. Retinoid chemoprevention: who can benefit? Current Pharmacol Rep. 2015;1(6):391–400.
- Brown G, Hughes P. Retinoid differentiation therapy for common types of acute myeloid leukemia. Leuk Res Treatment. 2012;2012:1–11.
- Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111–121.
- Kohrt HE, Coutre SE. Optimizing therapy for acute myeloid leukemia. J National Compr Cancer Network. 2008;6(10):1003–1016.
- Al-Hussaini M, DiPersio JF. Small molecule inhibitors in acute myeloid leukemia: from the bench to the clinic. Expert Rev Hematol. 2014;7(4):439–464.
- Johnson DE, Redner RL. An ATRActive future for differentiation therapy in AML. Blood Rev. 2015;29(4):263–268.
- Warrell RJ. Retinoid resistance in acute promyelocytic leukemia: new mechanisms, strategies, and implications [editorial; comment]. 1993.
- Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene. 2003;22(47):7305–7315.
- Bunaciu RP, MacDonald RJ, Gao F, et al. Potential for subsets of wt-NPM1 primary AML blasts to respond to retinoic acid treatment. Oncotarget. 2018;9(3):4134.
- Iriyama N, Yuan B, Hatta Y, et al. Lyn, a tyrosine kinase closely linked to the differentiation status of primary acute myeloid leukemia blasts, associates with negative regulation of all-trans retinoic acid (ATRA) and dihydroxyvitamin D3 (VD3)-induced HL-60 cells differentiation. Cancer Cell Int. 2016;16(1):1–11.
- Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23(48):7906–7909.
- Kim M, Park SI, Kopetz S, et al. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335(1):249–259.
- Johnson DE. Src family kinases and the MEK/ERK pathway in the regulation of myeloid differentiation and myeloid leukemogenesis. Adv Enzyme Regul. 2008;48:98.
- Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26(7):641–650.
- Superti-Furga G, Gonfloni S. A crystal milestone: the structure of regulated Src. Bioessays. 1997;19(6):447–450.
- Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997;7(6):777–785.
- Kropf P, Wang L, Zang Y, et al. Dasatinib promotes ATRA-induced differentiation of AML cells. Leukemia. 2010;24(3):663–665.
- Breitman T, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Nat Acad Sci. 1980;77(5):2936–2940.
- Kikuchi H, Yuan BO, Nishimura Y, et al. Cytotoxicity of Vitex agnus-castus fruit extract and its major component, casticin, correlates with differentiation status in leukemia cell lines. Int J Oncol. 2013;43(6):1976–1984.
- Collins SJ. The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. 1987.
- Guerrouahen BS, Futami M, Vaklavas C, et al. Dasatinib inhibits the growth of molecularly heterogeneous myeloid leukemias. Clin Cancer Res. 2010;16(4):1149–1158.
- Isfort S, Crysandt M, Gezer D, et al. Bosutinib: a potent second-generation tyrosine kinase inhibitor. In: Small Molecules in Hematology. Cham: Springer; 2018:87–108.
- Golas JM, Arndt K, Etienne C, et al. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63(2):375–381.
- Gambacorti-Passerini C, Coutre PL, Piazza R. The role of bosutinib in the treatment of chronic myeloid leukemia. Future Oncol. 2020;16(2):4395–4408.
- Keller-von Amsberg G, Koschmieder S. Profile of bosutinib and its clinical potential in the treatment of chronic myeloid leukemia. Onco Targets Ther. 2013;6:99.
- Boschelli DH, Ye F, Wang YD, et al. Optimization of 4-phenylamino-3-quinolinecarbonitriles as potent inhibitors of Src kinase activity. J Med Chem. 2001;44(23):3965–3977.
- Rashid A, Duan X, Gao F, et al. Roscovitine enhances all-trans retinoic acid (ATRA)-induced nuclear enrichment of an ensemble of activated signaling molecules and augments ATRA-induced myeloid cell differentiation. Oncotarget. 2020;11(12):1017–1036.
- Shen M, Yen A. c-Cbl interacts with CD38 and promotes retinoic acid–induced differentiation and G0 arrest of human myeloblastic leukemia cells. Cancer Res. 2008;68(21):8761–8769.
- Bunaciu RP, Yen A. Activation of the Aryl hydrocarbon receptor ahr promotes retinoic acid–induced differentiation of myeloblastic leukemia cells by restricting expression of the stem cell transcription factor Oct4. Cancer Res. 2011;71(6):2371–2380.
- Vultur A, Buettner R, Kowolik C, et al. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol Cancer Ther. 2008;7(5):1185–1194.
- MacDonald RJ, Bunaciu RP, Ip V, et al. Src family kinase inhibitor bosutinib enhances retinoic acid-induced differentiation of HL-60 leukemia cells. Leuk Lymphoma. 2018;59(12):2941–2951.
- Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6(10):587–595.
- Congleton J, Shen M, MacDonald R, et al. Phosphorylation of c-Cbl and p85 PI3K driven by all-trans retinoic acid and CD38 depends on Lyn kinase activity. Cell Signal. 2014;26(7):1589–1597.
- Kazim N, Yen A. Role for Fgr and Numb in retinoic acid-induced differentiation and G0 arrest of non-APL AML cells. Oncotarget. 2021;12(12):1147–1164.
- Jensen HA, Bunaciu RP, Ibabao CN, et al. Retinoic acid therapy resistance progresses from unilineage to bilineage in HL-60 leukemic blasts. PloS One. 2014;9(6):e98929.
- Congleton J, MacDonald R, Yen A. Src inhibitors, PP2 and dasatinib, increase retinoic acid-induced association of Lyn and c-Raf (S259) and enhance MAPK-dependent differentiation of myeloid leukemia cells. Leukemia. 2012;26(6):1180–1188.
- Yen A, Varvayanis S, Smith JL, et al. Retinoic acid induces expression of SLP-76: expression with c-FMS enhances ERK activation and retinoic acid-induced differentiation/G0 arrest of HL-60 cells. Eur J Cell Biol. 2006;85(2):117–132.
- Katzav S. Vav1: an oncogene that regulates specific transcriptional activation of T cells. Blood. 2004;103(7):2443–2451.
- Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20(5):1461–1477.
- Collins TL, Deckert M, Altman A. Views on Vav. Immunol Today. 1997;18(5):221–225.
- Bustelo XR. Vav family exchange factors: an integrated regulatory and functional view. Small GTPases. 2014;5(2):9.
- Wang J, Yen A. A MAPK-positive feedback mechanism for BLR1 signaling propels retinoic acid-triggered differentiation and cell cycle arrest. J Biol Chem. 2008;283(7):4375–4386.
- Yen A, Williams M, Platko JD, et al. Expression of activated RAF accelerates cell differentiation and RB protein down-regulation but not hypophosphorylation. Eur J Cell Biol. 1994;65(1):103–113.
- Yen A, Roberson MS, Varvayanis S, et al. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998;58(14):3163–3172.
- Katagiri K, Hattori S, Nakamura S, et al. Activation of Ras and formation of GAP complex during TPA-induced monocytic differentiation of HL-60 cells. 1994.
- Perez-Villar JJ, Whitney GS, Sitnick MT, et al. Phosphorylation of the linker for activation of T-cells by Itk promotes recruitment of Vav. Biochemistry. 2002;41(34):10732–10740.
- Bertagnolo V, Brugnoli F, Mischiati C, et al. Vav promotes differentiation of human tumoral myeloid precursors. Exp Cell Res. 2005;306(1):56–63.
- Ibabao CN, Bunaciu RP, Schaefer DMW, et al. The AhR agonist VAF347 augments retinoic acid-induced differentiation in leukemia cells. FEBS Open Bio. 2015;5(1):308–318.
- Tasseff R, Jensen HA, Congleton J, et al. An effective model of the retinoic acid induced HL-60 differentiation program. Sci Rep. 2017;7(1):1–21.
- Margolis B, Hu P, Katzav S, et al. Tyrosine phosphorylation of vav proto-oncogene product containing SH2 domain and transcription factor motifs. Nature. 1992;356(6364):71–74.
- Abbas R, Hsyu P-H. Clinical pharmacokinetics and pharmacodynamics of bosutinib. Clin Pharmacokinet. 2016;55(10):1191–1204.
- Elias D, Ditzel HJ. The potential of Src inhibitors. Aging (Albany Ny). 2015;7(10):734.
- Dos Santos C, Demur C, Bardet V, et al. A critical role for Lyn in acute myeloid leukemia. Blood J Am Soc Hematol. 2008;111(4):2269–2279.
- Shen M, Yen A. c-Cbl tyrosine kinase-binding domain mutant G306E abolishes the interaction of c-Cbl with CD38 and fails to promote retinoic acid-induced cell differentiation and G0 arrest. J Biol Chem. 2009;284(38):25664–25677.
- MacDonald RJ, Shrimp JH, Jiang H, et al. Probing the requirement for CD38 in retinoic acid-induced HL-60 cell differentiation with a small molecule dimerizer and genetic knockout. Sci Rep. 2017;7(1):1–11.
- Smith J, Bunaciu RP, Reiterer G, et al. Retinoic acid induces nuclear accumulation of Raf1 during differentiation of HL-60 cells. Exp Cell Res. 2009;315(13):2241–2248.
- Wallace AS, Supnick HT, Bunaciu RP, et al. RRD-251 enhances all-trans retinoic acid (RA)-induced differentiation of HL-60 myeloblastic leukemia cells. Oncotarget. 2016;7(29):46401.