
ABSTRACT
The maintenance of cellular cholesterol homeostasis is essential for normal cell function and viability. Excessive cholesterol accumulation is detrimental to cells and serves as the molecular basis of many diseases, such as atherosclerosis, Alzheimer’s disease, and diabetes mellitus. The peripheral cells do not have the ability to degrade cholesterol. Cholesterol efflux is therefore the only pathway to eliminate excessive cholesterol from these cells. This process is predominantly mediated by ATP-binding cassette transporter A1 (ABCA1), an integral membrane protein. ABCA1 is known to transfer intracellular free cholesterol and phospholipids to apolipoprotein A-I (apoA-I) for generating nascent high-density lipoprotein (nHDL) particles. nHDL can accept more free cholesterol from peripheral cells. Free cholesterol is then converted to cholesteryl ester by lecithin:cholesterol acyltransferase to form mature HDL. HDL-bound cholesterol enters the liver for biliary secretion and fecal excretion. Although how cholesterol is transported by ABCA1 to apoA-I remains incompletely understood, nine models have been proposed to explain this effect. In this review, we focus on the current view of the mechanisms underlying ABCA1-mediated cholesterol efflux to provide an important framework for future investigation and lipid-lowering therapy.
KEYWORDS:
1. Introduction
All mammalian cells require cholesterol that is essential for normal cell function, from maintenance of membrane fluidity to biogenesis of bile acids, steroid hormones and vitamin D. Excessive cholesterol, however, is toxic to the cells. It is well established that cholesterol accumulation in macrophages and vascular smooth muscle cells (VSMCs) leads to foam cell formation and atherogenesis [Citation1]. Atherosclerosis is the pathological basis of most cardiovascular disease (CVD), the leading cause of morbidity and mortality worldwide. A 25-year epidemiological study has demonstrated that CVD accounts for >17 million deaths globally every year, which is predicted to rise to >23 million by 2030 [Citation2]. Excessive cholesterol is also associated with occurrence and development of Alzheimer’s disease and diabetes mellitus [Citation3,Citation4]. Thus, the maintenance of optimal cellular cholesterol concentration is necessary for human healthy.
In mammals, cells obtain cholesterol through endogenous synthesis and exogenous uptake. The liver is the only organ than can convert cholesterol to bile acids for biliary secretion and fecal excretion. However, the peripheral cells, particularly macrophages and VSMCs in the arterial wall, do not possess this capability to metabolize cholesterol. To maintain cholesterol homeostasis and prevent toxicity, these cells must release surplus cholesterol into extracellular acceptors such as high-density lipoprotein (HDL) for removal. ATP-binding cassette transporter A1 (ABCA1), a transmembrane protein, is highly expressed in the liver, intestine, brain, and macrophages. Its major function is to mediate the transport of free cholesterol (FC) and phospholipids from cells to apolipoprotein A-I (apoA-I) for generating nascent HDL (nHDL) particles [Citation5]. Studies from our group and others have revealed that ABCA1-dependent cholesterol efflux plays a central role in blocking the transformation of macrophages and VSMCs into foam cells, the hallmark of early atherosclerotic lesions [Citation6–10]. Despite the detailed molecular mechanisms by which ABCA1 transfers cholesterol to apoA-I are not fully understood, a variety of models have been proposed to explain this effect. In the present review, we summarize the current knowledge about the structure and functions of ABCA1 with an emphasis on how it mediates cholesterol efflux.
2. Structural features of ABCA1
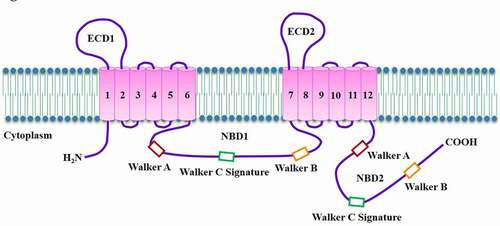
Human ABCA1 gene has been mapped to chromosome 9q31.1, spans 149 kb, and comprises 50 exons and 49 introns [Citation11]. The mature ABCA1 protein is composed of 2261 amino acid residues with a predicted molecular mass of 254 kDa. As an integral membrane protein, ABCA1 has two transmembrane domains (TMDs) linked covalently, each containing six transmembrane α-helices (TMs) [Citation12]. Unique to the ABCA subfamily, ABCA1 has two large extracellular domains (ECDs) and multiple membrane-spanning segments ()[Citation13]. ECD1 resides between TM1 and TM2, and ECD2 resides between TM7 and TM8. They are connected by two intramolecular disulfide bonds and serve as the binding sites for apoA-I [Citation14]. There are two nucleotide-binding domains (NBDs) in its intracellular region. Their amino acid sequences are highly conserved among ABC transporters. Each NBD contains two conserved peptide motifs known as Walker A and Walker B, and a Walker C signature unique to ABC proteins [Citation15]. Walker A is responsible for ATP binding while Walker B is associated with subsequent hydrolysis [Citation16]. It is worth noting that ATP can bind equally to both NBDs. The hydrolysis of ATP in these sites can alter ECD conformation, which is essential for apoA-I binding [Citation17]. In the NBD1, the PEST (proline, glutamic acid, serine, and threonine) sequence is responsible for ABCA1 degradation by calpain [Citation18]. However, whether Walker C signature is involved in nHDL biogenesis remains unclear and needs further characterization.
Figure 1. Topological diagram of ABCA1.
A recent study revealed the single-particle cryoelectron microscopy structure of human ABCA1 at 4.1 Å resolution [Citation19]. The overall structure resembles an elongated torch that is about 200 Å high. Both ECDs are the upward flaring flame, and other domains constitute the handle. There is a shallow pocket enclosed by the intracellular segments of TMs 1/2/5. The amino acid residues within these TMs are mostly polar and charged, which are responsible for binding to the polar heads of phospholipid molecules. Both TMD1 and TMD2 constitute a narrow chamber as an intracellular gate to move lipids from the inner leaflet to the outer leaflet of the cellular membrane. The helical domains in ECD1 and ECD2 together enclose an elongated hydrophobic tunnel that acts as a temporary storage and delivery passage for lipids. These structural features provide an important framework for better understanding ABCA1-mediated cholesterol efflux.
3. Biological roles of ABCA1
Cholesterol efflux is the first and rate-limiting step of reverse cholesterol transport (RCT), a process by which peripheral cholesterol is transferred by HDL to the liver for excretion into the bile and eventually feces [Citation20,Citation21]. Currently, four efflux pathways have been identified in the arterial wall cells [Citation22]. Two passive processes involve simple diffusion via the aqueous phase and facilitated diffusion mediated by scavenger receptor class B type I (SR-BI). Cholesterol flux is bidirectional and driven by FC concentration gradient between the cellular membrane and HDL particles [Citation23,Citation24]. When FC amount in the cellular membrane prevails over that in HDL particles, there is net cholesterol removal from cells. Despite these passive pathways occur ubiquitously, they are relatively inefficient. In contrast, two active processes mediated by ABCA1 and ATP-binding cassette transporter G1 (ABCG1) are thought to play a major role in exporting intracellular cholesterol. ABCA1 mediates initial transport of FC and phospholipid molecules to apoA-I for nHDL biogenesis, while ABCG1 facilitates subsequent continued FC efflux to these nHDL particles for further maturation [Citation25]. In cholesterol-loaded mouse peritoneal macrophages incubated with diluted human serum, ABCA1 and ABCG1 are responsible for 50 and 20% of cholesterol efflux, respectively [Citation26]. Thus, ABCA1 is the most important transporter of intracellular cholesterol efflux. ABCA1 is ubiquitously expressed, and most types of cells in the body do not possess the ability to degrade cholesterol. To maintain cholesterol homeostasis, ABCA1-dependent cholesterol efflux occurs in almost all cell types, particularly macrophages and VSMCs.
Mutations in the ABCA1 gene is known to cause Tangier disease, which is characterized by extremely low plasma levels of HDL cholesterol (HDL-C) and premature atherosclerosis [Citation27]. Macrophage-specific ablation of ABCA1 promotes foam cell formation, inhibits RCT, and increases plaque area in low-density lipoprotein receptor-deficient (Ldlr–/–) mice [Citation28,Citation29]. We also reported that heat shock protein 70, histone methyltransferase enhancer of zeste homolog 2, miR-19b, and pregnancy-associated plasma protein-A decrease ABCA1 expression and subsequent cholesterol efflux in THP-1 macrophage-derived foam cells and then accelerate the development of atherosclerosis in apolipoprotein E knockout (apoE–/–) mice [Citation30–33]. Conversely, macrophage ABCA1 overexpression markedly promotes the efflux of cholesterol from mouse peritoneal macrophages to apoA-I and alleviates atherosclerosis in Ldlr–/ – mice [Citation34]. Human ABCA1 transgenic mice lacking apoE develop dramatically smaller, less-complex lesions when compared with their apoE–/ – counterparts [Citation35]. In addition, studies from our laboratory showed that apoA-I binding protein and diosgenin elevate plasma HDL-C levels, promote RCT, and inhibit atherosclerosis progression by up-regulating ABCA1 expression in apoE–/ – mice [Citation36,Citation37]. Together, these findings provide strong evidence to support ABCA1 as an atheroprotective agent via its ability to remove intracellular cholesterol.
Although its major function is to mediate cholesterol efflux, ABCA1 acts as a multifunctional cell surface protein (). For example, knockout of ABCA1 leads to increased expression of proinflammatory cytokines and chemokines in mouse bone marrow-derived macrophages challenged with lipopolysaccharide (LPS) [Citation38]. In contrast, overexpression of ABCA1 attenuates interleukin-6 and tumor necrosis factor-α levels in bovine aortic endothelial cells stimulated with LPS [Citation39]. Previous studies from our group showed that treatment of THP-derived macrophages with apoA-I binding protein or apoA-I increases ABCA1 expression and consequently reduces proinflammatory cytokine secretion in response to LPS [Citation40,Citation41]. These observations suggest that ABCA1 exerts an anti-inflammatory action and acts as a critical link between inflammation and lipid metabolism. Additionally, ABCA1 can inhibit retinal ganglion cell apoptosis, promote colorectal cancer cell proliferation, enhance platelet reactivity, and stimulate insulin secretion from pancreatic β-cells [Citation42–45].
Table 1. The biological functions of ABCA1
4. Models for ABCA1-mediated cholesterol efflux
ABCA1 plays a central role in promoting intracellular cholesterol efflux. Currently, to the best of our knowledge, nine models have been proposed to reveal the molecular mechanisms of ABCA1-mediated cholesterol efflux ().
Table 2. The proposed models to explain ABCA1-mediated cholesterol efflux
4.1 The channel trafficking model
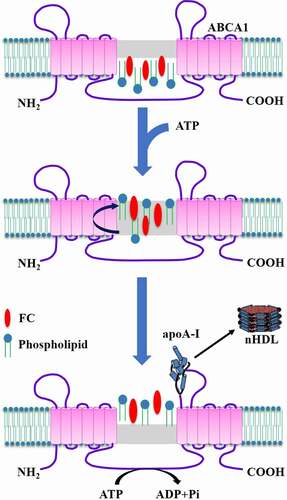
A channel is present between TMD1 and TMD2 of ABCA1, which plays a critical role in controlling lipid access [Citation19]. This suggests a channel trafficking model for ABCA1-mediated cholesterol efflux by using human embryonic kidney (HEK) 293 cells (). According to this model, the chamber is initially open at the bottom and close at the top. In the presence of lipid loading, accumulated phospholipids in the inner leaflet of plasma membrane are laterally transferred to the chamber by binding to amino acid residues within TMs 1/2/5. In this process, cholesterol is concurrently accessible to the chamber with the aid of phospholipids. ATP is then recruited to both NBDs, which leads to the dimerization of NBDs and consequently closes the chamber [Citation46]. ABCA1 then flops the trapped lipids to the outer leaflet of plasma membrane. The hydrolysis of ATP at both NBDs forms an ADP-bound intermediate that changes the conformation of TMD1 and TMD1 to open the chamber at the top. Cholesterol and phospholipids are egressed from the chamber to the elongated hydrophobic tunnel formed by both ECDs. The conformation of ECDs is also altered by ATP hydrolysis, allowing them to interact with apoA-I [Citation17]. Once bound to ABCA1, apoA-I takes up lipids from the elongated hydrophobic tunnel to assemble nHDL, which is then released from the cell surface. Following dissociation of ADP from NBDs, the chamber is restored to the initial open status for lipid uptake. Given that this is a structure-based model, more direct evidence needs to confirm its rationality.
Figure 2. Schematic illustration of the channel trafficking model.
4.2 The two-step process model
Fielding et al. and Wang et al. proposed a two-step mechanism for ABCA1-mediated nHDL biogenesis in VSMCs, fibroblasts and HEK293 cells () [Citation47,Citation48]. First, ABCA1 mediates the translocation of phospholipids from the inner leaflet to outer leaflet of plasma membrane. ATP is recruited to both NBDs of ABCA1. The hydrolysis of ATP induces a conformational change of ABCA1, allowing apoA-I to combine with ABCA1 [Citation49]. Phospholipids are loaded onto apoA-I to form a complex, which is a much better acceptor for cholesterol than apoA-I itself. Second, the phospholipid/apoA-I complexes enter the caveolae, 50 ~ 100 nm cellular membrane invaginations enriched in lipids. FC in the caveolae is transported to these complexes in an ABCA1-independent autocrine or paracrine fashion for nHDL production. It is noteworthy that when cells are pretreated with cyclodextrin to deplete intracellular cholesterol, phospholipid efflux and apoA-I binding are not affected while cholesterol removal is disappeared [Citation48]. This suggests that both steps are independent of each other. However, a later study showed that phospholipid efflux is tightly coupled with cholesterol efflux in RAW264.7 cells [Citation50]. Thus, additional work is needed to determine what causes the conflicting findings.
Figure 3. Schematic illustration of two-step process model.
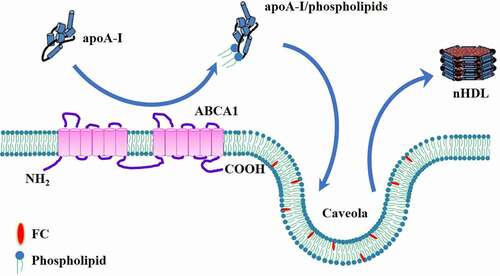
Caveolin-1, one of the major protein components of caveolae, can bind specifically to cholesterol [Citation51]. Accumulating evidence shows a positive correlation between the amount of caveolin-1 and intracellular cholesterol efflux [Citation52–54]. Overexpression of caveolin-1 in HepG2 cells markedly promotes cholesterol export to acceptors, human plasma and apoA-I [Citation55], whereas knockdown of caveolin-1 by small interfering RNA (siRNA) leads to a significant decrease in cholesterol efflux from RAW264.7 cells [Citation56]. In addition, caveolin-1 co-localizes with ABCA1 in the caveolae, which is required for ABCA1-mediated cholesterol efflux in rat aortic endothelial cells [Citation57,Citation58]. Given caveolin-1 as an important contributor to ABCA1-mediated cholesterol efflux, it is likely that caveolin-1 is responsible for the transfer of cholesterol from the caveolae to the phospholipids/apoA-I complex.
4.3 The mushroom-like protrusion model
Chemical cross-linking studies have demonstrated that the amphipathic α-helix of apoA-I can bind directly to the ECDs of ABCA1 when the latter is in its active conformation [Citation59,Citation60]. The interaction between apoA-I and ABCA1 increases the stability of ABCA1 at plasma membrane by protecting it from proteolytic degradation [Citation61]. Further analysis showed that apoA-I as a monomeric form interacts with ABCA1, and only about 10% of apoA-I associated with the plasma membrane is bound directly to ABCA1 [Citation62]. Thus, ABCA1 activity creates two apoA-I binding sites on the cell surface. One is low-capacity binding site for apoA-I/ABCA1 interaction. The other is high-capacity binding site for apoA-I/lipid interaction. Based on the apoA-I/ABCA1 reaction scheme, Vedhachalam et al. presented a model for nHDL biosynthesis in three steps in J774 mouse macrophages () [Citation63].
First, a small part of apoA-I is bound to cell surface ABCA1, known as low-capacity binding site. The direct interaction between the two molecules leads to a significant increase in ABCA1 translocase activity, which transfers FC and phospholipids from the inner leaflet to the outer leaflet of plasma membrane. This occurs in a chamber created by both TMDs of ABCA1 [Citation46]. Lipid accumulation in the outer membrane leaflet leads to the lateral compression of phospholipid molecules in this region. Accordingly, decreased lipid contents cause the lateral expansion of phospholipid molecules in the inner leaflet. The resultant asymmetric phospholipid packing across the bilayer leads to membrane strain. These protrusions are regarded as a critical site for apoA-I binding and nHDL biosynthesis.
Figure 4. Schematic illustration of mushroom-like protrusion model.
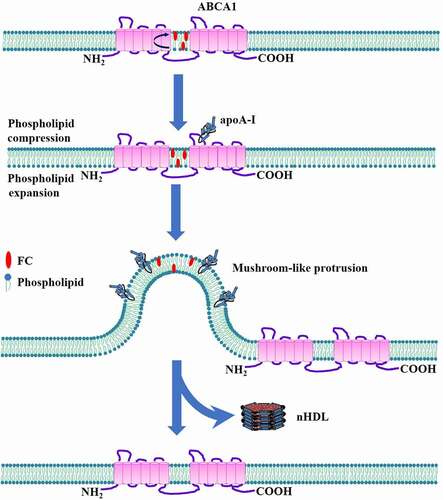
Second, apoA-I interacts with the mushroom-like protrusions. Because the phospholipid packing density in the outer leaflet is significantly higher than that in the inner leaflet, the mushroom-like protrusions are formed in the direction of extracellular space to relieve membrane strain. This idea is supported by a recent study showing that the vast majority of apoA-I is bound to the structures protruding from plasma membrane in cholesterol-loaded fibroblasts expressing ABCA1, whereas these protrusions are absent in fibroblasts isolated from a subject with Tangier disease [Citation64]. The mushroom-like protrusions, which are rich in FC and phospholipids but devoid of ABCA1, provide high-capacity binding sites for apoA-I [Citation65]. The hydrophobic C-terminal α-helix in apoA-I molecules then inserts into the phospholipid bilayer for lipid uptake [Citation66]. However, the detailed mechanisms underlying apoA-I interaction with the mushroom-like protrusions are still poor understood.
Third, lipids within mushroom-like protrusions are loaded onto apoA-I. Once apoA-I is bound to the mushroom-like protrusions, it receives a large amount of FC and phospholipids from these protrusions and is then spontaneously released into extracellular space. Concomitantly, plasma membrane restores normal structure. This process leads to the generation of three types of nHDL containing two, three, or four apoA-I molecules per particle [Citation67]. Moreover, these particles have distinct cholesterol amounts and phospholipid components [Citation67]. The heterogeneity of nHDL particles may result from the mushroom-like protrusions created by ABCA1 in distinct membrane environments.
4.4 The vacuolar ATPase (V-ATPase)-induced apoA-I unfolding model
As the acceptor of ABCA1-mediated cholesterol efflux, apoA-I exists in lipid-free, poor, and bound states. The former has a stronger capacity to accept cholesterol compared with the other two forms. During nHDL biogenesis, lipid-free apoA-I undergoes a variety of conformational changes, such as increased helicity and dimeric formation [Citation68,Citation69]. Recently, Wang et al. showed that ABCA1 can mediate the N-terminal unfolding of lipid-free apoA-I on the cell surface, which acts as an intermediate state and is essential for its lipidation [Citation70]. Thus, the ability to unfold apoA-I N-terminus is a third activity of ABCA1 besides apoA-I binding and plasma membrane remodeling.
V-ATPase, a member of the rotary ATPase protein family, is composed of a peripheral cytoplasmic domain (V1, 8 subunits) and an integral membrane domain (V0, 6 subunits). The V1 domain is responsible for the hydrolysis of ATP. The Vo domain then utilizes the released energy to translocate protons across the membrane, leading to the establishment of acidic environment in intracellular compartments or extracellular space [Citation71,Citation72]. V-ATPase is expressed in a variety of cell types, including macrophages, peptidergic cells, and melanoma cells [Citation73–75]. It has been reported that under acidic conditions, lipid-free apoA-I undergoes a conformational change to expose its hydrophobic α-helical structure, resulting in increased formation of reconstituted HDL from pure phosphatidylcholine liposomes [Citation76]. Another study showed that incubation of macrophage-derived foam cells with HDL at acidic pH markedly promotes cholesterol efflux due to HDL remodeling and lipid-poor apoA-I release [Citation77]. Given its acidic ability, it is likely that V-ATPase plays an important role in promoting ABCA1-dependent cholesterol efflux. As expected, overexpression of ABCA1 in baby hamster kidney (BHK) cells dramatically increases the cell surface levels of V-ATPase without altering its total levels [Citation78]. This finding suggests that ABCA1 promotes V-ATPase translocation to the plasma membrane instead of its transcription. As a consequence, V-ATPase acidifies not only plasma membrane to increase lipid fluidity but also apoA-I to unfold its N-terminus. The partially unfolded apoA-I dimerizes via the domain swapping around repeat 5 and then spontaneously inserts into the membrane [Citation79]. After being loaded with sufficient lipids, the apoA-I dimers are released from the cells to produce nascent HDL particles. This model reveals that recruitment of V-ATPase to the cell surface is required for the unfolding of apoA-I at its N-terminal domain by ABCA1, thereby providing a novel mechanistic insight into HDL biogenesis (). Enhancement of cell surface V-ATPase activity and apoA-I acidification could be valuable approaches to increase cholesterol efflux capability.
Figure 5. Schematic illustration of V-ATPase-induced apoA-I unfolding model.
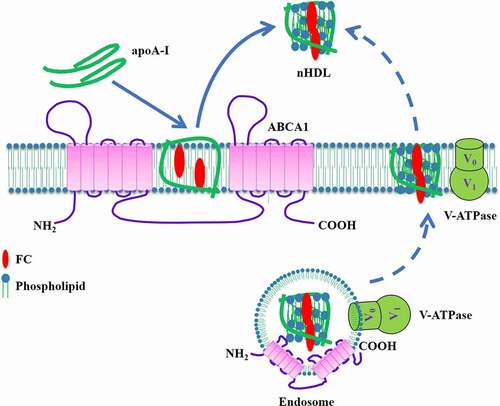
In addition to the plasma membrane, V-ATPase is located in the endosomes and normally pumps protons into the endosomal compartments to decrease their pH. It has been proposed that ABCA1 can promote cholesterol efflux to apoA-I via the retroendocytosis pathway [Citation80,Citation81]. Given that ABCA1, apoA-I, and V-ATPase are co-localized in endosomes, V-ATPase-induced acidification of the endosomal lumen may also contribute to apoA-I lipidation and subsequent nHDL secretion (). Future studies are needed to confirm this possibility.
4.5 The phosphatidylinositol 4,5-bisphosphate (PIP2)-induced apoA-I unfolding model
HDL is made up of 100 ~ 200 lipid molecules surrounded by two or more apoA-I molecules. The major phospholipid components within HDL are phosphatidylserine (PS), PIP2, phosphatidylethanolamine, and phosphatidylcholine (PC), which originate from lipid rafts in plasma membrane [Citation82]. Phospholipids are unequally distributed across the two leaflets of plasma membrane with an exclusive enrichment of PS in the inner leaflet [Citation83]. Like PS, PIP2 resides predominantly on the inner leaflet where it participates in a variety of biological processes, including endocytosis, exocytosis, protein transport, and endosomal recycling [Citation84–87]. PIP2 biogenesis is tightly modulated by myristoylated alanine-rich C-kinase substrate and small GTPases, such as Rab10 and Rab27a [Citation87–89].
Phospholipid flip-flop between two leaflets of a membrane bilayer is a bi-directional process that is essential for the maintenance of membrane lipid homeostasis. This transport process is known to rely on two classes of enzymes. The flippases mediate the inward translocation of phospholipids, while the floppases have an opposite effect [Citation83]. It is well established that ABCA1 promotes PS translocation to the outer leaflet, which is blocked by the mutations in its first large ECD [Citation90,Citation91]. Although the PS floppase activity of ABCA1 does not lead to direct interaction between apoA-I and the cell surface, it is required for efficient cholesterol efflux to apoA-I [Citation92].
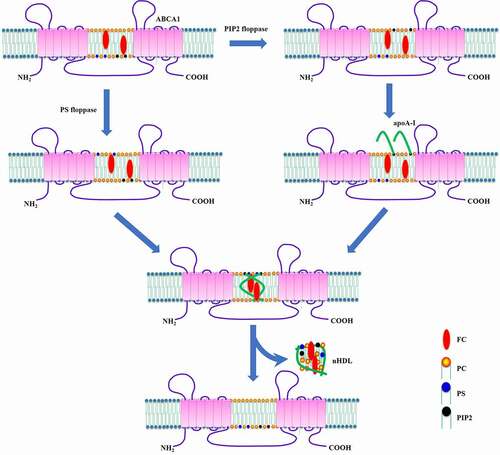
Cellular membrane contains various types of phospholipids. It is possible that ABCA1 translocates other phospholipids to the cell surface besides PS. Indeed, Gulshan et al. showed that the ECD2 of ABCA1 has PIP2 floppase activity, which is necessary for apoA-I binding and lipidation [Citation93]. Interestingly, these two floppase activities of ABCA1 are independent of each other [Citation93]. Thus, these authors proposed a novel model for ABCA1-dependent cholesterol efflux that includes PS and PIP2 in RAW264.7 cells (). In this model, ABCA1 flops PS to the outer leaflet, facilitating the remodeling of plasma membrane to increase its susceptibility to apoA-I. Meanwhile, it delivers PIP2 to the cell surface where PIP2 binds to apoA-I. The direct interaction between PIP2 and apoA-I promotes the unfolding of apoA-I N-terminus. The partially unfolded apoA-I forms a dimer that inserts into the membrane to assemble nHDL particles, which are released from the cells. This model provides another important mechanism for ABCA1-induced apoA-I unfolding beyond V-ATPase. Stimulating PIP2 translocation to the cell surface may be a promising strategy to promote cholesterol removal from cells and restore cellular cholesterol homeostasis.
Figure 6. Schematic illustration of PIP2-induced apoA-I unfolding model.
4.6 The ABCA1 dimerization model
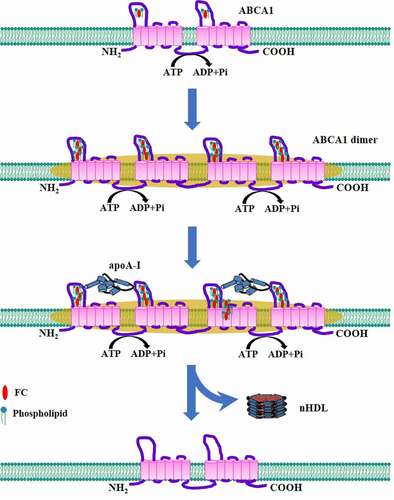
The majority of ABCA1 on the plasma membrane exists as an oligomeric complex, which constitutes the minimum functional unit required for apoA-I lipidation [Citation94]. Moreover, the formation of ABCA1 oligomerization is not dependent on apoA-I binding [Citation94]. On the basis of these findings, two later studies proposed a working model to reveal the mechanism underlying nHDL generation by using HeLa cells and BHK cells () [Citation95,Citation96]. In this model, ABCA1 monomers constantly transport FC and phospholipids from plasma membrane to their ECDs due to ATP hydrolysis. Following sequestration of sufficient lipids, these monomers undergo conformational changes to dimerize. The lipidated ABCA1 dimers interact with the membrane-skeletal actin filaments and other stable structures in plasma membrane, and become ready for apoA-I access. Lipid-free apoA-I combines with the ECDs of the ABCA1 dimers [Citation97,Citation98]. The lipids reserved by ABCA1 are then delivered to apoA-I. Lipid loading induces a conformational change of apoA-I, leading to its dissociation from ABCA1 and nHDL production [Citation95]. Upon release of the reserved lipids, the ABCA1 dimers transform into monomers, entering next cycle to receive lipids. This model suggests that ABCA1 dimerization is of critical importance to cholesterol export from cells. Promoting the conversion of ABCA1 monomers to dimers may be another effective strategy for inhibiting intracellular lipid accumulation.
Figure 7. Schematic illustration of ABCA1 dimerization model.
4.7 The apoA-I-free vesicle model
Two apoA-I monomers form a half-circle dimer as revealed by the crystal structure, which is necessary for lipid binding [Citation99]. Interestingly, dimerized apoA-I can acquire lipids from not only cellular membrane but also membrane-derived vesicles shed by ABCA1 during nHDL biogenesis in J774 mouse macrophages and HEK293 cells () [Citation67,Citation100]. In this process, ABCA1 first translocates FC and phospholipids to the outer leaflet independent of apoA-I. Higher phospholipid levels in this region create the mushroom-like protrusions in close proximity to ABCA1 for alleviating surface tension [Citation63]. These protrusions are released from plasma membrane as apoA-I-free vesicles, representing apoA-I-independent lipid efflux. Subsequently, apoA-I docks to plasma membrane through its hydrophobic C-terminus and binds to the ECDs of ABCA1 via its N-terminal domain. The interaction between both molecules leads to the unfolding of apoA-I N-terminus, allowing it to form a dimer. A large amount of FC and phospholipid molecules transported by ABCA1 are loaded onto dimerized apoA-I, leading to the production and release of nHDL. It is worth noting that nHDL can continue to gain lipids from apoA-I-free vesicles to form larger particles. This model is different from the mushroom-like protrusion model proposed by Vedhachalam et al. [Citation63], because it shows that upon creating a high-curvature structure, the interaction between apoA-I and ABCA1 is essential for nHDL generation.
Figure 8. Schematic illustration of apoA-I-free vesicle model.
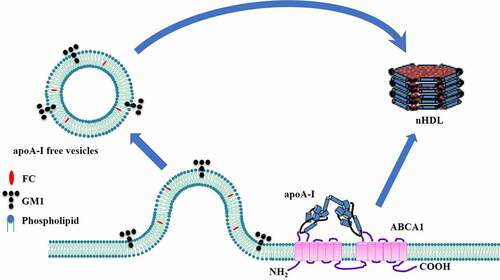
The formation of apoA-I-free vesicles is an important step in this model. It is well known that the lipid components of nHDL are similar to those of lipid rafts [Citation82]. Lipid analysis showed that monosialotetrahexosylganglioside (GM1), a lipid raft marker, is present in both apoA-I-free vesicles and nHDL [Citation100]. However, ABCA1 cannot transfer GM1 to apoA-I, excluding direct acquisition of GM1 from plasma membrane. A plausible explanation for this phenomenon is that GM1 is delivered to nHDL from apoA-I-free vesicles via a lipid exchange manner. Of note, flotillin, another lipid raft marker, does not appear in apoA-I-free vesicles [Citation100]. Thus, it remains largely unknown whether these vesicles are derived from raft-like regions of cellular membrane.
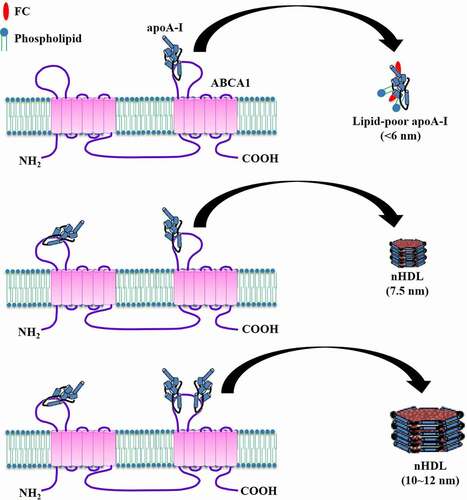
4.8 The sequential addition of apoA-I model
HDL is a heterogeneous population of particles with a range of sizes and molecular compositions [Citation101]. Recently, Sorci-Thomas et al. found that nHDL particles are produced as three relatively homogenous populations containing one, two, or three apoA-I molecules [Citation82]. Given that there is no precursor–product relationship between the smallest and largest nHDL particles [Citation102], the authors presented a model involving the sequential addition of apoA-I during nHDL assembly by using HEK293 cells (). If only one free-lipid apoA-I binds to ABCA1 on the cell surface, the <6 nm diameter nHDL is generated and released as poor-lipid apoA-I carrying four lipid molecules. When a second apoA-I is available, ABCA1 can transfer more trapped lipids to apoA-I, leading to the formation of the 7.5 nm diameter nHDL particle containing 24 lipid molecules. In the presence of a third apoA-I, apoA-I acquires maximal amounts of lipids to produce the 10 ~ 12 nm diameter nHDL with about 240 lipid molecules. Although this model provides a reasonable explanation for different-sized nHDL formation, future research is required to determine what causes the sequential addition of apoA-I to ABCA1.
Figure 9. Schematic illustration of sequential apoA-I addition model.
It is well accepted that apoA-I receives cholesterol transported by ABCA1 to generate nHDL, while ABCG1 and SR-BI facilitate subsequent cholesterol efflux to these particles to form mature HDL [Citation25]. Epidemiologic studies have demonstrated that plasma HDL-C levels are negatively correlated with the risk of CVD [Citation103,Citation104]. Recently, Du et al. showed that small dense HDL has a stronger capacity to receive cholesterol transported by ABCG1 than large HDL [Citation105]. Thus, HDL-raising treatments should aim to increase the concentrations of nHDL species with one or two apoA-I molecules.
4.9 The retroendocytosis model
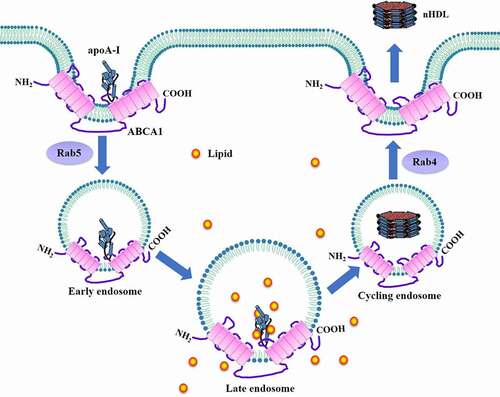
In addition to plasma membrane, the endosomes are regarded as an important reservoir of cellular cholesterol [Citation106]. ABCA1 resides not only on the cell surface, but also in the endosomal compartments [Citation107]. Consistently, apoA-I lipidation occurs at both sites. It has been proposed that apoA-I can be internalized to the endosomes for nHDL assembly in a pathway called retroendocytosis in RAW264.7 cells and mouse peritoneal macrophages () [Citation80,Citation81], which was first suggested by Schmitz et al. in 1985 [Citation108]. First, apoA-I binds to ABCA1 on the cell surface to form a complex, which then enters clathrin-coated pits. Second, the complex is endocytosed to early endosomes in a Rab5-dependent manner [Citation109]. ABCA1 located in the early endosomal membrane translocates lipids to the endosomal lumen for apoA-I lipidation. ApoA-I is further lipidated when early endosomes become larger late endosomes, leading to the biogenesis of nHDL particles. Forth, these particles are transferred to cycling endosomes, which then return to the cell surface with the help of Rab4 [Citation109]. After fusion with plasma membrane, nHDL is secreted into the extracellular space, and ABCA1 recycles to the cell surface. This model suggests that stimulating apoA-I internalization provides an alternative approach to enhance ABCA1-mediated cholesterol efflux.
Figure 10. Schematic illustration of the retroendocytosis model.
Calpain is known to cleave ABCA1 through interaction with its PEST sequence [Citation18]. Deletion of the PEST sequence (ABCA1-dPEST) significantly increases cell surface ABCA1 levels but decreases its localization to late endosomes. In cells stably expressing ABCA1-dPEST, overall cholesterol efflux is dramatically increased despite a reduction in cholesterol efflux from late endosomes [Citation110]. This suggests that increased apoA-I lipidation on the cell surface can fully compensate for the loss of retroendocytosis-associated cholesterol efflux. Another study showed that most apoA-I is lipidated on the cell surface of macrophages, and only a small portion of apoA-I is endocytosed and resecreted as nHDL particles [Citation111]. Consistent with this observation, Denis et al. reported that internalized apoA-I contributes to approximately 1.4% of total nHDL generation, and pharmacologic inhibition of its endocytosis with sucrose or cytochalasin D does no reduce intracellular cholesterol efflux [Citation112]. They also found that about 80% of internalized apoA-I is transported to the lysosomes for degradation and only the remaining part can produce nHDL [Citation112]. Thus, the retroendocytosis pathway may play a minor role in ABCA1-mediated cholesterol efflux, and nHDL is predominantly produced from the cell surface.
The mechanisms for nHDL biogenesis in cholesterol-loaded cells differ from those in cholesterol-depleted cells [Citation113]. When apoA-I internalization is blocked, cholesterol efflux is significantly decreased in cholesterol-loaded cells but increased in cells without cholesterol accumulation [Citation109]. In agreement with this, the amount of apoA-I endocytosed by cholesterol-normal cells is not sufficient to produce nHDL particles [Citation111]. Thus, the retroendocytosis pathway may contribute to nHDL formation only in cells with excessive cholesterol.
ABCA1 recycles rapidly between plasma membrane and the endosomal compartments, which has a significant effect on endosomal cholesterol removal. Of internalized ABCA1, only 30% is recycled back to plasma membrane, implying the presence of a degradative pathway [Citation114]. It has been proposed that ADP-ribosylation factor 6 (Arf6), a small GTPase, directs endocytosed ABCA1 into the lysosomes for degradation, leading to reduced efflux of intracellular cholesterol [Citation115]. Niemann-Pick type C (NPC) disease, an autosomal recessive condition, is characterized by excessive cholesterol accumulation in late endosomes [Citation116]. In fibroblasts isolated from NPC patients, overexpression of small GTPase Rab8 markedly inhibits late endosomal cholesterol deposition by translocating cholesterol to plasma membrane [Citation117]. Importantly, knockdown of Rab8 with siRNA attenuates cell surface ABCA1 expression and suppresses endosomal cholesterol efflux to apoA-I in primary human macrophages, suggesting Rab8 as a significant contributor to the retroendocytosis pathway [Citation118]. However, how Rab8 affects ABCA1 localization to the cell surface remains unknown. Palmitoylation, a reversible post-translational modification, plays an important role in promoting protein localization to specific membrane domains [Citation119]. Blockade of ABCA1 palmitoylation leads to a significant reduction in cell surface ABCA1 and cholesterol efflux, while overexpression of the palmitoyl transferase DHHC8 stimulates ABCA1-mediated cholesterol efflux [Citation120]. Collectively, these observations suggest that targeting Arf6, Rab8, or ABCA1 palmitoylation could be attractive strategies for promoting endosomal cholesterol efflux to apoA-I.
5. Conclusion and future directions
ABCA1 plays a central role in nHDL biogenesis by promoting the transport of FC and phospholipids to apoA-I. Decreased cholesterol efflux mediated by ABCA1 is closely associated with a variety of diseases, especially CVD. To date, nine models have been proposed to account for the mechanisms by which ABCA1 exports cholesterol from cells. It remains largely unknown, however, which model represents the dominant pathway for ABCA1-mediated cholesterol efflux. Thus, there is a great need for additional research to precisely define the relative contributions of each of these pathways to nHDL production. The two-step process model has demonstrated that ABCA1-dependent phospholipid efflux precedes FC efflux to apoA-I, while other models show a simultaneous translocation of two substrates. This paradoxical finding highlights the need for further research in this area. Both V-ATPase and PIP2 have been shown to unfold apoA-I at its N-terminal domain [Citation78,Citation93], while whether other factors can exert such an effect remains to be clarified. Given the lack of ABCA1 in the mushroom-like protrusions, additional work is required to determine how lipids in these membrane microdomains are transferred to apoA-I. In addition, the following issues need to be addressed. What are the biological roles of Walker C signature of ABCA1? How are apoA-I-free vesicles released from plasma membrane into extracellular environment? Does V-ATPase participate in the retroendocytosis pathway? Are there any other components involving the endocytosis of the ABCA1/apoA-I complex besides Rab5? In summary, answers to these questions will provide insightful knowledge about the mechanisms of ABCA1-dependent cholesterol efflux and promote the development of novel HDL-based therapies.
Acknowledgments
We thank Dr. Xiao-Hua Yu for helpful discussions and valuable suggestions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Yu XH, Fu YC, Zhang DW, et al. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245–252.
- Collaborators GBDRF. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388(10053):1659–1724.
- Benn M, Nordestgaard BG, Frikke-Schmidt R, et al. Low LDL cholesterol, PCSK9 and HMGCR genetic variation, and risk of Alzheimer’s disease and Parkinson’s disease: mendelian randomisation study. BMJ. 2017;357:j1648.
- Ebara S, Marumo M, Yamabata C, et al. Inverse associations of HDL cholesterol and oxidized HDL with d-dimer in patients with type 2 diabetes mellitus. Thromb Res. 2017;155:12–15.
- Li G, Gu HM, Zhang DW. ATP-binding cassette transporters and cholesterol translocation. IUBMB Life. 2013;65(6):505–512.
- Gao JH, Zeng MY, Yu XH, et al. Visceral adipose tissue-derived serine protease inhibitor accelerates cholesterol efflux by up-regulating ABCA1 expression via the NF-kappaB/miR-33a pathway in THP-1 macropahge-derived foam cells. Biochem Biophys Res Commun. 2018;500(2):318–324.
- Kuang HJ, Zhao GJ, Chen WJ, et al. Hsp27 promotes ABCA1 expression and cholesterol efflux through the PI3K/PKCzeta/Sp1 pathway in THP-1 macrophages. Eur J Pharmacol. 2017;810:57–62.
- Li CH, Gong D, Chen LY, et al. Puerarin promotes ABCA1-mediated cholesterol efflux and decreases cellular lipid accumulation in THP-1 macrophages. Eur J Pharmacol. 2017;811:74–86.
- Zhang M, Li L, Xie W, et al. Apolipoprotein A-1 binding protein promotes macrophage cholesterol efflux by facilitating apolipoprotein A-1 binding to ABCA1 and preventing ABCA1 degradation. Atherosclerosis. 2016;248:149–159.
- Wang H, Liu Y, Zhu L, et al. 17beta-estradiol promotes cholesterol efflux from vascular smooth muscle cells through a liver X receptor alpha-dependent pathway. Int J Mol Med. 2014;33(3):550–558.
- Santamarina-Fojo S, Peterson K, Knapper C, et al. Complete genomic sequence of the human ABCA1 gene: analysis of the human and mouse ATP-binding cassette A promoter. Proc Natl Acad Sci U S A. 2000;97(14):7987–7992.
- Bungert S, Molday LL, Molday RS. Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: identification of N-linked glycosylation sites. J Biol Chem. 2001;276(26):23539–23546.
- Kaminski WE, Piehler A, Wenzel JJ. ABC A-subfamily transporters: structure, function and disease. Biochim Biophys Acta. 2006;1762(5):510–524.
- Hozoji M, Kimura Y, Kioka N, et al. Formation of two intramolecular disulfide bonds is necessary for ApoA-I-dependent cholesterol efflux mediated by ABCA1. J Biol Chem. 2009;284(17):11293–11300.
- Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42(7):1007–1017.
- Hogue DL, Liu L, Ling V. Identification and characterization of a mammalian mitochondrial ATP-binding cassette membrane protein. J Mol Biol. 1999;285(1):379–389.
- Nagao K, Takahashi K, Azuma Y, et al. ATP hydrolysis-dependent conformational changes in the extracellular domain of ABCA1 are associated with apoA-I binding. J Lipid Res. 2012;53(1):126–136.
- Wang N, Chen W, Linsel-Nitschke P, et al. A PEST sequence in ABCA1 regulates degradation by calpain protease and stabilization of ABCA1 by apoA-I. J Clin Invest. 2003;111(1):99–107.
- Qian H, Zhao X, Cao P, et al. Structure of the Human Lipid Exporter ABCA1. Cell. 2017;169(7):1228–1239 e1210.
- van der Velde Ae. Reverse cholesterol transport: from classical view to new insights. World J Gastroenterol. 2010;16(47):5908–5915.
- Rader DJ, Alexander ET, Weibel GL, et al. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50:S189–194.
- Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289(35):24020–24029.
- Zhao Y, Van Berkel TJ, Van Eck M. Relative roles of various efflux pathways in net cholesterol efflux from macrophage foam cells in atherosclerotic lesions. Curr Opin Lipidol. 2010;21(5):441–453.
- Yancey PG, de la Llera-moya M, Swarnakar S, et al. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000;275(47):36596–36604.
- Yu XH, Zhang DW, Zheng XL, et al. Cholesterol transport system: an integrated cholesterol transport model involved in atherosclerosis. Prog Lipid Res. 2019;73:65–91.
- Adorni MP, Zimetti F, Billheimer JT, et al. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. 2007;48(11):2453–2462.
- Oram JF. Tangier disease and ABCA1. Biochim Biophys Acta. 2000;1529(1–3):321–330.
- van Eck M, Bos IS, Kaminski WE, et al. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci U S A. 2002;99(9):6298–6303.
- Wang MD, Franklin V, Marcel YL. In vivo reverse cholesterol transport from macrophages lacking ABCA1 expression is impaired. Arterioscler Thromb Vasc Biol. 2007;27(8):1837–1842.
- Zhao ZW, Zhang M, Chen LY, et al. Heat shock protein 70 accelerates atherosclerosis by downregulating the expression of ABCA1 and ABCG1 through the JNK/Elk-1 pathway. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863(8):806–822.
- Lv YC, Tang YY, Zhang P, et al. Histone methyltransferase enhancer of zeste homolog 2-Mediated ABCA1 promoter DNA methylation contributes to the progression of atherosclerosis. PLoS One. 2016;11(6):e0157265.
- Lv YC, Tang YY, Peng J, et al. MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1. Atherosclerosis. 2014;236(1):215–226.
- Tang SL, Zhao ZW, Liu SM, et al. Pregnancy-associated plasma protein-a accelerates atherosclerosis by regulating reverse cholesterol transport and inflammation. Circ J. 2019;83(3):515–523.
- Van Eck M, Singaraja RR, Ye D, et al. Macrophage ATP-binding cassette transporter A1 overexpression inhibits atherosclerotic lesion progression in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2006;26(4):929–934.
- Singaraja RR, Fievet C, Castro G, et al. Increased ABCA1 activity protects against atherosclerosis. J Clin Invest. 2002;110(1):35–42.
- Zhang M, Zhao GJ, Yao F, et al. AIBP reduces atherosclerosis by promoting reverse cholesterol transport and ameliorating inflammation in apoE(-/-) mice. Atherosclerosis. 2018;273:122–130.
- Lv YC, Yang J, Yao F, et al. Diosgenin inhibits atherosclerosis via suppressing the MiR-19b-induced downregulation of ATP-binding cassette transporter A1. Atherosclerosis. 2015;240(1):80–89.
- Yvan-Charvet L, Welch C, Pagler TA, et al. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118(18):1837–1847.
- Stamatikos A, Dronadula N, Ng P, et al. ABCA1 overexpression in endothelial cells in vitro enhances apoai-Mediated cholesterol efflux and decreases inflammation. Hum Gene Ther. 2019;30(2):236–248.
- Zhang M, Zhao GJ, Yin K, et al. Apolipoprotein A-1 binding protein inhibits inflammatory signaling pathways by binding to apolipoprotein a-1 in THP-1 macrophages. Circ J. 2018;82(5):1396–1404.
- Yin K, Deng X, Mo ZC, et al. Tristetraprolin-dependent post-transcriptional regulation of inflammatory cytokine mRNA expression by apolipoprotein A-I: role of ATP-binding membrane cassette transporter A1 and signal transducer and activator of transcription 3. J Biol Chem. 2011;286(16):13834–13845.
- Li L, Xu L, Chen W, et al. Reduced annexin a1 secretion by ABCA1 causes retinal inflammation and ganglion cell apoptosis in a murine glaucoma model. Front Cell Neurosci. 2018;12:347.
- Aguirre-Portoles C, Feliu J, Reglero G, et al. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness, and these effects can be ameliorated using the BET inhibitor apabetalone. Mol Oncol. 2018;12(10):1735–1752.
- Lhermusier T, Severin S, Van Rothem J, et al. ATP-binding cassette transporter 1 (ABCA1) deficiency decreases platelet reactivity and reduces thromboxane A2 production independently of hematopoietic ABCA1. J Thromb Haemost. 2016;14(3):585–595.
- Lyu J, Imachi H, Fukunaga K, et al. Angiotensin II induces cholesterol accumulation and impairs insulin secretion by regulating ABCA1 in beta cells. J Lipid Res. 2018;59(10):1906–1915.
- Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85(4):1343–1372.
- Fielding PE, Nagao K, Hakamata H, et al. A two-step mechanism for free cholesterol and phospholipid efflux from human vascular cells to apolipoprotein A-1. Biochemistry. 2000;39(46):14113–14120.
- Wang N, Silver DL, Thiele C, et al. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J Biol Chem. 2001;276(26):23742–23747.
- Chen J, Sharma S, Quiocho FA, et al. Trapping the transition state of an ATP-binding cassette transporter: evidence for a concerted mechanism of maltose transport. Proc Natl Acad Sci U S A. 2001;98(4):1525–1530.
- Smith JD, Le Goff W, Settle M, et al. ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein A-I. J Lipid Res. 2004;45(4):635–644.
- Murata M, Peranen J, Schreiner R, et al. VIP21/caveolin is a cholesterol-binding protein. Proc Natl Acad Sci U S A. 1995;92(22):10339–10343.
- Fielding CJ, Bist A, Fielding PE. Caveolin mRNA levels are up-regulated by free cholesterol and down-regulated by oxysterols in fibroblast monolayers. Proc Natl Acad Sci U S A. 1997;94(8):3753–3758.
- Hailstones D, Sleer LS, Parton RG, et al. Regulation of caveolin and caveolae by cholesterol in MDCK cells. J Lipid Res. 1998;39(2):369–379.
- Truong TQ, Aubin D, Falstrault L, et al. CD36, and caveolin-1 contribute positively to cholesterol efflux in hepatic cells. Cell Biochem Funct. 2010;28(6):480–489.
- Fu Y, Hoang A, Escher G, et al. Expression of caveolin-1 enhances cholesterol efflux in hepatic cells. J Biol Chem. 2004;279(14):14140–14146.
- Hu Q, Zhang XJ, Liu CX, et al. PPARgamma1-induced caveolin-1 enhances cholesterol efflux and attenuates atherosclerosis in apolipoprotein E-deficient mice. J Vasc Res. 2010;47(1):69–79.
- Lin YC, Ma C, Hsu WC, et al. Molecular interaction between caveolin-1 and ABCA1 on high-density lipoprotein-mediated cholesterol efflux in aortic endothelial cells. Cardiovasc Res. 2007;75(3):575–583.
- Kuo CY, Lin YC, Yang JJ, et al. Interaction abolishment between mutant caveolin-1(Delta 62-100) and ABCA1 reduces HDL-mediated cellular cholesterol efflux. Biochem Biophys Res Commun. 2011;414(2):337–343.
- Chroni A, Liu T, Fitzgerald ML, et al. Cross-linking and lipid efflux properties of apoA-I mutants suggest direct association between apoA-I helices and ABCA1. Biochemistry. 2004;43(7):2126–2139.
- Fitzgerald ML, Morris AL, Chroni A, et al. ABCA1 and amphipathic apolipoproteins form high-affinity molecular complexes required for cholesterol efflux. J Lipid Res. 2004;45(2):287–294.
- Yamauchi Y, Hayashi M, Abe-Dohmae S, et al. Apolipoprotein A-I activates protein kinase C alpha signaling to phosphorylate and stabilize ATP binding cassette transporter A1 for the high density lipoprotein assembly. J Biol Chem. 2003;278(48):47890–47897.
- Vedhachalam C, Ghering AB, Davidson WS, et al. ABCA1-induced cell surface binding sites for ApoA-I. Arterioscler Thromb Vasc Biol. 2007;27(7):1603–1609.
- Vedhachalam C, Duong PT, Nickel M, et al. Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high density lipoprotein particles. J Biol Chem. 2007;282(34):25123–25130.
- Lin G, Oram JF. Apolipoprotein binding to protruding membrane domains during removal of excess cellular cholesterol. Atherosclerosis. 2000;149(2):359–370.
- Iatan I, Bailey D, Ruel I, et al. Membrane microdomains modulate oligomeric ABCA1 function: impact on apoAI-mediated lipid removal and phosphatidylcholine biosynthesis. J Lipid Res. 2011;52(11):2043–2055.
- Tanaka M, Dhanasekaran P, Nguyen D, et al. Contributions of the N- and C-terminal helical segments to the lipid-free structure and lipid interaction of apolipoprotein A-I. Biochemistry. 2006;45(34):10351–10358.
- Duong PT, Collins HL, Nickel M, et al. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47(4):832–843.
- Davidson WS, Hazlett T, Mantulin WW, et al. The role of apolipoprotein AI domains in lipid binding. Proc Natl Acad Sci U S A. 1996;93(24):13605–13610.
- Silva RA, Hilliard GM, Li L, et al. A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry. 2005;44(24):8600–8607.
- Wang S, Gulshan K, Brubaker G, et al. ABCA1 mediates unfolding of apolipoprotein AI N terminus on the cell surface before lipidation and release of nascent high-density lipoprotein. Arterioscler Thromb Vasc Biol. 2013;33(6):1197–1205.
- Mazhab-Jafari MT, Rohou A, Schmidt C, et al. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature. 2016;539(7627):118–122.
- McGuire C, Stransky L, Cotter K, et al. Regulation of V-ATPase activity. Front Biosci (Landmark Ed). 2017;22(4):609–622.
- Xia Y, Liu N, and Xie X, et al. The macrophage-specific V-ATPase subunit ATP6V0D2 restricts inflammasome activation and bacterial infection by facilitating autophagosome-lysosome fusion. Autophagy. 2019;15(6):960–975.
- Rao VK, Zavala G, Deb Roy A, et al. A pH-sensitive luminal His-cluster promotes interaction of PAM with V-ATPase along the secretory and endocytic pathways of peptidergic cells. J Cell Physiol. 2019;234(6):8683–8697.
- Martins BX, Arruda RF, Costa GA, et al. Myrtenal-induced V-ATPase inhibition - A toxicity mechanism behind tumor cell death and suppressed migration and invasion in melanoma. Biochim Biophys Acta Gen Subj. 2019;1863:1–12.
- Fukuda M, Nakano M, Miyazaki M, et al. Conformational change of apolipoprotein A-I and HDL formation from model membranes under intracellular acidic conditions. J Lipid Res. 2008;49(11):2419–2426.
- Nguyen SD, Oorni K, Lee-Rueckert M, et al. Spontaneous remodeling of HDL particles at acidic pH enhances their capacity to induce cholesterol efflux from human macrophage foam cells. J Lipid Res. 2012;53(10):2115–2125.
- Lorkowski SW, Brubaker G, Gulshan K, et al. V-ATPase (vacuolar ATPase) activity required for ABCA1 (ATP-binding cassette protein A1)-Mediated cholesterol efflux. Arterioscler Thromb Vasc Biol. 2018;38(11):2615–2625.
- Gursky O, Jones MK, Mei X, et al. Structural basis for distinct functions of the naturally occurring Cys mutants of human apolipoprotein A-I. J Lipid Res. 2013;54(12):3244–3257.
- Takahashi Y, Smith JD. Cholesterol efflux to apolipoprotein AI involves endocytosis and resecretion in a calcium-dependent pathway. Proc Natl Acad Sci U S A. 1999;96(20):11358–11363.
- Chen W, Sun Y, Welch C, et al. Preferential ATP-binding cassette transporter A1-mediated cholesterol efflux from late endosomes/lysosomes. J Biol Chem. 2001;276(47):43564–43569.
- Sorci-Thomas MG, Owen JS, Fulp B, et al. Nascent high density lipoproteins formed by ABCA1 resemble lipid rafts and are structurally organized by three apoA-I monomers. J Lipid Res. 2012;53(9):1890–1909.
- Pomorski T, Menon AK. Lipid flippases and their biological functions. Cell Mol Life Sci. 2006;63(24):2908–2921.
- Mu L, Tu Z, Miao L, et al. A phosphatidylinositol 4,5-bisphosphate redistribution-based sensing mechanism initiates a phagocytosis programing. Nat Commun. 2018;9(1):4259.
- Walter AM, Muller R, and Tawfik B, et al. Phosphatidylinositol 4,5-bisphosphate optical uncaging potentiates exocytosis. Elife. 2017;6:e30203.
- Egea-Jimenez AL, Gallardo R, Garcia-Pino A, et al. Frizzled 7 and PIP2 binding by syntenin PDZ2 domain supports Frizzled 7 trafficking and signalling. Nat Commun. 2016;7(1):12101.
- Shi A, Grant BD. Interactions between Rab and Arf GTPases regulate endosomal phosphatidylinositol-4,5-bisphosphate during endocytic recycling. Small GTPases. 2013;4(2):106–109.
- Rohrbach TD, Shah N, Jackson WP, et al. The effector domain of marcks is a nuclear localization signal that regulates cellular PIP2 levels and nuclear PIP2 localization. PLoS One. 2015;10(10):e0140870.
- Gerber PP, Cabrini M, Jancic C, et al. Rab27a controls HIV-1 assembly by regulating plasma membrane levels of phosphatidylinositol 4,5-bisphosphate. J Cell Biol. 2015;209(3):435–452.
- Singaraja RR, Visscher H, James ER, et al. Specific mutations in ABCA1 have discrete effects on ABCA1 function and lipid phenotypes both in vivo and in vitro. Circ Res. 2006;99(4):389–397.
- Nagao K, Zhao Y, Takahashi K, et al. Sodium taurocholate-dependent lipid efflux by ABCA1: effects of W590S mutation on lipid translocation and apolipoprotein A-I dissociation. J Lipid Res. 2009;50(6):1165–1172.
- Gulshan K, Brubaker G, Wang S, et al. Sphingomyelin depletion impairs anionic phospholipid inward translocation and induces cholesterol efflux. J Biol Chem. 2013;288(52):37166–37179.
- Gulshan K, Brubaker G, Conger H, et al. PI(4,5)P2 Is Translocated by ABCA1 to the Cell Surface Where It Mediates Apolipoprotein A1 Binding and Nascent HDL Assembly. Circ Res. 2016;119(7):827–838.
- Denis M, Haidar B, Marcil M, et al. Characterization of oligomeric human ATP binding cassette transporter A1. Potential implications for determining the structure of nascent high density lipoprotein particles. J Biol Chem. 2004;279(40):41529–41536.
- Nagata KO, Nakada C, Kasai RS, et al. ABCA1 dimer-monomer interconversion during HDL generation revealed by single-molecule imaging. Proc Natl Acad Sci U S A. 2013;110(13):5034–5039.
- Ishigami M, Ogasawara F, Nagao K, et al. Temporary sequestration of cholesterol and phosphatidylcholine within extracellular domains of ABCA1 during nascent HDL generation. Sci Rep. 2018;8(1):6170.
- Fitzgerald ML, Morris AL, Rhee JS, et al. Naturally occurring mutations in the largest extracellular loops of ABCA1 can disrupt its direct interaction with apolipoprotein A-I. J Biol Chem. 2002;277(36):33178–33187.
- Oram JF, Lawn RM, Garvin MR, et al. ABCA1 is the cAMP-inducible apolipoprotein receptor that mediates cholesterol secretion from macrophages. J Biol Chem. 2000;275(44):34508–34511.
- Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286(44):38570–38582.
- Liu M, Mei X, Herscovitz H, et al. N-terminal mutation of apoA-I and interaction with ABCA1 reveal mechanisms of nascent HDL biogenesis. J Lipid Res. 2019;60(1):44–57.
- Rached FH, Chapman MJ, Kontush A. HDL particle subpopulations: focus on biological function. Biofactors. 2015;41(2):67–77.
- Mulya A, Lee JY, Gebre AK, et al. Minimal lipidation of pre-beta HDL by ABCA1 results in reduced ability to interact with ABCA1. Arterioscler Thromb Vasc Biol. 2007;27(8):1828–1836.
- Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79. 8–15.
- Prospective Studies C, Lewington S, Whitlock G, et al. Blood cholesterol and vascular mortality by age, sex, and blood pressure: a meta-analysis of individual data from 61 prospective studies with 55,000 vascular deaths. Lancet. 2007;370:1829–1839.
- Du XM, Kim MJ, Hou L, et al. HDL particle size is a critical determinant of ABCA1-mediated macrophage cellular cholesterol export. Circ Res. 2015;116(7):1133–1142.
- Mukherjee S, Zha X, Tabas I, et al. Cholesterol distribution in living cells: fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys J. 1998;75(4):1915–1925.
- Neufeld EB, Remaley AT, Demosky SJ, et al. Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem. 2001;276(29):27584–27590.
- Schmitz G, Robenek H, Lohmann U, et al. Interaction of high density lipoproteins with cholesteryl ester-laden macrophages: biochemical and morphological characterization of cell surface receptor binding, endocytosis and resecretion of high density lipoproteins by macrophages. EMBO J. 1985;4(3):613–622.
- Azuma Y, Takada M, Shin HW, et al. Retroendocytosis pathway of ABCA1/apoA-I contributes to HDL formation. Genes Cells. 2009;14(2):191–204.
- Chen W, Wang N, Tall AR. A PEST deletion mutant of ABCA1 shows impaired internalization and defective cholesterol efflux from late endosomes. J Biol Chem. 2005;280(32):29277–29281.
- Faulkner LE, Panagotopulos SE, Johnson JD, et al. An analysis of the role of a retroendocytosis pathway in ABCA1-mediated cholesterol efflux from macrophages. J Lipid Res. 2008;49(6):1322–1332.
- Denis M, Landry YD, Zha X. ATP-binding cassette A1-mediated lipidation of apolipoprotein A-I occurs at the plasma membrane and not in the endocytic compartments. J Biol Chem. 2008;283(23):16178–16186.
- Oram JF. The ins and outs of ABCA. J Lipid Res. 2008;49:1150–1151.
- Lu R, Arakawa R, Ito-Osumi C, et al. ApoA-I facilitates ABCA1 recycle/accumulation to cell surface by inhibiting its intracellular degradation and increases HDL generation. Arterioscler Thromb Vasc Biol. 2008;28(10):1820–1824.
- Mukhamedova N, Hoang A, Cui HL, et al. Small GTPase ARF6 regulates endocytic pathway leading to degradation of ATP-binding cassette transporter A1. Arterioscler Thromb Vasc Biol. 2016;36(12):2292–2303.
- Yu XH, Jiang N, Yao PB, et al. NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin Chim Acta. 2014;429:69–75.
- Linder MD, Uronen RL, Holtta-Vuori M, et al. Rab8-dependent recycling promotes endosomal cholesterol removal in normal and sphingolipidosis cells. Mol Biol Cell. 2007;18(1):47–56.
- Linder MD, Mayranpaa MI, Peranen J, et al. Rab8 regulates ABCA1 cell surface expression and facilitates cholesterol efflux in primary human macrophages. Arterioscler Thromb Vasc Biol. 2009;29(6):883–888.
- Charollais J, Van Der Goot FG. Palmitoylation of membrane proteins (Review). Mol Membr Biol. 2009;26(1–2):55–66.
- Singaraja RR, Kang MH, Vaid K, et al. Palmitoylation of ATP-binding cassette transporter A1 is essential for its trafficking and function. Circ Res. 2009;105(2):138–147.