
ABSTRACT
Pro-survival members of the BCL-2 family, including MCL-1, are emerging as important proteins during the development and therapeutic response of solid tumors. Notably, high levels of MCL-1 occur in breast cancer, where functional dependency has been demonstrated using cell lines and mouse models. The utility of restoring apoptosis in cancer cells through inhibition of pro-survival BCL-2 proteins has been realized in the clinic, where the first specific inhibitor of BCL-2 is approved for use in leukemia. A variety of MCL-1 inhibitors are now undergoing clinical trials for blood cancer treatment and application of this new class of drugs is also being tested in solid cancers. On-target compounds specific to MCL-1 have demonstrated promising efficacy in preclinical models of breast cancer and show potential to enhance the anti-tumor effect of conventional therapies. Taken together, this makes MCL-1 an extremely attractive target for clinical evaluation in the context of breast cancer.
Abbreviations: ADC (antibody-drug conjugate); AML (Acute myeloid leukemia); APAF1 (apoptotic protease activating factor 1); bCAFs (breast cancer associated fibroblasts); BCL-2 (B-cell lymphoma 2); BH (BCL-2 homology); CLL (chronic lymphocytic leukemia); EGF (epidermal growth factor); EMT (epithelial to mesenchymal transition); ER (estrogen receptor); FDA (food and drug administration); GEMM (genetically engineered mouse model); HER2 (human epidermal growth factor 2); IL6 (interleukin 6); IMM (inner mitochondrial membrane); IMS (intermembrane space); MCL-1 (myeloid cell leukemia-1); MOMP (mitochondrial outer membrane permeabilisation); MM (multiple myeloma); PDX (patient-derived xenograft); OMM (outer mitochondrial membrane); PROTAC (proteolysis-targeting chimeras) TNBC (triple negative breast cancer); UPS (ubiquitin mediated proteolysis system)
Introduction
Survival of patients diagnosed with breast cancer has improved dramatically in recent decades, largely owing to early detection through extensive screening programs, refinement of conventional treatments, and the advent of effective targeted and personalized therapies. Although there has been a ~ 40% decline in death rates of female breast cancer patients since the 1980s, the decreasing rate of mortality has slowed and it is estimated that global incidence will continue to rise steadily over the next 10–20 years [Citation1,Citation2]. Worldwide, breast cancer is the most prevalent tumor type and remains the leading cause of cancer-related death amongst women, highlighting the essential requirement for improved treatment strategies [Citation2]. During progression to lethality, solid tumors often overcome a series of key evolutionary bottlenecks including: primary tumor establishment; metastatic dissemination and seeding; and resistance to therapeutic intervention, and evasion of cell death is a prerequisite for survival in the presence of each of these selection pressures [Citation3].
Cell death can occur through several mechanisms but apoptosis, a form of regulated cell death, is the mechanism that is most commonly perturbed in cancer. Deregulation of processes that control intrinsic (mitochondrial) apoptosis is prominent in both tumorigenesis and during failed responses to cancer therapy [Citation4]. Activation of mitochondrial outer membrane permeabilisation (MOMP), a key event in intrinsic apoptosis, is governed by interactions within the multi-member BCL-2 (B-Cell Lymphoma 2) family of proteins. First characterized in hematological cancers, it is now recognized that alterations in the BCL-2 family are also frequent in solid tumors and that failed intrinsic (mitochondrial) apoptosis facilitates tumor survival throughout the oncogenic process [Citation4,Citation5].
Here, we focus on MCL-1 (Myeloid Cell Leukemia-1), a pro-survival BCL-2 family protein best characterized for its role in hematopoiesis and related malignancies of the blood. MCL-1 is required in many cell types as demonstrated by the embryonic lethality of germline Mcl1 deletion in mice, whilst conditional genetic ablation revealed a functional requirement of MCL-1 for survival of specific cell types such as hematopoietic stem cells and cardiomyocytes [Citation6–8]. It is now understood that MCL1 is frequently upregulated across a range of solid tumor types, including breast cancer [Citation9]. Importantly, dependency on MCL-1 in cancer cells may be exploited for therapeutic gain, and novel agents that specifically target MCL-1 are already undergoing clinical investigation as potential chemotherapeutics in acute myeloid leukemia (AML) and multiple myeloma (MM). Whilst elevation of MCL-1 may bestow a competitive advantage on breast cancer cells during tumor development and prevent tumor cell elimination in response to therapy, the reliance on MCL-1 for survival also represents a vulnerability that could be utilized to improve treatment outcome in breast cancer.
Induction of intrinsic apoptosis is controlled by the BCL-2 family
BCL-2 was first identified through mapping of the t[12:18] chromosomal translocation, which results in a constitutive BCL-2 expression from the immunoglobulin locus and often underpins follicular lymphoma [Citation10–12]. Initially, BCL-2 was considered functionally unique amongst known tumor-promoting genes as it supports oncogenesis by preventing tumor cell death rather than driving tumor cell proliferation [Citation13,Citation14]. Numerous proteins with regions of sequence homology to BCL-2 have since been identified and characterized as part of the BCL-2 family, and these fall into two categories with opposing pro-apoptotic and pro-survival functions [Citation15]. Protein–protein interactions between BCL-2 family members determine whether a cell undergoes apoptosis, and the relative quantity and activity of these proteins fluctuates throughout mammalian development to enable differential states of susceptibility to pro-apoptotic stimuli. The propensity of a cell to undergo apoptosis following the receipt of a stress stimulus is described as the extent to which it is “primed” [Citation16].
BCL-2 family proteins share at least one of four highly conserved BCL-2 homology (BH) regions. The pro-apoptotic BCL-2 family members can be subdivided into BH3-only proteins, so-called because they possess only the conserved alpha-helical BH3 domain, and multi-BH domain “effector” proteins, such as BAX/BAK (). Specifically, BH3-only “sensitizer” proteins (including BAD and NOXA) can indirectly enhance apoptosis through binding and occupying pro-survival proteins to ameliorate inhibitory sequestration of BAX/BAK [Citation17,Citation18]. As well as undertaking this function, BH3-only “activator” proteins (including BIM, PUMA, and BID) can bind directly to BAX/BAK to facilitate apoptosis [Citation17,Citation18] ().
Figure 1. BCL-2 family and intrinsic (mitochondrial) apoptosis. (A) Mitochondrial outer membrane permeabilisation (MOMP) is controlled by protein–protein interactions between members of the BCL-2 family. (B) Intrinsic apoptosis is initiated by internal or external stress stimuli such as DNA damage, oncogene activation or cytokine deprivation. These events activate intracellular signalling pathways which manifest by altering the balance of pro-apoptotic and pro-survival BCL-2 family proteins. In response to sufficiently toxic stress stimuli, pro-apoptotic BCL-2 family proteins are activated and overwhelm pro-survival proteins to liberate BAX/BAK, which initiates MOMP and the release of intermembrane space (IMS) proteins into the cytosol to activate apoptosome formation, caspase activation and degradation of intracellular material. (C) Members of the BCL-2 family of proteins bind to each other with highly selective affinity, and the electropositive surface within the BH3 binding groove of MCL-1 mediates strong affinity for the BH3-only proteins PUMA, BIM, and NOXA, but not BAD.
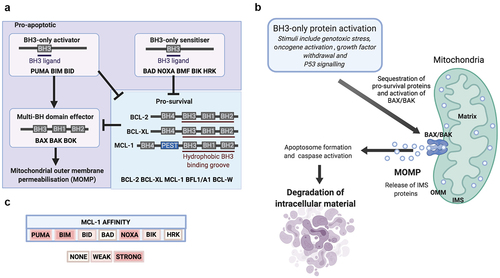
This process is antagonized by multi-BH domain pro-survival BCL-2 proteins (such as BCL-2, BCL-XL, and MCL-1) which maintain mitochondrial integrity through binding and sequestering pro-apoptotic members of the BCL-2 family (). Structural studies reveal that these associations are mediated by a hydrophobic binding groove (composed of BH1-3) which guides the interaction with hydrophobic residues of exposed alpha-helical BH3 domains [Citation19,Citation20]. The culmination of successful pro-apoptotic signaling is the activation of BAX/BAK, which oligomerize to form macropores and permeabilise the outer mitochondrial membrane (OMM) (). Mitochondrial outer membrane permeabilization (MOMP) enables the release of mitochondrial intermembrane space (IMS) proteins, such as cytochrome c, OMI and SMAC into the cytoplasm. Subsequently, cytochrome c can then complex with cytosolic apoptotic protease activating factor 1 (APAF-1) to form the apoptosome, a multimolecular protein complex, and this drives activation of the caspase-9/7/3 (cysteine-aspartic protease) cascade to trigger the destruction of cellular contents [Citation21–25] ().
Interaction with BH3-only proteins regulates MCL-1 activity
The sequence of MCL1 is most divergent from BCL2 amongst the pro-survival BCL-2 family members, as homology occurs only in the defined BH domains [Citation26,Citation27]. Much like other pro-survival BCL-2 family proteins, the BH1-3 domains of MCL-1 form a conserved hydrophobic groove that mediates interactions with the hydrophobic BH3 face of certain pro-apoptotic BCL-2 family members () [Citation28–30]. Distinct pro-survival BCL-2 family proteins have differential affinity for specific BH3 domain-containing pro-apoptotic proteins, and this is promoted by the unique amino acid residues that constitute the central alpha-helices. The electropositive surface within the BH3 binding groove of MCL-1 mediates a selective affinity for critical residues within the BH3 domain of BAK, BIM, BID, NOXA, and PUMA, but an inability to interact with BAD () [Citation17,Citation18,Citation31]. The binding interaction between MCL-1 and NOXA triggers proteasomal degradation of MCL-1, whereas binding to PUMA or BIM serve to stabilize MCL-1 in an inactive state without stimulating protein degradation [Citation30,Citation32,Citation33]. The BH3-only sensitizer protein NOXA is highly selective for MCL-1, and NOXA expression efficiently induces apoptosis in cells manipulated into MCL-1 dependency [Citation34]. Interestingly, high expression of NOXA mRNA has been shown to associate with improved survival in breast cancer and predict response to microtubule targeting chemotherapeutic agents [Citation35]. Furthermore, resistance to targeted therapies in HER-2 amplified breast cancer can be conferred by microRNA-4728-mediated suppression of NOXA which serves to prevent apoptosis of breast cancer cells in an MCL-1 dependent manner [Citation36]. Taken together, this highlights the intimate relationship between MCL-1 protein levels and NOXA expression which may be important for progression and drug resistance in breast cancer patients.
MCL1 amplification and expression in breast cancer
Genomic gain of chromosome 1q21.2, where MCL1 is located, is frequent across a range of tumor types and is particularly common in breast cancers [Citation9]. Breast cancer encompasses a spectrum of tumor subtypes with distinct morphological features, biological properties, and clinical implications. These can be broadly stratified by molecular and histopathological criteria including estrogen and progesterone receptor (ER/PR) status, amplification of ERBB2 (commonly referred to as human epidermal growth factor 2 [HER2]), and the transcriptomic landscape. The resultant classification system remains centered on the “classical” ER-positive /PR-positive, HER2-amplified and triple-negative breast cancer (TNBC) subtypes in the clinic, and this commonly informs the current therapeutic standard of care. However, breast cancer can be molecularly divided into a further series of subtypes including luminal A and B, HER2-enriched, basal-like, and claudin-low [Citation37,Citation38]. Amplification/gain of MCL1 persists across breast cancers independent of the tumor stratification approach and is observed in up to 72% of breast cancer cases in online genomic data repositories () [Citation39–43]. Notably, the frequency of MCL1 amplification and mRNA expression exceeds both BCL-2 (~9%) and BCL-XL (~27%) across all major subtypes of breast cancer, suggestive of a specific importance of MCL-1 in breast tumourigenesis [Citation39–43].
Figure 2. MCL-1 importance in breast cancer patients and breast cancer cell lines. (A) MCL1 is frequently increased in breast tumours. Genomic gain (red) or amplification (blue) of MCL1 occurs in up to 72% of breast cancer samples assayed for copy number alteration in non-redundant datasets composed of greater than 100 samples curated in cBioportal [Citation39–43]. (B) Essentiality of pro-survival BCL-2 family members in breast cancer. This graph illustrates the functional importance of pro-survival members of the BCL-2 family in a compendium of “CRISPR knockout screens” across breast cancer cell lines from the Cancer Cell Line Encyclopaedia (CCLE). TNBC cell lines are often split into Basal A and B subgroups, where Basal A represents a tumour subtype which is more representative of basal-like tumours and Basal B is increasingly claudin-low and stem/mesenchymal -like. A lower gene effect score indicates an increased likelihood that the gene is essential in a given cell line. A gene effect score of 0 infers non-essentiality, whereas a gene effect score of −1 represents the median of all pan-essential genes (https://depmap.org/portal/) DepMap 21Q2 dataset [Citation123].
![Figure 2. MCL-1 importance in breast cancer patients and breast cancer cell lines. (A) MCL1 is frequently increased in breast tumours. Genomic gain (red) or amplification (blue) of MCL1 occurs in up to 72% of breast cancer samples assayed for copy number alteration in non-redundant datasets composed of greater than 100 samples curated in cBioportal [Citation39–43]. (B) Essentiality of pro-survival BCL-2 family members in breast cancer. This graph illustrates the functional importance of pro-survival members of the BCL-2 family in a compendium of “CRISPR knockout screens” across breast cancer cell lines from the Cancer Cell Line Encyclopaedia (CCLE). TNBC cell lines are often split into Basal A and B subgroups, where Basal A represents a tumour subtype which is more representative of basal-like tumours and Basal B is increasingly claudin-low and stem/mesenchymal -like. A lower gene effect score indicates an increased likelihood that the gene is essential in a given cell line. A gene effect score of 0 infers non-essentiality, whereas a gene effect score of −1 represents the median of all pan-essential genes (https://depmap.org/portal/) DepMap 21Q2 dataset [Citation123].](/cms/asset/3a62ead1-7769-4243-9ed2-e06f33161f8a/kccy_a_2054096_f0002_oc.jpg)
MCL-1 protein abundance is the product of a series of upstream events, which may also be perturbed in breast cancer, such that quantification of MCL1 gene and mRNA copy number levels may underestimate the extent to which MCL-1 is upregulated [Citation44]. In a study of 428 treatment naïve breast cancers we observed a range of MCL-1 protein expression within all classical subtypes, and each subtype contained cases possessing high levels of MCL-1 [Citation45]. High MCL-1 protein expression was significantly associated with poor prognosis, increased invasive grade and lymph node positivity [Citation45]. Interestingly, elevated MCL-1 protein was predictive of poor outcome in both TNBC and non-TNBC patients but did not correlate with outcome in the HER2-amplified subtype, although HER2 positivity was correlated with increased MCL-1 expression [Citation45]. These studies support the widely available genomic and transcriptomic data detailing elevated levels of MCL-1 in breast cancer, and suggest that increased MCL-1 expression confers a selective advantage during breast tumorigenesis. It has been demonstrated using matched pairs of ER-positive breast cancers that the level of MCL1 alteration in primary and metastatic disease is comparable, so it remains to be determined whether MCL-1 expression is important for metastatic dissemination or if increases in MCL-1 protein occur at secondary tumor sites [Citation46].
The elevation of MCL-1 during tumor development may also impart an intrinsic resistance to cancer therapy. Indeed, high MCL-1 protein levels were indicative of poor response to a paclitaxel-containing treatment regime in a cohort of invasive breast cancer patients [Citation47]. Therapeutic resistance may also be acquired through alteration in MCL1 expression during treatment, and molecular profiling of post-adjuvant chemotherapy TNBC patient tumors revealed amplification of MCL1 in 54% of cases and showed that MCL1 amplification can arise during treatment [Citation48]. Furthermore, analysis of paired TNBC biopsy samples pre-/post-chemotherapy showed that MCL-1 protein levels were increased after treatment, suggesting that MCL-1 may become further up-regulated in response to chemotherapy [Citation49].
Transcriptional control of MCL-1 expression can be targeted for therapeutic effect in breast cancer
MCL-1 abundance is controlled at multiple levels including transcriptional, translational, and post-translational mechanisms, and each of these could be therapeutically targeted to reduce MCL-1 levels in breast cancer (). MCL1 was first identified as an immediate-early gene transcriptionally up-regulated in the ML-1 human myeloid leukemia cell line upon phorbol-ester induced differentiation [Citation26]. Signaling through MEK/ERK has been shown to induce transcription of MCL1 via ELK-1 and SRF transcription factors, and this pathway is commonly dysregulated during breast tumor development [Citation50,Citation51]. Various other signaling pathways and transcription factors have been implicated in stimulating MCL1 expression in response to diverse cytokines, growth factors, and stress stimuli, such as hypoxia, oncogene activation, and endoplasmic reticulum (ER) stress [Citation52]. For example, it has been illustrated that pro-inflammatory cytokine interleukin (IL)-6 potently stimulates JAK/STAT signaling in breast cancer cell lines, and it has also been shown that both IL-6 and JAK/STAT signaling can potentiate MCL1 transcription in multiple cancer subtypes [Citation53–56]. These mechanisms could be important in breast cancer where STAT3 is often constitutively active, and it has also been shown that higher levels of IL-6 in the blood of metastatic breast cancer patients correlates with poor prognosis [Citation57,Citation58].
Figure 3. Regulation of MCL-1 and mechanisms to reduce MCL-1 levels. MCL1 expression is stimulated following the receipt of diverse extracellular cytokines and growth factors. The signalling pathways which are predominantly responsible for inducing MCL-1 transcription, translation and protein stability include the PI3K/AKT pathway, JAK/STAT signalling and the MEK/ERK cascade. MCL1 mRNA is highly unstable and is particularly sensitive to widespread change in the rate of gene expression, protein synthesis and protein degradation. Potential pharmacological methods to decrease MCL-1 levels in cancer are indicated in red boxes. Inhibition of transcription-associated cyclin-dependent kinases (CDKs), such as CDK9, leads to decreased levels of short-lived mRNA and proteins, including MCL-1, and CDK inhibitors are undergoing clinical trials. MCL-1 protein normally has a short half life, and levels are strongly impacted by changes in the rate of translation. Inhibitors of mTOR may represent a viable approach to suppress cap-dependent translation and thereby reduce MCL-1 protein. MCL-1 stability can also be targeted by proteolysis-targeting chimeras (PROTACs) which selectively bind a target protein and recruit an E3 ligase to initiate proteosomal degradation. A series of deubiquitinases (DUBs) have been reported to remove ubiquitin moieties and thus stabilise MCL-1, and DUB inhibitors could also have the potential to prevent MCL-1 deubiquitination and thereby reduce MCL-1 protein levels.
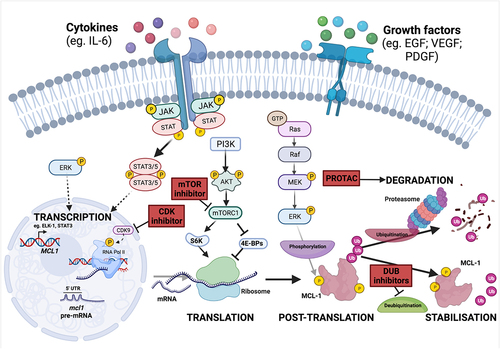
Cyclin-dependent kinase (CDK) inhibitors have been shown to down-regulate MCL1 transcription, and decreased MCL-1 expression is necessary for these agents to induce apoptosis. Whilst most CDKs primarily function through mediating cell-cycle transition, CDK7/8/9 are critical for transcription by RNA polymerase II. Indeed, inhibition of CDKs with dinaciclib has demonstrated promising clinical efficacy in pre-clinical models of lymphoma and HER2-amplified breast cancers [Citation59,Citation60]. This process is reliant upon inhibition of MCL1 transcription, liberation of pro-apoptotic BAK and subsequent BAK-induced mitochondrial apoptosis [Citation59,Citation60]. Interestingly, targeted CDK9 inhibitors, such as voruciclib, potently induce apoptosis through MCL-1 depletion in many hematologic cell lines and in a subset of breast cancer cell lines, and have recently entered clinical trials for the treatment of hematological malignancies () [Citation61,Citation62]. Indirect targeting of MCL-1 through CDK9 inhibition potently sensitizes non-Hodgkin Lymphoma and AML cells to the BCL-2 inhibitor venetoclax without significant toxicity to mice suggesting that this strategy to co-target multiple pro-survival BCL-2 proteins could be tolerable if applied to breast cancer in the clinic [Citation61,Citation63].
Translational control can be leveraged to reduce MCL-1
MCL-1 is a particularly short-lived protein (estimated half life ~1–3 hours) and MCL1 mRNA is considered “weak” due to its GC-rich and highly structured 5’ untranslated region (UTR) [Citation64,Citation65]. Taken together, this conspires to preferentially sensitize MCL-1 to alterations in the rate of global protein synthesis and offers the opportunity to indirectly target MCL-1 levels [Citation66]. As well as driving MEK/ERK-mediated phosphorylation of ELK-1 to activate MCL1 transcription, epidermal growth factor (EGF) induces PI3K pathway activation to stimulate mammalian target of rapamycin (mTOR) regulated cap-dependent translation, and inhibition of mTOR complex 1 (mTORC1) sensitizes multiple cancer cell lines and mouse models to apoptosis through decreased MCL-1 protein levels [Citation50,Citation65,Citation67–69]. Further to this, EGF signaling can also control MCL-1 protein abundance through mTOR-mediated phosphorylation and activation of ribosomal protein S6 kinase (S6K), a critical enzyme for ribosome recruitment during mRNA translation. Interestingly, Mcl1 deficiency impairs lactogenesis and delays reconstitution of mammary glands by mammary stem and progenitor cells in vivo, and independent inhibition of both EGF and mTOR serves to recapitulate this phenotype alongside an associated decrease in MCL-1 levels [Citation70].
These mechanisms may also be important in breast cancer as a series of breast cancer cell lines undergo cap-dependent translational increase in MCL-1 after BCL-2 and/or BCL-XL inhibition, and it has also been illustrated that a translational increase in MCL-1 protein can occur through the mTOR/4E-BP axis in PIK3CA-mutant breast cancers [Citation71]. Importantly, increased MCL-1 protein levels through PI3K/AKT signaling can enhance MCL-1 pro-survival functionality in the absence of genomic and transcriptomic MCL1 upregulation, indicating that targeting mTOR-mediated MCL-1 translation may provide an additional mechanism by which MCL-1 protein levels can be suppressed in some breast cancer subtypes () [Citation71]. Enhanced translation of MCL1 may be a common feature in ER-positive breast cancer where ERα has been shown to bind RNA and increase translation of specific mRNAs, including MCL1, to enhance cell survival and resistance to tamoxifen [Citation44].
Control of MCL-1 protein stability
MCL-1 is the largest pro-survival BCL-2 family protein due to the presence of an extended N-terminus rich in PEST (proline (P), glutamic acid (E), serine (S), and threonine (T)) motifs () [Citation26,Citation72]. PEST sequences commonly act as a scaffold for post-translational modifications, including phosphorylation and ubiquitination, and this facilitates the rapid turnover of MCL-1 protein when compared to BCL-2 and BCL-XL [Citation73,Citation74]. Indeed, the spatiotemporal activity of MCL-1 is closely regulated through post-translational modifications in a highly cell- and context-specific manner. A series of enzymes have been implicated in the post-translational control of MCL-1, including extracellular signal-regulated kinase (ERK), glycogen synthase kinase 3 (GSK-3), and CDK1, which have been reported to phosphorylate at least one of 10 residues in the MCL-1 PEST region. ERK-mediated phosphorylation enhances the stability of MCL-1 and appears to confer chemo-resistance in breast cancer cell lines, whereas CDK1-mediated phosphorylation targets MCL-1 for degradation during mitotic arrest, and GSK-3-mediated phosphorylation targets MCL-1 for proteosome-mediated degradation [Citation75–78]. Furthermore, GSK-3 protein expression is inversely correlated with MCL1 expression in primary breast cancer patient tissue, and increased levels of MCL-1 protein in this patient cohort are associated with worse prognosis [Citation79].
Multiple E3 ubiquitin ligases, including SCFβ-TrCP, FBW7, TRIM17, APC/CCdc20 and MULE (MCL-1 ubiquitin ligase E3), have been shown to link ubiquitin chains to MCL-1 in a phosphorylation – dependent or – independent manner to target MCL-1 for degradation [Citation52]. The relationship between MULE and MCL-1 is particularly interesting as MULE contains a BH3 domain that specifically interacts with MCL-1, and this interaction is abrogated when MCL-1 is occupied by BIM [Citation80,Citation81]. MCL-1 protein becomes stabilized upon MULE knock-down and this increases resistance to DNA-damaging agents [Citation80]. Both loss-of-function mutations and promoter hypermethylation of the E3 ubiquitin ligase FBW7 have been identified in breast tumors suggesting that the extent of MCL-1 protein upregulation in breast cancer may be as yet underappreciated [Citation82,Citation83].
A series of deubiquitinases (DUBs) functionally oppose E3 ligases by removing ubiquitin chains and have been reported to stabilize MCL-1 in a range of cancer types. These include Ku70, USP13, DUB3 (through MGMT), JOSD1, and USP9X, although specific roles in regulating MCL-1 during breast tumourigenesis have yet to be defined [Citation84–89]. Importantly, suppression of DUBs upstream of MCL-1 is an attractive approach by which MCL-1 levels could be decreased in cancers (), although the principal enzymes responsible for MCL-1 stabilization are highly cell-type specific and dependent on the wider proteomic profile of the target cell [Citation90].
Indirect inhibition of MCL-1 may also be achievable through targeted degradation by a novel class of therapeutic agents termed proteolysis-targeting chimeras (PROTACs). These hetero-bifunctional compounds harness endogenous degradation machinery by tagging a target protein with a linker group to promote interaction with a specific E3 ubiquitin ligase. In the case of MCL-1, it was recently demonstrated that the dMCL1-2 PROTACs, composed of the Cullin4A (CUL4A) -DNA-binding protein 1 (DDB1)- Cereblon (CRBN) E3 ubiquitin ligase complex, could effectively degrade MCL-1 and activate apoptosis, as evidenced by a concomitant increase of cleaved-caspase-3 [Citation91,Citation92].
Therefore, MCL-1 is tightly regulated at every step from gene expression to protein degradation, and the mechanisms involved may be harnessed to indirectly inhibit MCL-1 function during breast tumorigenesis, or serve as biomarkers for patients who could benefit from MCL-1 inhibition ().
Pharmaceutical targeting of MCL-1 with BH3-mimetics
During tissue homeostasis, various stress or developmental stimuli trigger alterations in the balance of proteins within the BCL-2 family. This can enable pro-apoptotic proteins to overwhelm pro-survival proteins and exceed the required threshold to execute apoptosis (). For example, P53 activation in response to DNA damaging agents such as γ-irradiation, etoposide or adriamycin, transcriptionally upregulates expression of pro-apoptotic BH3-only proteins to mediate DNA-damage induced apoptosis [Citation93–95]. However, these mechanisms are often deregulated in cancer where induction of pro-apoptotic BH3-only protein expression may fail and/or pro-survival BCL-2 family proteins are elevated, and the threshold for triggering apoptosis is not attained despite the receipt of significant cellular stress. As many of the upstream pathways that regulate expression of the BCL-2 family are disrupted in cancer, direct targeting of pro-survival BCL-2 proteins is an attractive way to restore apoptosis.
A class of drugs termed BH3-mimetics have been developed to phenocopy the function of BH3-only proteins by occupying the BH3-binding groove of pro-survival BCL-2 family proteins, liberating pro-apoptotic proteins to enable activation of BAX/BAK. The first BH3-mimetics, ABT737 and the orally bioavailable derivative ABT263 (navitoclax), mimic the BH3-only protein BAD by binding to BCL-2, BCL-XL, and BCL-W, and effectively induce apoptosis in cells which are dependent upon these pro-survival family members [Citation96,Citation97]. Clinical use of navitoclax has been limited due to thrombocytopenia, as platelets are heavily dependent on BCL-XL, but careful dosing can limit this side-effect and numerous clinical trials are investigating navitoclax as a potential combination agent for the treatment of solid cancers [Citation98,Citation99] (clinicaltrials.gov). Inhibition of BCL-2/BCL-XL has shown anti-cancer function in preclinical models of breast cancer where navitoclax inhibits TNBC patient derived xenograft (PDX) growth when administered in combination with docetaxel [Citation100]. Furthermore, an additional BCL-2/BCL-XL-specific BH3-mimetic that may avoid thrombocytopenia, called AZD0466, has recently entered clinical investigation [Citation101].
The first clinically approved BH3-mimetic, ABT199 (venetoclax), specifically targets BCL-2 and is currently used in chronic lymphocytic leukemia (CLL) and AML, which are often dependent upon BCL-2 for cancer cell survival [Citation102–104]. BCL2 is an ER target gene commonly upregulated in ER-positive breast cancer, and venetoclax enhances the response of ER-positive PDX models to hormone therapy [Citation105,Citation106]. Initial clinical evaluation of ventoclax in solid tumors was conducted in combination with tamoxifen to treat metastatic ER-positive breast cancers expressing BCL-2, where it was well tolerated and showed promising anti-cancer effect [Citation107]. The addition of venetoclax also enhanced the anti-tumor effect of fulvestrant and palbociclib (CDK4/6 inhibitor) in preclinical ER-positive breast cancer models, but a more complete assessment of the clinical value remains ongoing [Citation108,Citation109].
In recent years huge progress has been achieved in the development of MCL-1-specific BH3-mimetics with five independent compounds currently undergoing clinical evaluation in hematologic malignancies, and recruitment is underway for a further clinical trial in solid tumors, including breast cancer (NCT04837677) (). Whilst detail of the specificity and direct pro-apoptotic impact of some of these drugs is currently unknown, preclinical evidence strongly indicates that MCL-1 can be effectively inhibited to confer an anti-cancer effect in vitro and in vivo. Indeed, S63845 (a tool compound related to MIK665/S64315), AMG-176, AZD5991, and VU661013 effectively induce apoptosis in AML and MM cells in vitro and in xenograft mouse models, with clear synergy in combination with BCL-2 inhibition [Citation110–115]. Importantly, combinatorial targeting of BCL-2 and MCL-1 is tolerated in mice and is under investigation in clinical trials of hematological cancers (NCT03672695, NCT04702425) [Citation111–114].
Table 1. Clinical trials with MCL-1 specific BH3 mimetics. Data extracted from ClinicalTrials.gov.
As well as successfully inducing apoptosis in several models of hematological malignancies, subsets of breast cancer cells are sensitive to MCL-1-specific BH3-mimetics in vitro [Citation110–112,Citation115,Citation116]. For example, S63845 improves the response of the BT-474 HER2-amplified breast cancer cell line to lapatinib (HER2 tyrosine kinase inhibitor) in 2-D cell culture, and also inhibits growth of TNBC PDX cells in 3-D tumoursphere assays [Citation110,Citation117]. Further, it has been demonstrated that S63845 can successfully induce apoptosis when used in combination with either lapatinib, docetaxel, or trastuzumab (HER2-targeting monoclonal antibody) in another HER2-amplified breast cancer cell line, SK-BR-3, through liberation of pro-apoptotic BH3-only activator BIM [Citation117].
In vivo, S63845 illustrates single agent efficacy in restricting tumor growth in the MMTV-PyMT mouse model of breast cancer [Citation118]. Combining S63845 with docetaxel in two TNBC PDX mouse models, or trastuzumab in a HER2-amplified PDX mouse model, has been shown to significantly impair tumor growth and prolong survival, although monotherapy with the MCL-1 inhibitor failed to markedly prevent tumor growth [Citation117]. Similar results have been documented in a BRCA1-mutant TNBC PDX mouse model, where S63845 synergized with the PARP inhibitor olaparib [Citation124]. Finally, single-agent treatment with VU661013 effectively inhibited growth of TNBC xenografts (using HCC1187 and BT20 cell lines) and sensitized tumors to treatment with either docetaxel or doxorubicin [Citation115]. Taken together, these findings suggest that the most potent effects of MCL-1 inhibition in breast cancer may occur when used in combination with additional therapies that drive an increasingly “primed” apoptotic state [Citation16].
MCL-1 is required for breast cancer cell survival
The prevalence of MCL-1 upregulation in breast cancer suggests that it is required for tumor development, and associations between high MCL-1 and poor outcome may also indicate a role for MCL-1 in resistance to existing treatment regimen. Similar to the broad range of MCL-1 expression observed in breast cancer epithelial cells, human breast cancer cell lines representative of the major subtypes of disease express variable levels of MCL-1 and have provided a useful tool for assessing the essentiality of MCL-1 function in breast cancer [Citation45,Citation116,Citation117,Citation119,Citation120]. Indeed, studies utilizing breast cancer cell lines first indicated a requirement for MCL-1 to maintain cell viability, as genetic knock-down of MCL1 induces cell death in a subset of both TNBC and ER-positive breast cancer cell lines grown in 2-D monolayer [Citation119,Citation121,Citation122]. Whilst knock-down of MCL1 alone is sufficient to induce apoptosis in some TNBC and ER-positive breast cancer cell lines, functional buffering by BCL-XL can confer resistance to MCL-1 loss [Citation119,Citation121,Citation122]. This is in line with the analysis of breast cancer cell lines in Cancer Cell Line Encyclopedia data, which highlights a role for both MCL1 and BCL2L1 (BCL-XL) in maintenance of breast cancer cell line viability in 2-D culture, whereas BCL2 appears largely non-essential in this context () [Citation123,Citation125,Citation126].
The requirement for MCL-1 is exacerbated during 3-D tumoursphere growth in non-adherent culture conditions, where the functional redundancy often witnessed between MCL-1 and BCL-XL in 2-D monolayer growth is less profound [Citation118]. Genetic knock-down or pharmacological blockade of MCL-1 also restricts the growth of TNBC cells (MDA-MB-468) in xenograft mouse models [Citation45,Citation120]. It has been illustrated that the decreased viability upon MCL-1 knock-down/knock-out or pharmacological inhibition in breast cancer cell lines is associated with redistribution of BIM and the subsequent induction of apoptosis, as evidenced by cytochrome c release, cleavage of caspase-3 and/or PARP, whilst pan-caspase inhibition (with Q-VD-OPh) or deletion of the intrinsic apoptotic effectors BAX/BAK alleviates this effect [Citation45,Citation115,Citation116,Citation118,Citation119,Citation122,Citation127].
Alongside the requirement of MCL-1 for breast tumor growth, MCL-1 has also been implicated in acquired resistance to tamoxifen or fulvestrant treatment in ER-positive breast cancer cell lines [Citation128]. Furthermore, MCL-1 protein is elevated in post-chemotherapy biopsies of TNBC patients and increased MCL-1 has been shown in multiple in vitro models of acquired paclitaxel resistance, where knock-down of MCL1 restores the apoptotic response [Citation47–49]. MCL-1 has also emerged as an important resistance factor to BCL-2/BCL-XL specific BH3-mimetics in hematopoietic cancers, encouraging the investigation of combinatorial treatments to also target MCL-1. This could have relevance in ER-positive breast cancer where venetoclax is currently undergoing clinical trials, as MCL-1 expression is associated with resistance to BCL-2/BCL-XL inhibition in ER-positive breast cancer cell lines. Indeed, targeting of MCL-1 has been shown to sensitize cells and effectively inhibit tumor growth in vitro and in vivo when administered in combination with BCL-2/BCL-XL inhibitors [Citation122,Citation127]. Importantly, ER-positive breast cancer cell lines adapt to MCL-1 or BCL-XL inhibition through BIM sequestration by the respective uninhibited pro-survival BCL-2 family protein, and this could be harnessed therapeutically through sequential treatment to liberate BIM and stimulate activation of BAX/BAK [Citation129]. Interestingly, mono-therapeutic treatment with the MCL-1-specific BH3-mimetic VU661013 slowed the growth of HCC1187 and BT20 TNBC xenografts, and co-treatment with docetaxel or doxorubicin further enhanced the inhibition of tumor growth [Citation115].
Taken together, this indicates that MCL-1 inhibition can sensitize different breast cancer subtypes to a range of therapies, though it is likely that refined treatment strategies will be required to circumvent significant toxicity in normal tissues. For example, MCL-1 is known to be important during homeostatic cardiomyocyte functionality [Citation7,Citation8,Citation130]. Whilst the pro-tumor role of MCL-1 in models of breast cancer is highly reliant on BH3-dependent functions of MCL-1, there may be important non-apoptotic roles for MCL-1 in tissues outside the breast and the ability of MCL-1-targeting strategies to impact or spare these alternative functions must be considered given the fatality of complete Mcl1 deletion in mice [Citation6,Citation118,Citation131]. Potential toxicity could be countered using antibody-drug conjugates (ADCs) to selectively target tumor cells. Indeed, targeted inhibition of BCL-XL with Mirzotamab clezutoclax (ABBV-155), composed of a BCL-XL inhibitor linked to a monoclonal anti-B7H3 antibody, is currently undergoing phase I clinical trials for the treatment of B7H3-expressing relapsed/refractory solid tumors (NCT03595059) [Citation132].
As well as the documented tumor cell-intrinsic role for MCL-1, tumor explant and co-culture experiments have also highlighted a pro-tumor function for MCL-1 within the stromal microenvironment. Breast cancer associated fibroblasts (bCAFs) are dependent upon MCL-1 for survival and through secretion of IL-6, bCAFs can induce upregulation of MCL1 mRNA and protein in luminal breast cancer cells to reduce sensitivity to BCL-2/XL inhibition [Citation133]. Genetically engineered mouse models (GEMM) allow investigation of tumor cells within an intact tumor microenvironment and have revealed a key role for MCL-1 in tumor development and maintenance of mammary cancer in vivo (). The MMTV-PyMT mouse is molecularly and histopathologically identified as representing human luminal B breast tumors and faithfully recapitulates the tumorigenic process from hyperplasia to invasion of the lung and lymph nodes [Citation134,Citation135]. Genetic deletion of Mcl1 in the mammary epithelium of MMTV-PyMT mice reveals an absolute requirement for MCL-1 in mammary tumor development and outgrowth [Citation45]. Dramatically, when Mcl1 is conditionally ablated in the tumor epithelium of mice possessing allografted mammary tumors 8/9 tumors regressed, and in 4/9 cases this regression was complete and underpinned long-term tumor-free survival [Citation118]. These genetic studies reveal an exquisite dependency of MMTV-PyMT mammary tumors on MCL-1 for tumor maintenance and development ().
Table 2. Summary of genetically engineered mouse models revealing a requirement for Mcl1 in mammary cancer.
The importance of MCL-1 in breast cancer has also emerged from new models that recapitulate the genetic events in BRCA1-mutant tumors, where loss-of-function mutations in TP53 and amplification of MYC occur in 65% and 44% of cases, respectively [Citation124]. In elegant mouse models, deletion of Brca1 and Trp53, alongside concurrent amplification of MYC, results in mammary tumors that molecularly resemble basal-subtype human breast cancers. Comparative oncogenomics of these mouse tumors and human breast cancers identifies common amplification of the MCL1 locus, and MCL-1 overexpression was shown to accelerate mammary tumor development in this mouse model (). Knock-down of Mcl1 in GEMM-derived organoids further highlighted a requirement for MCL-1 expression, thus confirming the necessity of MCL-1 in a genetic model of BRCA1-mutant basal-like TNBC [Citation124]. In line with these studies, which demonstrate that genetic manipulation of MCL1 alters tumor development, pharmaceutical targeting of MCL-1 with the BH3-mimetic S63845 can also inhibit growth of established MMTV-PyMT tumors in a BAX/BAK-dependent manner, indicating on-target efficacy of pharmacological inhibition of the canonical MCL-1 function to inhibit mammary tumor growth [Citation118].
PDX models of TNBC have shown that a reversible cell state enriched in a cancer stem-like cell (CSC) gene expression signature arises in drug-refractory breast cancer, and as elevation of MCL-1 occurs in treatment-resistant samples it is tempting to speculate that MCL-1 may facilitate this process [Citation48,Citation49,Citation136,Citation137]. Indeed, knock-down of MCL1 can restrict tumor initiation in vivo when tested in limiting dilution transplantation assays with the SUM159PT TNBC cell line, whilst MCL-1 overexpression increases the stemness of MDA-MB-468 cells (TNBC) as determined by an increased CD44hi/CD24lo cell-surface profile and enhanced tumorsphere-forming capacity [Citation49]. Furthermore, analysis of extensive breast cancer datasets reveals an association between MCL1 expression and markers of both stemness and an epithelial-to-mesenchymal transition (EMT) [Citation118].
It has been suggested that the primary function of MCL-1 in breast CSCs is due to non-canonical activity at the inner mitochondrial membrane where it functionally cooperates with MYC to elevate mitochondrial biogenesis and oxidative phosphorylation, although this non-canonical function was not disrupted when MCL-1 was inhibited pharmacologically using the VU0659158 BH3-mimetic in MDA-MB-436 or SUM159PT TNBC cell lines [Citation49]. Recent work in our group suggests that a key function of MCL-1 in conferring stem-like behavior in breast cancer is through its canonical role within the BCL-2 family. We demonstrated that deletion or pharmacological inhibition of MCL-1 with either S63845 or A1210477 BH3-mimetics impaired the tumorsphere-forming capacity of breast cancer cells, and that genetic ablation of pro-apoptotic effector proteins BAX and BAK abrogates this effect [Citation118]. There are subtle differences in the interactions between MCL-1 and distinct BH3-mimetics, and a more complete characterization of the functional consequence of each MCL-1 inhibitor is required [Citation115]. For effective anti-cancer therapy, BH3-mimetic drugs must counteract the pro-tumor functions of MCL-1 that occur in vivo. Promisingly, S63845 effectively mimics the anti-tumor impact of MCL1 deletion in a mouse model of breast cancer and the restricted tumor growth was dependent upon the presence of BAX/BAK, suggesting that the major anti-apoptotic function of MCL-1 in breast cancer cell survival can be effectively targeted with BH3-mimetics [Citation118]. Advances in BH3-profiling techniques have the potential to direct BH3 mimetic use in the clinic. Indeed, the ability of cancer therapeutics to prime individual cancers for apoptosis can be gauged ex vivo using an adapted BH3-profiling protocol [Citation138]. The utility of inhibiting different pro-survival BCL-2 proteins in combination with chemotherapy has been demonstrated using freshly isolated non-small cell lung cancer patient samples [Citation139]. It can be envisioned that such technologies could pave the way for personalized use of BH3 mimetics in breast cancer.
Concluding remarks
Whilst preclinical work has validated MCL-1 as a relevant target in breast cancer, the essentiality of MCL-1 in many normal cell types may necessitate careful administration of MCL-1 targeting drugs to facilitate a therapeutic window. To achieve tumor-specific cell death, MCL-1 inhibition may work best as a combination therapy, where it could be employed to exacerbate the effect of conventional cytotoxic and targeted treatment approaches. Distinct BCL-2 family proteins may play differentially prominent roles in individual cancers, and biomarkers to reveal potential sensitivity to MCL-1 inhibition would allow effective tailoring of treatment. It is possible that inhibition of multiple pro-survival BCL-2 family proteins is required to efficiently restore apoptosis in some patients, and this provides a compelling argument for further research into understanding which subsets of patients could benefit from MCL-1-specific inhibition; the optimal timing for prospective treatment; the potential value of co-administration of additional therapies; and in-depth characterization of the bona fide role that MCL-1 plays in breast development and tumorigenesis.
Acknowledgments
We thank Karen Blyth, Stephen Tait and Laura Martinez-Escardo for insightful comments and critical reading of this manuscript, and Catherine Winchester for assistance with manuscript preparation. Figures created using BioRender.com and GraphPad Prism. This work was supported by Cancer Research UK (grant number A29799).
Disclosure statement
KJC is a former employee of the Walter and Eliza Hall Institute, which receives milestne payments and royalities from the sale of venetoclax.
Data availability statement
References are provided for publically available data that was accessed through cBioportal (https://www.cbioportal.org) and DepMap (https://depmap.org).
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: A Cancer Journal for Clinicians. 2020;70(1):7–30.
- Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
- Campbell KJ, Tait SWG. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018;8(5):180002.
- Campbell KJ, Leung HY. Evasion of cell death: a contributory factor in prostate cancer development and treatment resistance. Cancer Letters. 2021;520:213–221.
- Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307(5712):1101–1104.
- Wang X,Bathina M, Lynch J, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27(12):1351–1364.
- Thomas RL,Roberts DJ,Kubil DA, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27(12):1365–1377.
- Beroukhim R,Porter D,Wei G, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905.
- Tsujimoto Y, Finger LR, Yunis J, et al. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226(4678):1097–1099.
- Bakhshi A, Jensen JP, Goldman P, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell. 1985;41(3):899–906.
- Cleary ML, Sklar J. Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci U S A. 1985;82(21):7439–7443.
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335(6189):440–442.
- Hockenbery D, Nuñez G, Milliman C, et al. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348(6299):334–336.
- Adams JM, Cory S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018;25(1):27–36.
- Certo M, Moore VDG, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9(5):351–365.
- Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17(3):393–403.
- Kuwana T,Bouchier-Hayes L,Chipuk JE, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17(4):525–535.
- Sattler M, Liang H, Nettesheim D, et al. Structure of Bcl-x L-bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275(5302):983–986.
- Liu XQ, Dai S, Zhu Y, et al. The structure of a Bcl-x(L)/Bim fragment complex: implications for bim function. Immunity. 2003;19(3):341–352.
- Zou H, Henzel WJ, Liu X, et al. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90(3):405–413.
- Kluck RM, Bossy-Wetzel E, Green DR, et al. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275(5303):1132–1136.
- Korsmeyer SJ, Wei MC, Saito M, et al. Pro-apoptotic cascade activates Bid, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7(12):1166–1173.
- Letai A, Bassik MC, Walensky LD, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2(3):183–192.
- Goldstein JC,Waterhouse NJ,Juin P, et al. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol. 2000;2(3):156–162.
- Kozopas KM,Yang T, Buchan HL, et al. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90(8):3516–3520.
- Zhou P, Qian L, Kozopas KM, et al. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89(2):630–643.
- Sattler M, Liang H, Nettesheim D, et al. Structure of Bcl-x L -Bak Peptide Complex: recognition Between Regulators of Apoptosis. Science. 1997;275(5302):983–986.
- Liu X, Dai S, Zhu Y, et al. The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity. 2003;19(3):341–352.
- Czabotar PE,Lee EF, van Delft MF, et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci U S A. 2007;104(15):6217–6222.
- Day CL,Chen L,Richardson SJ, et al. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J Biol Chem. 2005;280(6):4738–4744.
- Mei Y,Du W, Yang Y, et al. Puma(*)Mcl-1 interaction is not sufficient to prevent rapid degradation of Mcl-1. Oncogene. 2005;24(48):7224–7237.
- Lee EF,Czabotar PE,Van Delf MF, et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180(2):341–355.
- van Delft MF,Wei AH,Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399.
- Karbon G,Haschka MD,Hackl H, et al. The BH3-only protein NOXA serves as an independent predictor of breast cancer patient survival and defines susceptibility to microtubule targeting agents. Cell Death Dis. 2021;12(12):1151.
- Floros KV,Lochmann TL,Hu B, et al. Coamplification of miR-4728 protects HER2-amplified breast cancers from targeted therapy. Proc Natl Acad Sci U S A. 2018;115(11):E2594–e2603.
- Sørlie T,Perou CM,Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874.
- Prat A,Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68.
- Cerami E,Allen GI,Liu Z, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404.
- Gao J,Aksoy BA,Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):1.
- Pereira B,Chinn SF,Rueda OM, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479.
- Lefebvre C, et al. Mutational Profile of Metastatic Breast Cancers: a Retrospective Analysis. PLoS Med. 2016;13(12):e1002201.
- Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352.
- Xu Y, et al. ERα is an RNA-binding protein sustaining tumor cell survival and drug resistance. Cell; 2021.
- Campbell KJ, et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018;9(2):19.
- Jeselsohn R, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757–1767.
- Gasca J, et al. Loss of FBXW7 and accumulation of MCL1 and PLK1 promote paclitaxel resistance in breast cancer. Oncotarget; 2016.
- Balko JM, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–245.
- Lee KM, et al. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017;26(4):633–647.e7.
- Booy EP, Henson ES, Gibson SB. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene. 2011;30(20):2367–2378.
- Townsend KJ, et al. Regulation of MCL1 through a serum response factor/Elk-1-mediated mechanism links expression of a viability-promoting member of the BCL2 family to the induction of hematopoietic cell differentiation. J Biol Chem. 1999;274(3):1801–1813.
- Senichkin VV, et al. Saga of Mcl-1: regulation from transcription to degradation. Cell Death Differ. 2020;27(2):405–419.
- Cavarretta IT, et al. Mcl-1 is regulated by IL-6 and mediates the survival activity of the cytokine in a model of late stage prostate carcinoma. Adv Exp Med Biol. 2008;617:547–555.
- Berishaj M, et al. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007;9(3):R32.
- Siersbæk R, et al. IL6/STAT3 signaling hijacks estrogen receptor α enhancers to drive breast cancer metastasis. Cancer Cell. 2020;38(3):412–423.e9.
- Jourdan M, et al. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death Differ. 2000;7(12):1244–1252.
- Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: a review. Int J Cancer. 2016;138(11):2570–2578.
- Salgado R, et al. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int J Cancer. 2003;103(5):642–646.
- Gregory GP, et al. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia. 2015;29(6):1437–1441.
- Floros KV, et al. Targeting transcription of MCL-1 sensitizes HER2-amplified breast cancers to HER2 inhibitors. Cell Death Dis. 2021;12(2):179.
- Cidado J, et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin Cancer Res. 2020;26(4):922–934.
- Dey J, et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci Rep. 2017;7(1):18007.
- Phillips DC, et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia. 2020;34(6):1646–1657.
- Liu H, et al. Stabilization and enhancement of the antiapoptotic activity of mcl-1 by TCTP. Mol Cell Biol. 2005;25(8):3117–3126.
- Mills JR, et al. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A. 2008;105(31):10853–10858.
- Tailler M, et al. By reducing global mRNA translation in several ways, 2-deoxyglucose lowers MCL-1 protein and sensitizes hemopoietic tumor cells to BH3 mimetic ABT737. Cell Death Differ. 2019;26(9):1766–1781.
- Vega F, et al. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res. 2006;66(13):6589–6597.
- Wei G, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10(4):331–342.
- Wendel HG, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21(24):3232–3237.
- Fu NY, et al. EGF-mediated induction of Mcl-1 at the switch to lactation is essential for alveolar cell survival. Nat Cell Biol. 2015;17(4):365–375.
- Anderson GR, et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci Transl Med. 2016;8(369):369ra175.
- Germain M, Duronio V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. J Biol Chem. 2007;282(44):32233–32242.
- Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234(4774):364–368.
- Yang T, Kozopas KM, Craig RW. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol. 1995;128(6):1173–1184.
- Ding Q, et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27(11):4006–4017.
- Ding Q, et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008;68(15):6109–6117.
- Domina AM, et al. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23(31):5301–5315.
- Harley ME, et al. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. Embo J. 2010;29(14):2407–2420.
- Ding Q, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007;67(10):4564–4571.
- Zhong Q, et al. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–1095.
- Warr MR, et al. BH3-ligand regulates access of MCL-1 to its E3 ligase. FEBS Lett. 2005;579(25):5603–5608.
- Balko JM, et al. molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–245.
- Akhoondi S, et al. Inactivation of FBXW7/hCDC4-β expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010;12(6):R105.
- Schwickart M, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463(7277):103–U114.
- Wu X, et al. JOSD1 inhibits mitochondrial apoptotic signalling to drive acquired chemoresistance in gynaecological cancer by stabilizing MCL1. Cell Death Differ. 2020;27(1):55–70.
- Wu X, et al. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc Natl Acad Sci U S A. 2019;116(8):2961–2966.
- Kabir S, et al. The CUL5 ubiquitin ligase complex mediates resistance to CDK9 and MCL1 inhibitors in lung cancer cells. Elife. 2019;8.
- Wang B, et al. Role of Ku70 in deubiquitination of Mcl-1 and suppression of apoptosis. Cell Death Differ. 2014;21(7):1160–1169.
- Zhang S, et al. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat Commun. 2018;9(1):215.
- Wu X, Luo Q, Liu Z. Ubiquitination and deubiquitination of MCL1 in cancer: deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis. 2020;11(7):556.
- Wang Z, et al. Proteolysis targeting chimeras for the selective degradation of mcl-1/bcl-2 derived from nonselective target binding ligands. J Med Chem. 2019;62(17):8152–8163.
- Papatzimas JW, et al. From Inhibition to Degradation: targeting the antiapoptotic protein myeloid cell leukemia 1 (MCL1). J Med Chem. 2019;62(11):5522–5540.
- Oda E, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288(5468):1053–1058.
- Shibue T, et al. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17(18):2233–2238.
- Villunger A, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302(5647):1036–1038.
- Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681.
- Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428.
- Mason KD, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186.
- Wilson WH, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11(12):1149–1159.
- Oakes SR, Vaillant F, Lim E, et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc Natl Acad Sci U S A. 2012;109(8):2766–2771.
- Balachander SB, et al. Abstract 56: AZD0466, a nanomedicine of a potent dual Bcl2/Bcl-xl inhibitor, exhibits anti–tumor activity in a range of henatological and solid tumor models. Cancer Research. 2020;80(16_Supplement):56.
- Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322.
- Seymour JF, Ma S, Brander DM, et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. 2017;18(2):230–240.
- DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629.
- Perillo B, Sasso A, Abbondanza C, et al. 17beta-estradiol inhibits apoptosis in mcf-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol. 2000;20(8):2890–2901.
- Vaillant F, Merino D, Lee L, et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell. 2013;24(1):120–129.
- Lok SW, Whittle JR, Vaillant F, et al. A phase ib dose-escalation and expansion study of the bcl2 inhibitor venetoclax combined with tamoxifen in er and bcl2-positive metastatic breast Cancer. Cancer Discov. 2019;9(3):354–369.
- Whittle JR, Vaillant F, Surgenor E, et al. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res. 2020;26(15):4120–4134.
- Lindeman GJ, Bowen R, Jerzak KJ, et al. Results from VERONICA: a randomized, phase II study of second-/third-line venetoclax (VEN) + fulvestrant (F) versus F alone in estrogen receptor (ER)-positive, HER2-negative, locally advanced, or metastatic breast cancer (LA/MBC). J clin oncol. 2021;39(15_suppl):1.
- Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–482.
- Tron AE, Belmonte MA, Adam A, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9(1):5341.
- Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a selective mcl1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8(12):1582–1597.
- Ramsey HE, Fischer MA, Lee T, et al. A novel mcl1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566–1581.
- Moujalled DM, Pomilio G, Ghiurau C, et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia. 2019;33(4):905–917.
- Lee T, et al. Discovery of potent myeloid cell leukemia-1 (mcl-1) inhibitors that demonstrate in vivo activity in mouse xenograft models of human cancer. J Med Chem. 2019;62(8):3971–3988.
- Xiao Y, Nimmer P, Sheppard GS, et al. MCL-1 is a key determinant of breast cancer cell survival: validation of mcl-1 dependency utilizing a highly selective small molecule inhibitor. Mol Cancer Ther. 2015;14(8):1837–1847.
- Merino D, Whittle JR, Vaillant F, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med. 2017;9(401).
- Campbell KJ et al Breast cancer dependence on MCL-1 is due to its canonical anti-apoptotic function, et al. Cell Death Differ. 2021;28(9): 2589–2600.
- Goodwin CM, Rossanese OW, Olejniczak ET, et al. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015;22(12):2098–2106.
- Young AI, Law AMK, Castillo L, et al. MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast Cancer Res. 2016;18(1):125.
- Petrocca F, Altschuler G, Tan S, et al. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell. 2013;24(2):182–196.
- Williams MM,Lee, L, Hicks, D. J, et al. Key Survival Factor, Mcl-1, Correlates with Sensitivity to Combined Bcl-2/Bcl-xL Blockade. Mol Cancer Res. 2017;15(3):259–268.
- Tsherniak A. Defining a cancer dependency map. Cell. 2017;170:564–576 e16.
- Annunziato S,Henneman, L, Brambillasca, C. S, et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat Commun. 2019;10(1):397.
- Meyers RM, Bryan JG, McFarland JM, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–1784.
- Dempster JM,Rossen, J., Kazachkova, M, et al. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. BioRxiv. 2019;720243.
- Williams MM, Elion DL, Rahman B, et al. Therapeutic inhibition of mcl-1 blocks cell survival in estrogen receptor-positive breast cancers. Oncotarget. 2019;10(52):5389–5402.
- Thrane S, Pedersen AM, Thomsen MBH, et al. A kinase inhibitor screen identifies mcl-1 and aurora kinase A as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene. 2015;34(32):4199–4210.
- Alcon C, Gómez Tejeda Zañudo JGT, Albert R, et al. ER+ breast cancer strongly depends on mcl-1 and bcl-xl anti-apoptotic proteins. Cells. 2021;10(7):1659.
- Rasmussen ML,Taneja, N., Neininger, A. C, et al. MCL-1 inhibition by selective bh3 mimetics disrupts mitochondrial dynamics causing loss of viability and functionality of human cardiomyocytes. Science. 2020;23(4):101015.
- Opferman JT, Letai A, Beard C, et al. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426(6967):671–676.
- Tolcher AW, et al.
- Louault K, Bonneaud TL, Séveno C, et al. Interactions between cancer-associated fibroblasts and tumor cells promote MCL-1 dependency in estrogen receptor-positive breast cancers. Oncogene. 2019;38(17):3261–3273.
- Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163(5):2113–2126.
- Lim E,Wu, D, Pal, B, et al. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast Cancer Res. 2010;12(2):R21.
- Creighton CJ,Li, X, Landis, M, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–13825.
- Echeverria GV,Ge, Z., Seth, S, et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci Transl Med. 2019;11(488).
- Montero J, Sarosiek K, DeAngelo J, et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015;160(5):977–989.
- Potter DS, Du R, Bhola P, et al. Dynamic BH3 profiling identifies active BH3 mimetic combinations in non-small cell lung cancer. Cell Death Dis. 2021;12(8):741.