
ABSTRACT
A hallmark of cellular senescence is proliferation-like activity of growth-promoting pathways (such as mTOR and MAPK) in non-proliferating cells. When the cell cycle is arrested, these pathways convert arrest to senescence (geroconversion), rendering cells hypertrophic, beta-Gal-positive and hyperfunctional. The senescence-associated secretory phenotype (SASP) is one of the numerous hyperfunctions. Figuratively, geroconversion is a continuation of growth in non-proliferating cells. Rapamycin, a reversible inhibitor of growth, slows down mTOR-driven geroconversion. Developed two decades ago, this model had accurately predicted that rapamycin must extend life span of animals. However, the notion that senescent cells directly cause organismal aging is oversimplified. Senescent cells contribute to organismal aging but are not strictly required. Cell senescence and organismal aging can be linked indirectly via the same underlying cause, namely hyperfunctional signaling pathways such as mTOR.
“How wonderful that we have met with a paradox. Now we have some hope of making progress.” Niels Bohr
Preface
The activity of mitogen-activated and growth-promoting pathways such as PI3K/mTOR and ERK/MAPK is unremarkable in senescent cells compared to proliferating cells. But this seemingly “unremarkable” lack of the difference is remarkable in and of itself: proliferation-like activity of growth-promoting pathways in non-proliferating cells [Citation1]. But why should we compare apples and oranges? The most relevant counterpart to senescence is quiescence: cells are arrested but not senescent (most cells in the organism). In such a comparison, a difference emerges: the activity of growth-promoting pathways, such as mTOR and MAPK pathways, is high in senescent cells, as if senescent cells proliferate. However, they do not.
There are many paradoxes in the field of cell senescence. For example, as we will discuss later, hyperfunctional mitogenic signaling (MEK/ERK/MAPK) can cause senescence instead of accelerated proliferation [Citation2–7]. In addition, forced activation of the cell cycle in quiescent cells may lead to apoptosis, also instead of proliferation [Citation7].
Seemingly paradoxically, all known anti-aging interventions in animals from nutrient restriction to rapamycin (an mTOR inhibitor) can slow down cell proliferation.
How and why does rapamycin inhibit cell proliferation but maintains proliferative potential in the arrested cells?
Also, discussed here is the notion that senescent cells contribute to age-related diseases, but are not required. In the organism, senescent cells are a subset of gerogenic cells, which do not look like senescent cells. Finally, this article attempts to link cell senescence and organismal aging.
Conventional view on cell senescence is inadequate
For most scientists, the term “cellular senescence” means irreversible cell cycle arrest or, more precisely, permanent loss of proliferative potential. This is known as the Golden Marker of cell senescence. However, all attempts to link cell cycle arrest to organismal aging have not been successful. The oldest people and animals do not die from bone marrow failure or intestinal atrophy due to cessation of cell proliferation. In contrast, aging is associated with hyper-proliferative conditions such as cancer and leukemia, organ hypertrophy (e.g. prostate and heart hypertrophy), atherosclerotic plaques, tissue fibrosis, obesity, and others.
For example, myocardial infarction, a common cause of death in humans, is caused by neither cell cycle arrest nor DNA damage. It is cell growth and excessive functions (hyperfunction) that, via multiple steps, lead to atherosclerosis, hypertension, vasoconstriction and thrombosis, culminating in myocardial infarction.
Increasingly, despite the definition, the link between cell senescence and organismal aging becomes viewed via SASP or hyper-inflammatory phenotype of senescent cells [Citation8–18]. This is a step in the right direction, but still insufficient. SASP is a typical hyper-function (secretion is a normal function of many cell types), but it is not the only one. Most functions are tissue-specific. For example, functions of blood platelets are adhesion and aggregation, for macrophages – phagocytosis, including phagocytosis of oxidized LDL. Hyperfunction of arterial smooth muscle cells (SMC) results in cell hypertrophy, vessel stiffness, and vasoconstriction, which all contribute to hypertension (systemic hyperfunction) and atherosclerosis (see Ref. [Citation19]) (of note, beta-Gal staining is a marker of hyperfunctional lysosomes [Citation20,Citation21] and both increased p16 and beta-Gal staining are markers of hyperfunctional macrophages [Citation22–24]).
Also to be discussed is the seemingly paradoxical notion that despite permanent loss of proliferative potential, senescence is not a form of arrest, it is a form of growth [Citation25]. Cell senescence cannot be understood without the concept of geroconversion – a conversion from non-senescence to senescence or a continuation of growth, when actual growth is completed [Citation1,Citation25]. Let us start from the beginning.
Proliferation, quiescence and senescence in cell culture
Proliferation:
In cell culture, growth factors (GF) and nutrients stimulate growth-promoting pathways such as MEK/MAPK and PI3K/mTOR (). These pathways stimulate cellular mass growth and induce cyclin D1, which initiates cell cycle progression. Mass growth is balanced by cell division [Citation1].
Figure 1. Representation of proliferation, quiescence and senescence.
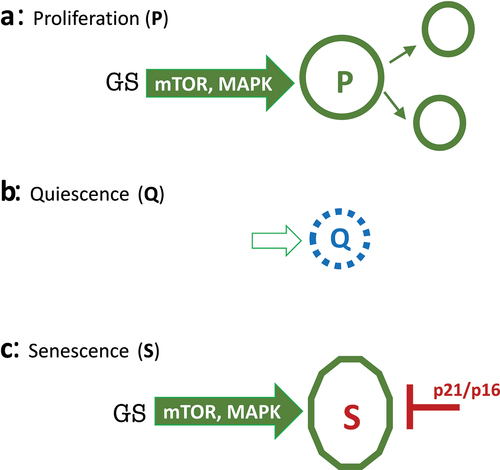
Quiescence:
In the absence of growth stimulation, MAPK and mTOR are deactivated, cyclins are not induced, the cell cycle slows down and cells stop proliferating – a condition known as quiescence (Q) (). In this state, cells neither grow in size nor cycle. Quiescent cells are small and metabolic processes are downregulated. In cell culture, quiescence can be caused by withdrawal of serum, growth factors and nutrients, thus deactivating MAPK and mTOR and causing, in turn, cell cycle arrest [Citation1]. (Note: Some cancer cells tend to die rather than become quiescent for the reason discussed in the last section.) Quiescence is reversible by growth stimulation, such as the readdition of growth factors or serum.
Contact inhibition also causes quiescence [Citation26]. In contact-inhibited cells, MAPK and mTOR pathways are deactivated, halting both cell mass growth and cell cycle (26). Splitting contact-inhibited cells into a lower density culture re-activates MAPK and mTOR pathways and releases the cell cycle. The cells then re-start proliferation.
Senescence:
Once again, in quiescence, cell cycle arrest is caused by the deactivation of growth-promoting (MAPK, mTOR, etc.) pathways. However, the cell cycle can be blocked directly by induction of p21 and p16, which in turn can be induced by numerous stressors (). When the cell cycle is blocked in such a manner, mTOR and MAPK remain fully activated, driving unbalanced growth without inducing division and cyclins D. This induction is futile, however, because the cell cycle is blocked by p21 and p16. The cell is frozen in a hypermitogenic state [Citation1]. Cells have a large and flat morphology and are hyperactive and hyperfunctional (e.g, SA-beta-Gal staining is lysosomal hyperfunction and SASP is secretory hyperfunction).
Thus, a combination of molecular markers of senescence is as follows: p16 and p21 (markers of cell cycle block) plus phospho-S6 (a convenient marker of mTOR activity) and cyclin D1 (a marker of overstimulation (1).
Figuratively, geroconversion is “twisted” growth when actual growth is completed [Citation1,Citation25,Citation27]. mTOR drives geroconversion, rendering cells hypertrophic and hyperfunctional (e.g. SASP) and thus leading eventually to age-related pathologies [Citation27–29].
The car analogy: push brake and gas at the same time
When a driver pushes the gas pedal (analogous to growth stimulation), the car drives. This is proliferation (). When the gas pedal is released, the car slows down and stops (). This is a reversible quiescence. In cell senescence, the cell cycle is blocked by powerful brakes, such as p16 and/or p21, in the presence of growth stimulation (). In this senescence analogy, the brakes and accelerator are pushed simultaneously. Eventually, the car will be out of order. This analogy is applicable not only to cellular senescence but also to organismal aging [Citation30].
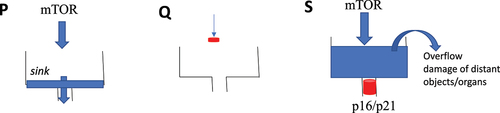
The clogged sink analogy
Senescent cells can be damaging to the organism. This is not molecular damage but organ damage and mostly to the distant organs. Consider the clogged sink analogy (). In proliferation, the water comes into the sink and gets out. If you close the faucet, water does not come at all (quiescence) and the sink is empty, as well. However, if the sink is clogged and the faucet is still turned on (cell senescence), then the problem arises. The water overflows the sink and may damage distant objects, such as a laptop on the floor. Similarly, hyperfunctional cells in the arterial walls, liver, adipose tissue, and the hematopoietic system can eventually cause atherosclerosis and thrombosis, leading to a stroke, damaging the brain.
Figure 2. The clogged sink analogy for proliferation, quiescence and senescence.
From the model of geroconversion to the hyperfunction theory
The hyperfunction theory is a translation of the rules of geroconversion to organismal aging [Citation31].
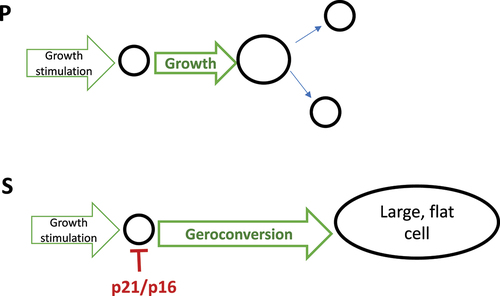
Geroconversion is the continuation of a growth program when actual growth is restricted by cell cycle arrest. Geroconversion is a quasi-program of growth (). Organismal aging is a continuation of developmental growth, when actual growth is completed. Aging is a quasi-program of developmental growth [Citation27,Citation31–37].
Figure 3. Geroconversion is a form of “twisted” growth.
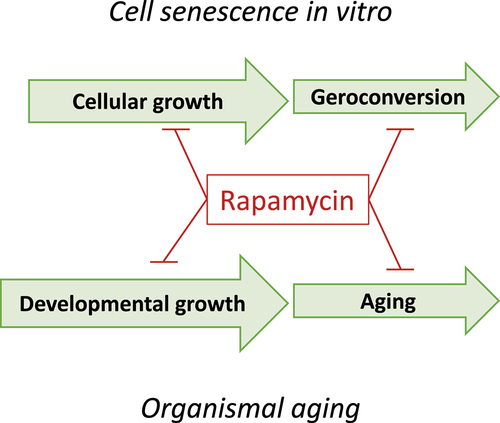
Both geroconversion and organismal aging are driven in part by the mTOR pathway (). Like cell senescence, organismal aging is primarily associated with hypertrophy and hyperfunction that eventually lead to diseases of aging, organ damage and secondary loss of function. Examples of organismal hyperfunctions include hypertension, hyperglycemia, hyperlipidemia, hyperinsulinemia, hypercoagulation, prostate hypertrophy autoimmunity, osteoarthritis and early stages of all diseases. Hyperfunctions eventually lead to organ and system damage and loss of functions [Citation27]. This creates the illusion that aging is primarily functional decline. Since it is more difficult to restore function than to suppress hyperfunctions, rapamycin should be most effective before organ damage occurs [Citation27].
Figure 4. The analogy between cell senescence and organismal aging.
Oversimplified model of cellular senescence in organismal aging
exemplifies a conventional and oversimplified model linking cellular senescence to organismal aging:
Figure 5. Linking cell senescence to organismal aging.
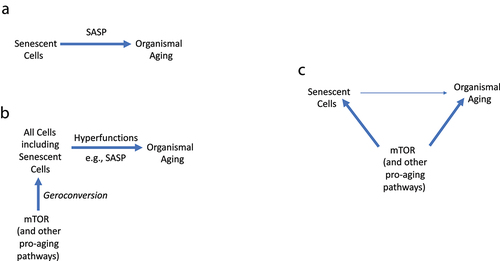
Senescent cells-SASP-Aging ().
However, this simple schema (Senescent cells-SASP-Aging) cannot substitute the complex pathogenesis of age-related diseases, involving multiple cell types and different hyperfunctions. All achievements of biomedical sciences should not be thrown away.
Notably, SASP is just one of the numerous cellular hyperfunctions, and its suppression may not always be sufficient for life extension. For example, glucocorticoids (corticosterone and cortisol) inhibit SASP [Citation12]; however, glucocorticoids do not extend life span. In contrast, very high levels of glucocorticoids in untreated Cushing’s disease are associated with a poor prognosis; the median survival is about five years [Citation38].
From an oversimplified model to hyperfunctional model
Let us return to the example of myocardial infarction. The atherosclerotic plaque consists of hypertrophic and hyperplastic SMC, hyperfunctional macrophages (foam cells), and calcification and lipid accumulation by foam cells. Hyperfunctional (activated) endothelial cells attract hyperfunctional blood platelets with a higher propensity to adhere and aggregate. Atherosclerosis, hypercoagulation, platelet overactivation, and hypertension myocardial hypertrophy all together may lead to myocardial infarction.
According to the hyperfunction theory, the sequence of events is:
Constitutively active signaling pathways (in the absence of growth) – geroconversion – Hyperfunctional gerogenic cells (including a few senescent cells) – Tissue-specific hyperfunctions (including SASP) – Age-related diseases, whose sum is aging ().
In the organism, geroconversion creates a few senescent cells but makes all cells hyperfunctional (gerogenic), causing age-related diseases – which are deadly manifestations of aging. More generally, senescent cells may contribute to organismal aging but are not required. Even this model is not sufficient to fully understand the link between cellular senescence and organismal aging.
The ultimate hyperfunction model
According to the view, cell senescence may or may not be involved in organismal aging. In this scenario, both cell senescence and organismal aging result from hyperfunction by the same signal-transduction pathways such as mTOR, without a necessarily causative link ( C). More on this in the last section: “The origin of the hyperfunction theory: personal perspective.”
Consider analogy. What is the link between yeast aging and organismal aging? There is no direct link. Our body is not built of yeast cells. Yeast aging and our aging are linked only by analogy. mTOR is involved in both yeast and organismal aging. No mechanistic link is observed. Since rapamycin slows down geroconversion, one may expect that it should slow aging and delay age-related diseases [Citation27].
How to measure the “Golden marker” of senescence: technical section
To study geroconversion using “golden marker” (permanent loss of proliferative potential), one first needs to induce reversible arrest and then observe how it becomes irreversible over time [Citation25,Citation39]. The arrest should be easily switched on and off, without damaging the cell.
As described by Dr Roninson and coworkers, the HT-1080 fibrosarcoma cell line has been permanently transfected with IPTG-inducible p21 and p16 [Citation40–42].
Importantly, p21 and p16 do not cause DNA damage and do not inhibit mTOR and MEK/ERK/MAPK [Citation25,Citation39,Citation43]. These CDK inhibitors just cause cell cycle arrest, which can be easily reversed by switching p21 and p16 off, by adding and removing IPTG. These cell lines are called HT-p21 and HT-p16, respectively.
To study geroconversion, these cells must be plated at low cell density to give them space to become large and flat during geroconversion (Note: High cell density inhibits geroconversion by itself [Citation26]). The culture medium should be fresh to avoid mTOR deactivation.
The addition of IPTG to the culture medium rapidly induces super-high levels of p21 (or p16), causing cell cycle arrest in 100% of cells after 16 hours [Citation42].
When (after 16 hours) IPTG is removed by media change, the cells resume proliferation. So, at that time, cells are not senescent. But if IPTG is washed out (change of the media) after 3–4 days, then most cells cannot restart proliferation. While still entering the S phase of the cell cycle (because p21 and p16 are switched off), senescent cells cannot complete the cell cycle and die. This means that they lost the potential to proliferate, form colonies, regenerate cell culture, and reverse senescent morphology. Cell senescence is irreversible even when cell cycle arrest is reversible [Citation1].
The longer the arrest before IPTG is washed out, the lower the proliferative (regenerative) potential of the cell cultures [Citation39,Citation44]. The same effect was observed with other reversible CDK inhibitors [Citation44].
Rapamycin (when added together with IPTG) slows down geroconversion, so most cells can resume proliferation, when both IPTG and rapamycin are washed out (Note: Rapamycin should be removed because it inhibits proliferation on its own). Rapamycin slowed down geroconversion approximately 3-fold [Citation39]. It also decreased cellular hypertrophy, which can be measured as a ratio of protein per cell and moderately decreases beta-Gal staining [Citation45].
Convenient markers of geroconversion to senescence
IPTG and butyrate cause cell cycle arrest that can be reversed by a simple change of the media [Citation39]. However, in many models, the method of induction of cell cycle arrest (DNA damaging drugs such as doxorubicin) results in permanent loss of the proliferative potential (PLPP) because arrest cannot be reversed by change of the media. Such models are not suitable to measure the golden marker of senescence.
During geroconversion, cells grow in size (large morphology), express super-high levels of cyclin D1 and are characterized by beta-Gal staining (lysosomal hyperfunction) and SASP (hypersecretion).
For example, during cell cycle arrest by IPTG, the ratio of protein per cell increased exponentially for 3–4 days [Citation45]. Rapamycin slows down exponential growth in size (decreased protein per cell ratio), which can be easily determined by measuring total protein in the well and the cell count in parallel wells. This is perhaps the best way to measure effects of gerostatics such as rapamycin. Another marker of geroconversion is a combined expression of cyclin D1 and phospho-S6 in arrested cells with p21/p16 expression [Citation1].
Gerostatics
The concept of geroconversion allowed us to discover gerostatics (a subgroup of cytostatic drugs that slow down not only cell proliferation but also geroconversion). Rapamycin and other rapalogs are prototypical gerostatics. All gerostatics may induce cell cycle arrest on their own, but in the presence of p21/p16-induced cell cycle arrest, their gerostatic effects become apparent. For example, nutlin-3 (an Mdm-2 inhibitor and indirect p53 inducer) induces p53/p21 and reversible quiescence in HT-p21 and HT-p16 cells. But why does it not induce senescence, despite p53/p21 induction? This is because nutlin-3 simultaneously inhibits mTOR and slows down geroconversion [Citation46]. In fact, when added to cells arrested by IPTG-inducible ectopic p21 and p16, nutlin-3 suppresses geroconversion, sustains quiescence, prevents loss of proliferative potential, and renders cells smaller and elongated instead of flat (46). Rapamycin makes cells smaller, but they are still flat (Note: in some cell lines, nutlin-3 does not sufficiently inhibit mTOR and therefore induces senescence [Citation47]).
In agreement with our findings, the work from different laboratories demonstrated that rapamycin decreases senescent markers including SASP [Citation5,Citation16,Citation48–60].
Inhibitors of MEK and PI3K (43), double PI3K/mTOR and pan-mTOR inhibitors [Citation61,Citation62] are gerostatics, which slow down proliferation and geroconversion.
Pan-mTOR inhibitors are even more effective than rapamycin to suppress senescent phenotype [Citation61,Citation62]. They are slightly superior to rapamycin (and other rapalogs) in inhibiting hypertrophy (preventing large cell morphology) and beta-Gal staining and oil red staining (fatty droplets). Unlike rapamycin, they prevent flat morphology. Their advantage is that they inhibit rapamycin-insensitive functions of mTORC1. However, pan-mTOR inhibitors are cytotoxic at higher doses [Citation62].
I suggest a combination of low doses of pan-mTOR or PI3K/mTOR inhibitors plus high doses of rapamycin (or everolimus). At low doses, these inhibitors are expected to moderately suppress mTORC1 functions without toxicity, while high doses of rapamycin may inhibit rapamycin-sensitive function profoundly also without toxicity.
Unfortunately, pan-mTOR inhibitors have not yet been tested on life extension in animals. MEK inhibitor (in combination with rapamycin) was tested in Drosophila only [Citation63]).
Replicative senescence
In specific conditions, rapamycin can also delay replicative senescence [Citation60,Citation64–66] and prevent senescence during cell reprogramming [Citation67]. The mechanism is still unclear. Replicative senescence (also known as the Hayflick limit) is an ineffective method of causing cell cycle arrest, which is induced by varying mechanisms after varying number of divisions and at varying times for individual cells. Replicative senescence is an inconvenient model that has no relevance to the organism. Therefore, we will not discuss replicative senescence further.
The notion of geroconversion is somewhat underappreciated
The notion of geroconversion adds the second dimension to distinguish between senescence (S) and quiescence (Q). The second dimension reveals a reverse relationship between proliferation and geroconversion in primary normal cells ().
Figure 6. Two-dimensional representation of proliferation P, quiescence Q and senescence S in normal cells.
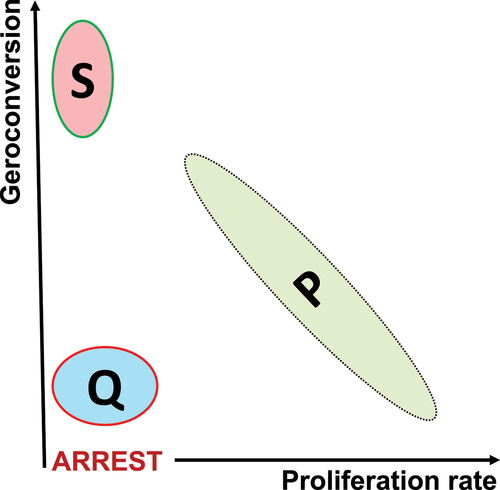
Noteworthy, geroconversion was described in the organism [Citation68–71].
DNA damage causes cell cycle arrest in the first 16 hours, but the senescent phenotype develops slowly during next 3–6 days. It is obvious there is a separate process apart from cell cycle arrest. This separate process is geroconversion. Without geroconversion, the arrested cell would be unchanged over time. Despite the obvious, the notion of geroconversion is underappreciated for several reasons:
1.The confusion between proliferation and proliferative potential. Note: Rapamycin inhibits proliferation but preserves proliferative potential in non-proliferating cells.
2.The confusion between irreversible cell cycle arrest and loss of proliferative potential. Note: Rapamycin does not reverse cell cycle arrest, it causes it (in the form of quiescence), but it maintains the proliferative potential in quiescent cells.
3.The use of replicative senescence models is complicated by cytostatic effects of rapamycin on cellular proliferation.
4.Beta-Gal staining is a marker of both senescence and quiescence caused by serum withdrawal and contact inhibition.
To study the effects of gerostatics on the golden marker, the culture model should meet several requirements:
a.To render arrest reversible, the inducer of cell cycle arrest at the beginning should be easily removable (e.g. by changing media). IPTG-inducible p21/p16, sodium butyrate and pharmacological CDK inhibitors are good examples. In contrast, most DNA damaging drugs, such as doxorubicin, are not removable and damage the cell.
b.The inducer of cell cycle arrest should not damage the cell.
c.The inducer should cause rapid arrest in all (or almost all) cells in the dish. Otherwise, non-arrested cells will take over.
Gerostatics (e.g. rapamycin) should be added simultaneously (not later) with the inducer of cell cycle arrest.
The origin of hyperfunction theory: personal perspective
Paradoxes may herald a paradigm shift. At the end of the last millennium, it was paradoxically shown that, in the absence of growth factors, forced activation of the cell cycle (e.g. by transfection with c-myc and E2F) in quiescent cells can cause apoptosis instead of proliferation [Citation7]. This happens because growth-promoting (upstream pathways) are deactivated, but the cell cycle (downstream) is forcefully activated.
So, there are three scenarios (): (i) growth signaling is ON and the cell cycle is ON (proliferation), (ii) growth signaling is OFF and cell cycle is OFF (quiescence), and (iii) growth signaling is OFF and cell cycle is ON (apoptosis). But what about the ON/OFF combination?
Figure 7. Four combinations of growth and cycling: formal approach.
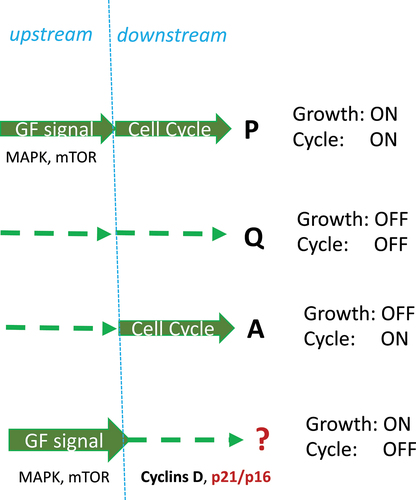
Here, the second paradox is helpful. In proliferating cells, strong mitogenic/growth-promoting signaling such as hyper-activated MEK, Ras, Raf and Akt can cause senescence, instead of proliferation [Citation2–7]. Strong growth stimulation simultaneously induces cyclins and CDK inhibitors (p21, p16), thus causing cell cycle arrest because inhibitors are dominant [Citation7].
As suggested in 2003, this condition (Growth ON/Cycle OFF) leads to cellular senescence [Citation7] in the process later named geroconversion [Citation72]. In fact, in the absence of cell division, growth becomes imbalanced, and cells become hypertrophic, beta-gal-positive (lysosomal hyperfunction), hypersecretory and so on. Growth factor/insulin resistance develops in compensation [Citation7].
By 2005, numerous deactivating mutations were identified that could extend the life span of model organisms. Among them were components of the mTOR pathway. Since the same pathways are involved in cell senescence and organismal aging, the hyperfunction theory of quasi-programmed aging was developed [Citation27]. Given that rapamycin was available for human use and mTOR was involved in most human diseases, to be useful, the theory became mTOR-centric [Citation27].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Blagosklonny MV. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle. 2018;17(24):2655–2665.
- Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602.
- Lin AW, Barradas M, Stone JC, et al. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12(19):3008–3019.
- Blagosklonny MV. The mitogen-activated protein kinase pathway mediates growth arrest or E1A-dependent apoptosis in SKBR3 human breast cancer cells. Int J Cancer. 1998;78(4):511–517.
- Deschenes-Simard X, Gaumont-Leclerc MF, Bourdeau V, et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27(8):900–915.
- Deschenes-Simard X, Kottakis F, Meloche S, et al. ERKs in cancer: friends or foes? Cancer Res. 2014;74(2):412–419.
- Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003;4(4):358–362.
- Wiley CD, Campisi J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab. 2021;3(10):1290–1301.
- Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. 2020;288(5):518–536.
- Coppe JP, Desprez PY, Krtolica A, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
- Velarde MC, Demaria M. Targeting senescent cells: possible implications for delaying skin aging: a mini-review. Gerontology. 2016;62(5):513–518.
- Laberge RM, Zhou L, Sarantos MR, et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell. 2012;11(4):569–578.
- Campisi J, Andersen JK, Kapahi P, et al. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21(6):354–359.
- Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972.
- Wang R, Sunchu B, Perez VI. Rapamycin and the inhibition of the secretory phenotype. Exp Gerontol. 2017;94:89–92.
- Wang R, Yu Z, Sunchu B, et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell. 2017;16(3):564–574.
- Bent EH, Gilbert LA, Hemann MT. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016;30(16):1811–1821.
- Christy B, Demaria M, Campisi J, et al. p53 and rapamycin are additive. Oncotarget. 2015;6(18):15802–15813.
- Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012;181(4):1142–1146.
- Lee BY, Han JA, Im JS, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5(2):187–195.
- Kurz DJ, Decary S, Hong Y, et al. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113(Pt 20):3613–3622.
- Frescas D, Hall BM, Strom E, et al. Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle. 2017;16(16):1526–1533.
- Hall BM, Balan V, Gleiberman AS, et al. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY). 2016;8(7):1294–1315.
- Hall BM, Balan V, Gleiberman AS, et al. p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). 2017;9(8):1867–1884.
- Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7(21):3355–3361.
- Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A. 2014;111(24):8832–8837.
- Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006;5(18):2087–2102.
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493(7432):338–345.
- Blagosklonny MV. Rapamycin for longevity: opinion article. Aging (Albany NY). 2019;11(19):8048–8067.
- Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. 2009;8(24):4055–4059.
- Blagosklonny MV. The hyperfunction theory of aging: three common misconceptions. Oncoscience. 2021;8:103–107.
- Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010;9(16):3151–3156.
- Gems D, de la Guardia Y. Alternative perspectives on aging in Caenorhabditis elegans: reactive oxygen species or hyperfunction? Antioxid Redox Signal. 2013;19(3):321–329.
- Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013;75:621–644.
- Ezcurra M, Benedetto A, Sornda T, et al. C. elegans eats its own intestine to make yolk leading to multiple senescent pathologies. Curr Biol. 2018;28(16):2544–56 e5.
- Wang H, Zhang Z, Gems D. Monsters in the uterus: teratoma-like tumors in senescent C. elegans result from a parthenogenetic quasi-program. Aging (Albany NY). 2018;10(6):1188–1189.
- Gems D. The hyperfunction theory: an emerging paradigm for the biology of aging. Ageing Res Rev. 2022;74:101557.
- Plotz CM, Knowlton AI, Ragan C. The natural history of cushing’s syndrome. Am J Med. 1952;13(5):597–614.
- Demidenko ZN, Zubova SG, Bukreeva EI, et al. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8(12):1888–1895.
- Chang BD, Watanabe K, Broude EV, et al. Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: implications for carcinogenesis, senescence, and age-related diseases. Proc Natl Acad Sci U S A. 2000;97(8):4291–4296.
- Chang BD, Broude EV, Fang J, et al. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19(17):2165–2170.
- Shtutman M, Chang BD, Schools GP, et al. Cellular Model of p21-Induced senescence. Methods Mol Biol. 2017;1534:31–39.
- Leontieva OV, Demidenko ZN, Blagosklonny MV. MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ. 2013;20(9):1241–1249.
- Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12(18):3063–3069.
- Demidenko ZN, Blagosklonny MV. Quantifying pharmacologic suppression of cellular senescence: prevention of cellular hypertrophy versus preservation of proliferative potential. Aging (Albany NY). 2009;1(12):1008–1016.
- Demidenko ZN, Korotchkina LG, Gudkov AV, et al. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010;107(21):9660–9664.
- Korotchkina LG, Leontieva OV, Bukreeva EI, et al. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY). 2010;2(6):344–352.
- Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–1061.
- Herranz N, Gallage S, Mellone M, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205–1217.
- Chen C, Liu Y, Liu Y, et al. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009;2(98):ra75.
- Iglesias-Bartolome R, Patel V, Cotrim A, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11(3):401–414.
- Mercier I, Camacho J, Titchen K, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012;181(1):278–293.
- Houssaini A, Breau M, Kebe K, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018;3(3). DOI:https://doi.org/10.1172/jci.insight.93203.
- Summer R, Shaghaghi H, Schriner D, et al. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am J Physiol Lung Cell Mol Physiol. 2019;316(6):L1049–L60.
- Luo Y, Li L, Zou P, et al. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014;97(1):20–29.
- Hinojosa CA, Mgbemena V, Van Roekel S, et al. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol. 2012;47(12):958–965.
- Gu Z, Tan W, Ji J, et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016;8(5):1102–1114.
- Nie D, Zhang J, Zhou Y, et al. Rapamycin treatment of tendon stem/progenitor cells reduces cellular senescence by upregulating autophagy. Stem Cells Int. 2021;2021:6638249.
- Gao C, Ning B, Sang C, et al. Rapamycin prevents the intervertebral disc degeneration via inhibiting differentiation and senescence of annulus fibrosus cells. Aging (Albany NY). 2018;10(1):131–143.
- Kolesnichenko M, Hong L, Liao R, et al. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11(12):2391–2401.
- Walters HE, Deneka-Hannemann S, Cox LS. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY). 2016;8(2):231–244.
- Leontieva OV, Blagosklonny MV. Gerosuppression by pan-mTOR inhibitors. Aging (Albany NY). 2016;8(12):3535–3551.
- Castillo-Quan JI, Tain LS, Kinghorn KJ, et al. A triple drug combination targeting components of the nutrient-sensing network maximizes longevity. Proc Natl Acad Sci U S A. 2019;116(42):20817–20819.
- Pospelova TV, Leontieva OV, Bykova TV, et al. Suppression of replicative senescence by rapamycin in rodent embryonic cells. Cell Cycle. 2012;11(12):2402–2407.
- Pospelova TV, Bykova TV, Zubova SG, et al. Rapamycin induces pluripotent genes associated with avoidance of replicative senescence. Cell Cycle. 2013;12(24):3841–3851.
- Antonioli E, Torres N, Ferretti M, et al. Individual response to mTOR inhibition in delaying replicative senescence of mesenchymal stromal cells. PLoS One. 2019;14(1):e0204784.
- Menendez JA, Vellon L, Oliveras-Ferraros C, et al. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle. 2011;10(21):3658–3677.
- Sousa-Victor P, Gutarra S, Garcia-Prat L, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506(7488):316–321.
- Sousa-Victor P, Perdiguero E, Munoz-Canoves P. Geroconversion of aged muscle stem cells under regenerative pressure. Cell Cycle. 2014;13(20):3183–3190.
- Yue F, Bi P, Wang C, et al. Conditional loss of PTEN in myogenic progenitors leads to postnatal skeletal muscle hypertrophy but age-dependent exhaustion of satellite cells. Cell Rep. 2016;17(9):2340–2353.
- Rodgers JT, King KY, Brett JO, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature. 2014;510(7505):393–396.
- Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012;4(3):159–165.