
ABSTRACT
Ferroptosis is a form of iron-dependent cell death caused by an excessive accumulation of reactive oxygen species and lipid peroxidation. The importance of ferroptosis in the occurrence and progression of various diseases is gradually being recognized; however, the exact biological effects and potential mechanisms of endothelial cell ferroptosis remain unclear. The endothelium forms the innermost layer of the blood vessels and lymphatic vessels. It acts as an important functional interface, responds to various pathological stimuli and causes endothelial dysfunction. Here, we review recent findings to elucidate the role of ferroptosis in endothelial cells under different pathophysiologic settings.
Introduction
Ferroptosis is a form of programmed cell death (PCD) in which intracellular iron and lipid reactive oxygen species (ROS) accumulate, eventually leading to the irreversible destruction of the structure and function of the plasma membrane. Ferroptosis, which is associated with specific morphological changes and different biochemical characteristics, is involved in many pathophysiological processes. Ferroptosis is regulated by signaling pathways related to various diseases as well as a variety of metabolic pathways, including amino acid, lipid, and iron metabolism and mitochondrial activity[Citation1].
Recently, several studies have confirmed the close relationship of ferroptosis to endothelial cell (EC) dysfunction (ECD) [Citation2–4]. ECs are widely distributed in human tissues and organs and play an important role in pathophysiological processes. Broadly, ECD includes a series of non-adaptive changes in functional phenotypes, which are of great significance to vascular permeability. ECD is also critical to the regulation of thrombosis and hemostasis, angiogenesis, local vascular tension and redox balance, and acute and chronic inflammatory reactions in arterial walls.
ECD is a form of endothelial injury caused by hemodynamic changes (such as increased vascular pressure caused by hypertension) or harmful stimuli (such as fine particulate matter [PM] composition, oxidized cholesterol, and hyperglycemia)[Citation5]. Interestingly, ferroptosis in ECs has been confirmed to contribute to various pathological processes such as stroke[Citation2], atherosclerosis[Citation3], and cancer[Citation4]. Here, we review the current knowledge on the molecular mechanisms of ferroptosis based on metabolic processes and describe how ferroptosis is involved in ECD in the development of different diseases.
Search strategy
An extensive literature review was carried out on PubMed, Google Scholar and Web of Science for relevant publications from January 2010 to March 2022. The keywords used for the literature search were “endothelium or endothelial cell or vascular or vessel” and “ferroptosis or iron or programmed cell death or reactive oxygen species or ROS”. Irrelevant, repetitive and non-English publications were excluded after analyzing the title and abstract. Following preliminary screening, the full text of the selected articles were read and the relevant references were further explored to identify other potentially related studies.
An overview of ferroptosis
Iron metabolism
Serum transferrin (Tf) carrying Fe3+ binds to the transferrin receptor (TFRC) in the cell membrane and is transported into cells. The six-transmembrane epithelial antigen of the prostate 3 (STEAP3) metal reductase in the endosome reduces Fe3+ to Fe2+, and then releases Fe2+ from the endosome to the cytoplasm, which is mediated by the solute carrier family 11 member 2/divalent metal transporter 1 (SLC11A2/DMT1) to form iron pools or bind to ferritin (Fn) as an iron reserve. Similar to Tf, lactoferrin also regulates ferroptosis by increasing iron intake[Citation6].
Fn, composed of heavy chain 1 (FTH1) and light chain (FTL) subunits, is the main iron storage protein in cells. Heavy chains have ferrous oxidase activity, which can oxidize Fe2+ to Fe3+ and store it in Fn, and contribute to intracellular iron homeostasis[Citation7]. Fn can also be encapsulated by autophagy-lysosomes, mediated by nuclear receptor coactivator protein 4 (NCOA4), and then degrade to release a large amount of Fe2+ into the labile iron pool[Citation8]. Fe2+ plays a key role in metabolic and biochemical processes, including the production of iron-sulfur proteins, oxygen transport in the mitochondria, and energy metabolism.
Additionally, iron can increase the activities of enzymes responsible for lipid peroxidation and oxygen homeostasis, such as arachidonate lipoxygenase (ALOX) or egl-9 family hypoxia inducible factor (EGLN) proline hydroxylase[Citation9]. Excessive iron produces ROS through an iron-dependent Fenton reaction or activates iron-containing enzymes (e.g. ALOX) to promote subsequent lipid peroxidation[Citation7].
Alternatively, multivesicular bodies and exosomes in Fn secrete iron into the extracellular space in the form of Fn[Citation10]. Global and local dynamic regulation of iron alters cell sensitivity to ferroptosis. Cellularly, iron chelation effectively prevents lipid peroxidation and associated ferroptosis. Iron homeostasis is mainly controlled by the iron regulatory protein hepcidin, which is involved in the post-transcriptional regulation of intracellular iron storage and release as well as import and export-related genes[Citation11].
Main regulatory signaling pathways of ferroptosis
Considering the critical role of ferroptosis in cell survival and death, it is not surprising that ferroptosis is finely regulated primarily through the cyst(e)ine/glutathione (GSH)/GSH peroxidase 4 (GPX4) axis, nicotinamide adenine dinucleotide phosphate (NADPH)/ferroptosis suppressor protein 1 (FSP1)/coenzyme Q10 (CoQ10) system, and the GTP cyclohydrolase-1 (GCH1)/tetrahydrobiopterin (BH4)/dihydrofolate reductase (DHFR) system, which are involved in numerous metabolic pathways[Citation7].
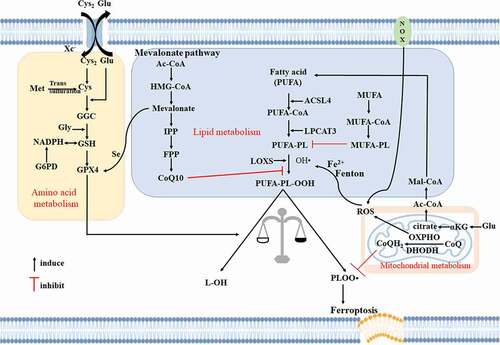
The main substrates of lipid peroxidation in ferroptosis are polyunsaturated fatty acids (PUFAs), including arachidonic acid and adrenic acid. Acyl-coenzyme A (Acyl-CoA) synthetase long-chain family member 4 (ACSL4), an important biomarker and driver of ferroptosis, catalyzes the combination of PUFA with CoA to form derivatives PUFA-CoA. The esterification of PUFA-CoA to phospholipid (PL) is promoted by lysophosphatidylcholine acyltransferase 3 (LPCAT3), forming PUFA-PL. Finally, ALOX mediates lipid peroxidation under different conditions to produce hydroperoxides, PUFA-PL-OOH, which promote ferroptosis[Citation12]. Fatty-acid ACSL3 protects cells from ferroptosis by converting monounsaturated fatty acids (MUFAs) into their acyl-CoA esters and merging them into membrane phospholipids[Citation13]. Although lipid droplets are not essential for exogenous MUFA-mediated ferroptosis inhibition, Rab7A-associated autophagy (called lipophagy) promotes free fatty acid production through selective degradation of lipid droplets and increases lipid peroxidation and subsequent ferroptosis. Therefore, increasing lipid storage inhibits ferroptosis, and vice versa[Citation13]. Thus, the regulation of ACSL4 expression may be an important mechanism in desensitizing cells to ferroptosis under various pathophysiological conditions[Citation14]. The mevalonate pathway regulates ferroptosis on several levels through the production of isopentenyl pyrophosphate (IPP), squalene, CoQ10, and cholesterol[Citation7]. The mevalonate pathway is a target of statins, which inhibit selenoprotein biosynthesis to decrease CoQ10 production and GPX4 function, thereby increasing cell sensitivity to ferroptosis[Citation15].
As a phospholipid hydroperoxidase, GPX4 catalyzes the formation of phospholipid hydroperoxide (PUFA-PL-OOH) to the corresponding phospholipid alcohol. The expression or activity of GPX4 is modulated by GSH, selenium, and some small molecular compounds (such as RSL3, ML162, ML210, FIN56, and FINO2), but some compounds also cause GPX4 protein degradation[Citation16]. L-selenocysteine (Sec)-tRNA is positively regulated by IPP and contributes to the formation of GPX4[Citation17]. GSH is enzymatically synthesized from cysteine, glycine, and glutamic acid and the available cysteine (endogenous and exogenous) is the main rate limiting factor in this process.
Endogenous cysteine is mainly produced through cysteine reduction mediated by GSH – or thioredoxin reductase 1. In mammalian cells, the sodium-independent, chloride dependent cystine-glutamate antiporter known as system XC− consists of two subunits, solute carrier family 7 member 11 (SLC7A11) and the 4F2 cell surface antigen heavy chain. System XC− introduces the oxidized form of cysteine (cystine) into cells to provide raw materials for GSH biosynthesis. Therefore, system XC− may be considered an important upstream regulator of ferroptosis. The expression and activity of SLC7A11 are regulated by various nuclear transcription factors[Citation18]. Moreover, OUT deubiquitylase, ubiquitin aldehyde binding 1 (OTUB1) directly binds to and stabilizes SLC7A11, reducing its ubiquitination level and thereby inhibiting its degradation in a proteasome-dependent manner[Citation19]. Another source of GSH is the transsulfuration pathway, which is negatively modulated by the aminoacyl-tRNA synthase family[Citation20].
Mitochondrial metabolic abnormalities are also involved in ferroptosis, and α-ketoglutarate (αKG) is produced by glutamate via glutamic-oxaloacetic transaminase 1 -mediated ammonia transfer, which promotes ferroptosis through at least two mechanisms[Citation21]. ATP citrate lyase catalyzes the conversion of αKG-derived citric acid to acetyl-CoA[Citation22], which is an anabolic precursor of acetyl-CoA carboxylase alpha and fatty acid synthase for lipid biosynthesis. In addition to ROS derived from NADPH oxidase (NOX) at the cell membrane, αKG generates mitochondrial ROS through dihydrolipoamide dehydrogenase[Citation23]. Thus, αKG is an important mitochondrial metabolic intermediate that induces ferroptosis by producing mitochondrial ROS or lipids.
The inner mitochondrial membrane is a unit membrane enclosing the mitochondrial matrix and located on the inner side of the outer mitochondrial membrane. It contains enzymes responsible for oxidative phosphorylation[Citation24]. Although GPX4 is a classical ferroptosis inhibitor, dihydroorotate dehydrogenase (DHODH) in the mitochondrial intima is considered a new ferroptosis inhibitor independent of GPX4. DHODH inhibits lipid peroxidation and iron-induced death by reducing ubiquinone (CoQ) to ubiquinol (CoQH2) in the mitochondrial inner membrane[Citation25]. Considering the important role of the mitochondria in tumors, this discovery provides a potential new strategy for targeting ferroptosis in cancer therapy.
FSP1 and GCH1 are two other non-GPX4-dependent ferroptosis inhibitors. FSP1 is mainly located in lipid droplets and the plasma membrane where it removes harmful lipid hydroperoxides. FSP1 directly reduces CoQ to the lipophilic radical-trapping antioxidant (RTA) ubiquinol (CoQH2) on the cell membrane, or indirectly via recycling of another natural lipophilic RTA, α-tocopherol, leading to inhibition of lipid peroxidation[Citation26]. GCH1 inhibits ferroptosis through its metabolism to BH4 and dihydrobiopterin BH2. BH4 may protect phospholipids containing two PUFA tails from oxidative damage through a dual mechanism, either as a lipophilic RTA or through its involvement in ubiquinone synthesis[Citation27].
Therefore, there are at least four ferroptosis defense systems with different subcellular localizations: GPX4 in the cytoplasm and mitochondria, FSP1 on the cell membrane, DHODH in the mitochondria, and GCH1 in the cytoplasm. Currently, ferroptosis inducers mainly include those targeting the cystine/glutamate reverse transporter, GPX4, GSH, Fe2+, and ROS production. Ferroptosis inhibitors play a role mainly by eliminating free radicals, inhibiting enzymes that produce lipids or lipid peroxides, and reducing Fe2+.
As a hydrogen carrier, NADPH plays an important role in the regulatory signaling pathways of ferroptosis, which can originate from many sources including the pentose phosphate pathway, nicotinamide-adenine dinucleotide (NAD) kinase-catalyzed phosphorylation of the reduced form of NAD, and the conversion of isocitrate to α-KG by NADP-dependent isocitrate dehydrogenase[Citation28]. Thus, the sensitivity of cells to ferroptosis can be affected by the NADP/NADPH ratio. These ferroptosis-resistant cells may have higher NADPH basal levels or lower NADP/NADPH ratios[Citation29].
In] conclusion, the regulatory pathway of ferroptosis forms a network that is precisely regulated in cell survival and death. The regulatory networks of ferroptosis are detailed in references 1, 7and28. shows a brief regulatory pathway for ferroptosis.
Figure 1. Ferroptosis mainly involves amino acid metabolism, fat metabolism and mitochondrial metabolism. ACSL4, acyl-CoA synthetase long-chain family member 4; αKG, α-ketoglutarate; Cys, cysteine; Cys2, cystine; DHODH, dihydroorotate dehydrogenase; FPP, farnesyl pyrophosphate; G6PD, glucose-6-phosphate dehydrogenase; Glu, glutamate; GPX4, glutathione peroxidase 4; GSH, glutathione; Gly, glycine; HMG-CoA, 3-hydroxy-3-methyl glutaryl coenzyme A; IPP, isoprene pyrophosphate; LOX, lipoxygenase; LPCAT3, lysophosphatidylcholine acyltransferase 3; Met, methionine; MUFA, monounsaturated fatty acid; NOX, NADPH oxidase; OXPHO, oxidative phosphorylation; PLOO•, peroxyl radical; PUFA, polyunsaturated fatty acid; PUFA-PL, phospholipid containing polyunsaturated fatty acid chain; ROS, reactive oxygen species.
Differences between ferroptosis and apoptosis
Ferroptosis is morphologically, biologically, and genetically distinct from apoptosis. It is characterized by cell swelling, the loss and/or disruption of plasma membrane integrity, and shrunken mitochondria[Citation30], whereas apoptosis mainly involves cell shrinkage, chromatin condensation, and formation of apoptotic bodies without inflammatory reaction[Citation31]. The signaling pathways involved in apoptosis are different from those involved in ferroptosis and comprise three alternative pathways: (1) the extrinsic pathway, which involves interaction of the tumor necrosis factor receptor family with a ligand, followed by procaspase-8 binding leading to activation of caspase-3; (2) the intrinsic pathway, which employs alterations in the inner mitochondrial membrane for induction of apoptosis; and (3) the perforin/granzyme pathway[Citation32]. With regard to genetic differences, apoptosis is strictly controlled by polygenes, such as those of the Bcl-2 family, caspase family, oncogenes including c-myc and tumor suppressor gene p53Citation[33]; whereas, ferroptosis includes iron metabolism-related genes and regulatory genes for mitochondrial function, GSH steady-state and redox function[Citation34]. The molecular markers of ferroptosis include upregulation of the proteins ACSL4, TFRC, prostaglandin endoperoxide synthase 2 (PTGS2) and cation transport regulator homolog 1 and the downregulation of ferritin, GPX4 and voltage-dependent anion-selective channel 2/3. However, in apoptosis, the Fas receptor and its ligand, Bcl2 family of proteins, p53 transcription factor, and caspase cleavage and activity are the key markers[Citation35].
Vascular ECD
Healthy ECs are not only the main place for blood supply and nutrient exchange, but also act as a barrier to ensure the selective exchange of substances in blood and tissue fluid. In addition, the endothelium also functions in synthesis, secretion, metabolism, and immunity and can regulate immune responses, hemodynamics, and peripheral organ response. ECs have obvious organ and species heterogeneity in their function, morphology, gene expression, and antigen composition[Citation36].
ECs are important targets of metabolic substances and hemodynamic signals in regulating the microcirculation in vascular injury. EC injury may be exogenous and endogenous (circulating substances in the blood), which destroys the normal physiological function of the vascular endothelium. EC injury leads to EC death, increased vascular permeability, vascular barrier dysfunction, inflammatory reactions, vasoconstriction, and perfusion defects occur[Citation3].
Role of ferroptosis in ECD
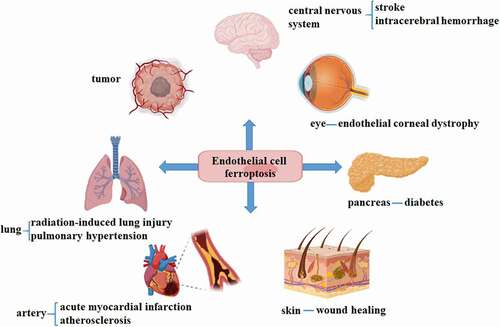
In view of the important role of ECs in the body and the insufficient understanding of ferroptosis under physiological conditions, the next section will focus on the main role of ECs ferroptosis under disease conditions. summarizes the diseases of various organs associated with ECs ferroptosis.
Figure 2. Endothelial ferroptosis involves different pathological processes, including eyes, blood vessels, brain, pancreas, skin, tumor and lung.
Ferroptosis in atherosclerotic endothelium
Atherosclerosis is a well-known chronic inflammatory disease associated with a variety of risk factors, such as hypertension, hypercholesterolemia, and diabetes. Arterial subcutaneous lipid accumulation and hyperglycemia stimulation lead to inflammatory effects, endothelial dysfunction, and extracellular matrix remodeling, ultimately leading to calcification and vulnerable plaque formation[Citation37]. The relationship between iron, atherosclerosis, and coronary heart disease has been investigated for decades; however, the results have been inconclusive. Two recent human population studies have shown a positive association between iron metabolism and atherosclerosis in postmenopausal women and healthy children, respectively[Citation38,Citation39]. Iron overload was shown to aggravate atherosclerotic plaque formation in hypercholesterolemic rabbits and homozygous apolipoprotein E knockout (ApoE−/−) mice[Citation40]. Furthermore, iron deficiency or iron chelation diets attenuated atherosclerosis in animals[Citation41], even without iron overload. In contrast, another study showed that feeding ApoE−/− mice with levels of high iron did not cause atherosclerosis or even reduce its progressions[Citation42]. Although data on the relationship between iron and atherosclerosis are contradictory, different amounts and administration routes of iron could explain these discrepancies[Citation43].
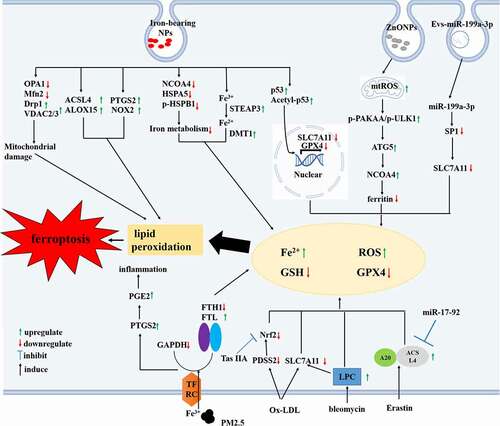
Iron overload causes elevated non-transferrin bound iron levels and ECD, including increased ROS, monocyte adhesion, and damage to EC-derived nitric oxide production, eventually leading to endothelial injury/death and increased vascular permeability[Citation44]. ECD was also manifested as acetylcholine-induced endothelial-dependent relaxation damage and enhanced vasoconstriction response to phenylephrine in iron overload ApoE−/− mice[Citation43]. The underlying mechanisms may be iron overload upregulated prostanoids through promoting the expression of cyclooxygenase-2 (COX2) in aorta and reduced vascular NO bioavailability and the catalase and superoxide dismutase activity, leading to the imbalance between oxidation and antioxidant factors[Citation43]. In contrast, the ferroptosis inhibitor ferrostatin-1 (Fer-1) alleviated atherosclerotic lesions in a high-fat diet (HFD)-fed ApoE−/− mouse model by inhibiting the levels of lipids in the blood, reversing the expression of SLC7A11 and GPX4 in the thoracic aorta, and reducing iron accumulation and lipid peroxidation[Citation3]. Fer-1 also inhibited EC death and angiogenesis in the thoracic aorta of ApoE−/− mice fed an HFD. In vitro, Fer-1 increased EC viability, reduced cell death, downregulated the expression of adhesion molecules (intercellular adhesion molecule-1 and vascular cell adhesion molecule-1), attenuated the expression of vascular endothelial growth factor (VEGF) A, and upregulated endothelial nitric oxide synthase (NOS) by inhibiting ferroptosis in mouse aortic ECs exposed to oxidized low-density lipoprotein (ox-LDL)[Citation3]. Interestingly, ox-LDL is involved in numerous atherogenetic factors and its contribution to ferroptosis may be mediated through multiple downstream molecules. In ox-LDL-treated human coronary artery ECs, prenyl diphosphate synthase subunit 2 (PDSS2), the key enzyme involved in the synthesis of CoQ10, was also decreased[,][Citation45]. Overexpression of PDSS2 activates the antioxidant nuclear factor erythroid-2 related factor 2 (NRF2) and suppresses the release of ROS, promoting the proliferation of human coronary artery ECs and inhibiting their ferroptosis[Citation45].
In addition to oxLDL, as another harmful stimulus to ECs, many epidemiological and experimental studies have shown that fine PM promotes cardiovascular disease and may be involved in the mechanisms of inflammation, epigenetics, and oxidative stress[Citation46]. Wang et al. reported that the internalization of PM of a diameter < 0.25 µm (PM2.5) in ECs increased their iron content through the aberrant expression of iron-related genes such as TFRC, FTL, and FTH1. Moreover, it also increased ROS production, depleted GSH, and decreased GPX and NADPH. Consequently, ferroptosis and an increase in proinflammatory cytokines (prostaglandin E synthase 2/prostaglandin E 2) were induced in PM2.5-treated cells. Ferroptosis inhibitors (such as Fer-1) and iron-chelating agents (deferoxamine mesylate) alleviate these effects pharmacologically. Surprisingly, increased protein expression of SLC7A11 and GPX4 may be involved in PM2.5-induced ferroptosis in ECs, which is thought to be a negative feedback mechanism initiated in response to the rapid depletion of GHS by PM2.5[Citation47]. Intriguingly, cigarette smoke, a risk factor for atherosclerosis, contains compounds that have the potential to activate endothelial ROS, and its extract has been demonstrated to induce ferroptosis of vascular smooth cells (VSMCs) rather than ECs. This might be due to the difference between the threshold damage required to induce ferroptosis in VSMCs and ECs[Citation48].
Metal nanoparticles (NPs), including metals, metal alloys, and metal oxide particles, are one of the most widely used nanomaterials and are known to exhibit endothelial toxicity[Citation49]. Qin et al. found that zinc oxide NPs (ZnONPs) induced ferroptosis of human umbilical vein ECs (HUVECs) and HUVEC fusion cell line EA.hy926 cells by increasing intracellular iron levels and promoting lipid peroxidation, which could be reversed by Fer-1 and the iron-chelating agent deferiprone. The underlying mechanisms involved NCOA4-mediated autophagic degradation of the major intracellular iron storage protein Fn, which was mitigated by an autophagy pharmacological inhibitor or by autophagy-related 5 (ATG5) gene knockout. NCOA4-mediated ferritinophagy induced by ZnONPs may be involved in increasing the production of mitochondrial ROS (mtROS) and upregulating the phosphorylation of protein kinase AMP-activated catalytic subunit alpha and unc-51 like autophagy activating kinase 1 (Ser555). This protein, in turn, upregulated downstream autophagy-related signaling molecules in HUVECs and EA.hy926 cells. The in vivo results indicated that exposure of mouse lung tissue to ZnONP could induce pulmonary vascular inflammation, which might be related to ferroptosis induced by ZnONP-mediated ferritinophagy, thus further suggesting its harmful effects[Citation50]. Similarly, iron-bearing NPs (γ-Fe2O3 and Fe3O4) induced HUVECs ferroptotic responses by the upregulation of intrinsic (ACSL4 and ALOX15) and extrinsic (NOX2) enzymes. HUVECs showed an increase in intracellular Fe2+ levels after phagocytosis of γ-Fe2O3 and Fe3O4 NPs through upregulation of ferrireductase STEAP3 and DMT1. Beside, γ-Fe2O3 and Fe3O4 NPs downregulated the expression of following iron metabolism-related proteins, heat shock protein family A member 5 (HSPA5), HSPB1, phosphorylated HSPB1 (p-HSPB1), and NCOA4 in HUVECs. Therefore, exposure of ECs to γ-Fe2O3 and Fe3O4 NPs promoted iron release and inhibited iron metabolism, eventually leading to the cellular accumulation of iron[Citation51]. At the same time, The abnormal expression of mitochondrial division and fusion proteins (mitochondrial fusion protein-2, motility related protein-1, optic atrophy protein) and voltage dependent anion channel 2/3 caused the changes of mitochondrial morphology and dysfunction in γ-Fe2O3 and Fe3O4 NPs treated HUVECs. Thus, multiple factors implicate in ECs ferroptosis induced by iron-bearing NPs. Together, the studies mentioned above suggest that NPs-induced ECs ferroptosis is a risk factor for atherosclerosis. However, the investigation of ferroptosis in ECs by NPs constitutes only a part of related toxicity studies. The safety assessment of nanomaterials and the chemical modification of biotoxic nanomaterials should be a focus of future research.
There is, in addition, mounting evidence that epigenetic modifications also play a crucial role in the regulation of EC ferroptosis. As a multifunctional oncogenic micro RNA (miRNA) cluster, the overexpression of miRNA-17-92 in ECs targeted Zinc lipoprotein A20, which contributed to the decreased expression of ACSL4 and upregulation of GPX4. Therefore, it protects HUVECs from erastin-induced ferroptosis by inhibiting ROS generation[Citation52]. miRNA-199a-3p and miR-30e-5p, which are contained in extracellular vesicles derived from circulating progenitor cells, are also capable of inhibiting ferroptosis upon binding to specificity protein 1 following uptake by ECs[Citation53,Citation54]. In fact, there are abundantly expressed miRNAs in ECs, which could be regulated by systemic stimuli, such as hyperlipidemia, local inflammation and disturbed flow[Citation55]. Given the dual role of miRNAs in ECs death and survival[Citation55], we are just now starting the studies of the complex regulatory patterns of miRNAs in ECs ferroptosis in atherogenic process.
Circulating ECs (CECs) are involved in the metabolism of the vascular endothelium, which is associated with capillary ischemia, the perfusion state, and vasoconstriction functions. Chen et al. established a random forest diagnosis model of acute myocardial infarction based on ferroptosis-related genes of CECs using different bioinformatics technologies (gene annotation enrichment and protein-protein interaction analysis), which are helpful in determining whether patients are in the process of developing microvascular occlusion and the degree[Citation56]. Thus, circulating markers of ferroptosis are expected to provide predictive and diagnostic significance for the occurrence of coronary heart disease.
Many regulatory effects of natural compounds on iron metabolism and homeostasis have been identified, which are related to their mechanisms of action in the treatment of ferroptosis-related diseases[Citation57]. α-Tocopherol (vitamin E) and brown rice extracts have been shown to attenuate lipid peroxidation, cytotoxicity, and delay proliferation induced by GPX4 knockdown[Citation58]. Despite the above-mentioned findings that vitamin E induced onset of EC ferroptosis can have protective effects on atherosclerosis in vivo and in vitro, clinical studies revealed contradictory results [Citation59,Citation60]. The inconsistent results of the clinical trials may be owing to variations in the selection criteria of the subjects and the dose and duration of vitamin E supplementation. Additionally, the test subjects could have gene polymorphisms in vitamin E uptake- and transport-related proteins[Citation61]. Another noteworthy product is Tanshinone IIA, a rhizome extract of the plant Salvia miltiorrhiza from the family Labiatae. Tanshinone IIA has been shown to protect human coronary artery ECs from erastin/RSL3-induced ferroptosis by promoting nuclear translocation of NRF2[Citation62]. This effect on NRF2, in turn, reduces the excessive accumulation of total cellular ROS, inhibits lipid peroxidation, and decreases GSH and free intracellular iron levels[Citation62]. Besides, astragaloside IV (AS-IV), a natural compound from Astragalus membranaceus, was reported to reverse the ferroptosis induced by bleomycin in HUVECs. The upregulation of lysophosphatidylcholine in the bleomycin-treated ECs contributes to the reduction of anti-ferroptotic factors (GPX4 AND SLC7A11) and the increase in pro- ferroptotic factors (iron ions and lipid ROS). These effects could be reversed by AS-IV[Citation63]. Also, Tongxinluo (TXL) extracted from Chinese medicine mixture, protected pulmonary microvascular barrier dysfunction induced by cigarette smoke in HFD mice partially through inhibiting ferroptosis in human pulmonary microvascular ECs, indicating by increased expression of GPX4 and FSP1 and decreased level of ACSL4[Citation64]. Although studies on cell culture and animal models have identified the regulatory effects of these bioactives, further clinical investigations are necessary to validate the ferroptosis-related cardiovascular protective effects of these products.
In summary, current literature suggests that ECs ferroptosis contributes to cell viability, inflammation, and angiogenesis and, thus, is implicated in the pathology of atherosclerosis. The mechanisms and signaling pathways involved in ECs ferroptosis in atherosclerosis are shown in . However, more investigations need to be conducted for additional clarification. In addition to ECs, a variety of cells such as smooth muscle and inflammatory cells are involved in atherosclerosis and have been shown to also exhibited ferroptosis[Citation65]. However, whether these cells exhibit crosstalk with ECs during ferroptosis has not been determined. Moreover, the possible roles of ECs ferroptosis in early and advanced arteriosclerosis have not been clarified. At last, the susceptibility of blood vessels in different parts of the body to atherosclerosis differs[Citation66]. Whether the propensity for ferroptosis to occur in the vascular ECs of various parts also differs or the same is unknown.
Figure 3. The molecular mechanisms of ferroptosis occur in endothelial cells (ECs) during atherosclerosis. It leads to the accumulation of iron and ROS, the reduction of GPX4 and GSH levels when ECs are subjected to harmful stimuli through various effector molecules, resulting in lipid peroxidation and ferroptosis. ACSL4, Acyl – CoA synthetase long-chain family member 4; ALOX, arachidonate lipoxygenase; Drp1, dynamin-related protein 1; DMT1, divalent metal transporter 1; FTH1, ferritin heavy chain 1; FTL, ferritin light chain; HSPA5, heat shock protein family A member 5;HSPB1, heat shock protein family B member 1; mtROS, mitochondrial ROS; LPC, lysophosphatidylcholine; Mfn2, mitofusin 2; NRF2, nuclear factor erythroid-2 related factor 2; NCOA4, nuclear receptor coactivator protein 4; PDSS2, prenyl diphosphate synthase subunit 2;PGE2, prostaglandin E2; PTGS2, prostaglandin-endoperoxide synthase 2; PAKKA, protein kinase AMP-activated catalytic subunit alpha; SP1, specificity protein 1; STEAP3, six-transmembrane epithelial antigen of the prostate 3; SLC7A11, solute carrier family 7 member 11; ULK1, unc-51 like autophagy activating kinase 1; VDAC, voltage-dependent anion-selective channel protein 2.
Ferroptosis in sepsis
Sepsis refers to severe systemic inflammatory response induced by infection and trauma, which can cause secondary damage to tissues and organs. Animal models of sepsis can be established via intravenous injection of lipopolysaccharide (LPS) or cecal ligation and puncture (CLP)[Citation67]. The pathogenesis of sepsis is complex, involving effects on the immune, nervous, and endocrine systems and blood coagulation[Citation68]. Recent evidence indicates that ferroptosis plays an important role in sepsis-induced multiorgan damage. Several studies have demonstrated inhibition of ferroptosis to markedly attenuate organ damage, as evidenced by lowered incidence of cardiac injury[Citation69], acute kidney injury[Citation70], acute lung injury (ALI)[Citation71], and sepsis-associated encephalopathy[Citation72]. Sepsis causes systemic bacterial infection and inflammatory imbalance leading to endothelial barrier dysfunction, vascular leakage, tissue edema, and ultimately, organ failure[Citation73]. LPS-induced ferroptosis in ECs, which is indicated by increased levels of lipid peroxidation and malondialdehyde (MDA), can cause decreased expression of GPX4 and the loss of mitochondrial cristae leading to fragmental morphology[Citation74,Citation75]. Additionally, it induces stress on brain microvascular ECs, resulting in the disruption of BBB permeability[Citation72]. Dexmedetomidine (Dex), a known, selective α2-adrenoceptor agonist with sedative effects, has recently been found to inhibit ferroptosis in ECs. Dex ameliorates CLP-induced vascular leakage via modulation of metabolic reprogramming and improving mitochondrial dysfunction[Citation74]. Mechanistically, Dex promotes the expression of NRF2 and upregulation of the downstream molecular target glucose-6-phosphate dehydrogenase, subsequently activating the pentose phosphate pathway that results in increased production of GSH and reduced lipid peroxidation[Citation74]. Moreover, Dex inhibits sepsis-induced mitochondrial fission, thus reducing the levels of ROS and protecting ECs against ferroptosis[Citation74]. Acute myocardial injury has been shown to play a key role in poor prognosis of patients with sepsis. A recent study demonstrated that, LPS-induced cardiac injury in mice induced ferroptosis, as indicated by increased levels of PTGS2, MDA, and lipid ROS as well as mitochondrial damage. Besides, Fer-1 and DXZ rescued these changes, weakened LPS-mediated cardiac dysfunction, and decreased mortality rate[Citation69]. Studies have been conducted to elucidate the potential molecular mechanism of ferroptosis involved in LPS-induced myocardial injury. While LPS promotes the expression of NCOA4, which can degrade ferritin in a ferritinophagy-dependent manner, it also increases intracellular levels of Fe2+. Cytoplasmic Fe2+ further enhances the expression of ferrite protein on the mitochondrial membrane, which then induces the transport of cytoplasmic Fe2+ to mitochondria, leading to mitochondrial ROS production and ferroptosis[Citation69]. These findings represent a novel link between ferroptosis and sepsis-induced cardiac injury and provides a potential therapeutic target in patients. Ferroptosis also plays a pivotal role in sepsis-related renal injury[Citation70]. In diabetic mice fed a high-fat diet, oxidative stress levels (marked by MDA, 4-hydroxy-2-nonenal, ROS, 8-hydroxy-2 deoxyguanosine and NADPH oxidase) were increased and ferroptosis-related proteins (including ACSL4, FTH1 and GPX4) were altered by injection of LPS. These effects could then be reversed by the inhibition of NADPH which restored sepsis-induced renal dysfunction[Citation70]. Taken together, these findings indicate that ferroptosis is an important step in sepsis-related organ injury by mitochondrial dysfunction and redox imbalance. Therefore, the possibility of treating sepsis by targeting ferroptosis should be further explored.
Ferroptosis in tumor endothelium
Tumor endothelial cells (TECs), which line the blood vessels that supply blood to tumors, have unique characteristics. They are different from inflammatory vascular ECs and normal tissues with respect to genetics[Citation76], structure[Citation77], and cell function[Citation78]. Like tumor cells, tumor endothelium can be genetically unstable. The genetic markers of tumor endothelium are mainly cell surface molecules with unknown function[Citation76]. Instead of the organized hierarchy of a normal vascular bed, the tumor vasculature comprises a chaotic network of structurally and functionally defective vessels. The ECs of the tumor vessels lose their normal monolayer structural arrangement and are loosely connected to the pericytes[Citation77]. Due to this vascular disorder, there is high resistance and reduced blood flow, poor tumor perfusion and tissue hypoxia, large vascular endothelial gaps, and endothelial barrier leakage, which promote cancer cell metastasis[Citation78]. Immune cells rely on adhesion molecules on vascular ECs to infiltrate into tumor tissues and exert antitumor effects. They can also regulate tumor angiogenesis and lymphangiogenesis by secreting chemokines and cytokines. Hence, they play an important role in the process of tumor hematogenous metastasis[Citation79]. Inflammatory ECs upregulate their surface adhesion molecules and promote leukocyte adhesion and attachment to the wall of vessels, in order to kill pathogens and degrade necrotic tissue in the inflammatory site. In this process, leukocytes also cause tissue damage by releasing proteolytic enzymes, chemical mediators, and oxygen free radicals[Citation80].
As a ferroptosis activator, erastin exposure increased the generation of ROS and lipid peroxides, enhanced the depletion of GSH, and downregulated genes involved in lipid peroxide scavenging and GSH synthesis, which could be reversed using propranolol through an increase in hydrogen sulfide (H2S) production. Moreover, the anti-angiogenic effect of propranolol was partially related to the inhibition of HUVEC proliferation, migration, and vessel-like structure formation induced by erastin. More importantly, propranolol improves the dissociation of vascular endothelial-cadherin adherens junctions and further inhibits the adhesion of cancer cells to ECs and transendothelial migration through suppression of erastin-induced ferroptosis[Citation4].
Beyond its major effects on endothelial functional phenotype, ferroptosis can also be modulated at an epigenetic level in tumor ECs. Long chain noncoding RNAs (lncRNAs) are a RNA molecules with ≥ 200 nucleotide transcripts. They do not directly participate in gene coding and protein synthesis, but can regulate gene expression epigenetically, transcriptionally, and post-transcriptionally[Citation81]. The lncRNA MEG8 was found to be upregulated in infantile hemangioma. Knockdown of MEG8 inhibited the proliferation of hemangioma ECs and induced their ferroptosis via upregulation of miR-497-5p and downregulation of Notch receptor 2 (Notch2), which is associated with EC cell death[Citation82]. This effect downregulated SLC7A11 and GPX4 expression. Therefore, targeting the MEG8/miR-497-5p/Notch2 signaling pathway may be a potential method for promoting the death of hemangioma cells[Citation82]. Increasing preclinical evidence supports the notion that targeting ferroptosis may be an effective strategy for tumor treatment or improving tumor resistance to therapies[Citation83]. However, treatment targeting ferroptosis of tumor ECs still requires extensive research. As previously reported, three criteria (iron level, gene expression, and mutation) can be used to determine patients who are most likely to benefit from the promotion of ferroptosis[Citation83]. In addition, according to the characteristics of tumor ECs, the purpose of modulating EC ferroptosis should be to normalize tumor blood vessels, reduce metastasis by tightening the vascular barrier, and improve chemotherapy response by providing input pathways for chemotherapy drugs instead of inducing ECs death[Citation84].
Ferroptosis in diabetic endothelium
Diabetic angiopathy includes microvascular and macrovascular diseases, which affect numerous critical organs. ECD is closely involved with the development of diabetic vascular complications. Excessive oxidative stress induced by a high-glucose environment and insulin resistance in diabetes is an important cause of ECD[Citation85].
The analysis of gene expression profiles showed that ferroptosis is a likely important mode of cell death in diabetic ECs[Citation86]. A decrease in the XC− system and the presence of de-endothelialization were observed in the aortic endothelium of db/db diabetic mice. In vitro studies have shown that high glucose (HG) and interleukin (IL)-1β increased lipid ROS and decreased the viability of HUVECs. This results in ferroptosis whereas, conversely, deferoxamine (an iron chelating agent, DFO) and the ferroptosis inhibitor Fer-1 can reverse these effects. Mechanistically, the activation of transcription factor p53 mediated by HG and IL-1β suppressed the substrate-specific subunit of system the XC− and finally led to the downregulation of GPX4 and GSH[Citation87]. In line, HG combined with high lipids induced ferroptosis through upregulating heme oxygenase 1, the rate-limiting enzyme in the heme catabolism of iron porphyrin compounds, in human ECs[Citation88]. Diabetic retinopathy is one of the most common microvascular complications of diabetes and is characterized by retinal microvascular leakage and hyperglycemia-induced obstruction[Citation89]. Tripartite motif containing 46 (TRIM46), a member of theE3 ubiquitin ligase family TRIM, is upregulated and promotes the ubiquitination and degradation of GPX4 in HG-treated human retinal capillary ECs, which induces ferroptosis and cell growth inhibition[Citation89]. Thereby, intensive glucose control and reduced related risk factors such as dyslipidemia could protect ECs from ferroptosis.
Additionally, patients with diabetes have a slow or even nonunion wound, and the complex underlying mechanism includes oxidative stress, chronic long-term inflammation, and reduced neovascularization[Citation90]. In an in vitro model, HG decreased survival and migration rates of HUVECs by inducing ferroptosis, which could be improved by Fer-1. Furthermore, in a streptozotocin-induced diabetic rat model, Fer-1 promoted the expression of angiogenesis-associated protein VEGF and reduced inflammatory cell infiltration by activating the anti-inflammatory phosphatidylinositol 3-kinase/protein kinase B signaling pathway, ultimately accelerating wound healing. Therefore, the pharmacological effects of ferroptosis attenuate inflammation in diabetic wounds, promote the production of angiogenic factors, and improve wound healing[Citation91]. Interestingly, ECD may precede the development of diabetes mellitus and is associated with insulin resistance[Citation92]. Whether ferroptosis occurs in ECs during insulin resistance requires further study. Furthermore, an abnormal increase of mitochondrial ROS and excessive free fatty acids are associated with the pathogenesis of diabetic vasculature[Citation85]. Thus, the mechanisms of ferroptosis in diabetic ECs may be far beyond what has been currently identified.
Ferroptosis in neurological diseases
The blood-brain barrier (BBB) is an important physiological barrier that separates the central nervous system from inflammatory mediators and effector immune cells in the peripheral circulation[Citation93]. It is composed of cerebral vascular ECs, peripheral vascular cells, extracellular matrix membrane, and astrocytes[Citation94]. The integrity of the BBB structure and function is related to a variety of nervous system diseases, such as acute ischemic stroke and Alzheimer’s disease[Citation95].
Iron mainly mediates BBB damage by producing ROS, which induce tight junction protein degradation[Citation96]. Individuals with diabetes have a higher risk of ischemic stroke. Moreover, diabetic mice exhibited an exacerbated BBB breakdown[Citation97]. The LncRNA MEG3 is highly expressed in oxygen and glucose deprived (OGD) and hyperglycemia-co-treated rat brain microvascular ECs, which upregulated the expression of p53. P53 transcriptionally inhibited GPX4 activity to induce GSH depletion and in turn, suppressed cell viability and promoted ferroptosis of brain microvascular ECs[Citation98]. Secondary bleeding in the brain after ischemic brain injury, known as hemorrhagic transformation (HT), occurs more commonly in diabetes, which contributes to poor recovery[Citation99]. In diabetic rats, HT led to the accumulation of iron in the brain parenchyma and deterioration of functional recovery. In contrast, iron chelation with DFO after thromboembolic stroke improved sensorimotor and cognitive function by preventing vascular dropout and improving neurovascular integrity. The poor outcomes in diabetes partially resulted from the occurrence of ECs ferroptosis and vascular loss, which could also be attenuated by DFO[Citation2]. Therefore, EC ferroptosis plays a vital role in the BBB dysfunction associated with in HT after acute ischemic stroke. Although ECs are the main targets in angiogenesis, microglia and pericytes are also involved[Citation97]. More studies are needed to investigate the underlying mechanisms of DFO-induced angiogenesis.
Cerebral hemorrhage is another common neurological disease that involves destruction of the BBB. LncRNA H19 is highly expressed in the OGD/hemin-treated (cell model of intracerebral hemorrhage) in brain microvascular ECs, followed by downregulation of the expression of miR-106b-5p and upregulation of ACSL4[Citation100]. These effects, in turn, suppressed the viability and promoted ferroptosis of brain microvascular ECs[Citation100]. However, the death of cerebral vascular ECs may trigger the coagulation cascade reaction of blocked damaged blood vessels, which only increases the temporary barrier permeability[Citation101]. Therefore, the extent to which this occurrence in endothelial monoculture studies reflects in vivo function is a matter of concern. A recent study by Gao et al., found brain injury in murine subarachnoid hemorrhage (SAH) models to be correlated to the upregulation of ALOX15 levels in brain ECs and microglia, which promotes ferroptosis in these cells. In contrast, CEP, a bisbenzylisoquinoline alkaloid that possesses antioxidative and anti-inflammatory properties, protects the brain from SAH related damage by inhibiting cell ferroptosis[Citation102].
Ferroptosis in ALI
ALI is a serious pulmonary disease, which involves progressive injury of alveolar epithelium and endothelial barrier, filling of alveolar chamber and neutrophil infiltration, increased alveolar membrane permeability, and refractory hypoxemia of respiratory failure[Citation103]. Ferroptosis plays a crucial role in radiation-induced lung injury (RILI), and the lungs of an RILI mouse model showed typical ferroptosis characteristics[Citation104]. ECs are sensitive to ionizing radiation (IR) damage[Citation105]. Piezo1 expression in pulmonary ECs were shown to be upregulated in vitro after IR. Increased expression or activation of Piezo1 could activate Ca2+/calpain signaling, which promotes the cleavage of VE-cadherin[Citation106]. These effects decrease the expression of GPX4 and SLC7A11, and increase levels of DMT1, thereby promoting ferroptosis[Citation106]. Acute respiratory distress syndrome (ARDS), a severe form of ALI, is the pulmonary manifestation of the systemic inflammatory response syndrome. Inflammatory mediators and cytokines released by macrophages, neutrophils, vascular ECs, and platelets mediate inflammation in the lungs of ARDS[Citation107]. High levels of ROS and inflammatory factors trigger an imbalance between oxidative and antioxidant systems, leading to a disturbance of the local microenvironment in the lung and resulting in a series of inflammatory responses[Citation107]. Zhou et al., demonstrated iron overload, GSH deficiency, and downregulation of ferritin expression, as well as mitochondrial contraction and membrane disruption in the lung cells of oleic acid-induced septic mice. Moreover, Fer-1 alleviates LPS-induced downregulation of GPX4 and SLC7A11, and lipid peroxidation in human bronchial epithelial cells and improves lung inflammatory response, indicating that ferroptosis plays an important role in the development and progression of ARDS[Citation108]. Li et al reported that CYY4137 offered protection against CLP-induced ALI in mice through inhibition of ferroptosis in lung tissue and increased survival rate. CYY4137, which is a stable H2S donor, has a unique role in vascular tension and anti-inflammatory properties. It inhibits the activation of autophagy by suppressing phosphorylation of mammalian target of rapamycin, leading to the downregulation of NOX and COX2 and increased expression of GPX4 and SLC7A11[Citation71]. Additionally, 4-OI, a cell permeable derivative of endogenous itaconate has been reported to inhibit LPS-induced lipid peroxidation in THP-1 macrophages through activation and accumulation of NRF2[Citation109]. Conversely, the depletion of NRF2 abolished the inhibition of ferroptosis, thus abolishing the protection of 4-OI in LPS-induced ALI. [Citation109] These findings provide the connection between ferroptosis and ALI, offering scope for novel therapeutics of targeting ferroptosis in ALI. However, clinical studies on the relationship between ferroptosis and ALI are minimal; therefore, further research is needed to elucidate the role of ferroptosis in the disease progression of ALI patients and the clinical benefits of targeting ferroptosis in them.
Endothelial ferroptosis in other diseases
Fuchs’ endothelial corneal dystrophy (FECD) is characterized by progressive damage to the corneal endothelium, and dysregulation of corneal EC oxidative stress is considered an important cause[Citation110]. Peroxiredoxin 1 (PRDX1) expression was low in corneal ECs derived from FECD patients. Similarly, cumene hydroperoxide (CH) downregulated PRDX1 in human corneal ECs or B4G12-CEnCs and increased lipid peroxidation[Citation111]. These effects, in turn, led to cell death, whereas Fer-1 restored the viability of CH-treated ECs[Citation111]. In addition, ferroptosis is associated with age-related macular degeneration (AMD)[Citation112]. The underlying mechanism of AMD involves excessive accumulation of retinal iron and lipid peroxidation[Citation112]. Studies have shown that GSH deficiency can induce soluble and lipid ROS generation in a spontaneously immortalized cell line of human retinal pigment epithelium (ARPE-19) cells. It can promote cell ferroptosis and autophagy, and mediate aging and death of ARPE-19 cells[Citation113]. Moreover, Fer-1 and DFO inhibited the death of ARPE-19 cells induced by tert-butyl hydroperoxide through inhibition of lipid peroxidation and iron accumulation and reduction of GSH depletion[Citation114]. Although existing studies have elucidated a potential role of ferroptosis in AMD, more mechanistic investigations are needed in future. Pulmonary hypertension is characterized by pulmonary vascular remodeling and increased pulmonary vascular resistance[Citation115]. Xie et al demonstrated the physio-pathologic effects and molecular mechanism of ferroptosis in PH and found that in MCT-injected rat, a classical animal model of PH, ferroptosis markers were increased in pulmonary arterial endothelial cells (PAECs), whereas, pretreatment by Fer-1 attenuated hemodynamics, right ventricular hypertrophy, and pulmonary remodeling in these rats. Mechanistically, ferroptosis of PAECs promoted the release of the high-mobility group box 1 protein, which is a key regulatory molecule of inflammation[Citation116], This in turn activates toll-like receptor 4/NLRP3 inflammasome signaling in macrophages and mediates inflammation. However, Fer-1 is capable of reversing the above-mentioned effects[Citation117]. In conclusion, these findings provide a research basis for therapy targeting diseases related to abnormal ferroptosis.
Conclusion
Ferroptosis is an important mode of cell death, involving complex regulatory networks. Exogenous or transporter-dependent pathways and endogenous or enzymes are involved in the regulation of ferroptosis. High sensitivity of PUFA and lipids containing PUFA to oxidation by enzymes (such as ALOX) and non-enzymatic reactions (such as the iron-dependent Fenton reaction) act as central factors in ferroptosis.
Ferroptosis is involved in ECD including increased vascular permeability, inflammation, vasoconstriction, and angiogenesis. The treatment intervention using EC ferroptosis needs to consider the disease background, and inhibiting or inducing EC ferroptosis has therapeutic potential in different environments. Although the role and mechanism of EC ferroptosis is slowly being elucidated in different disease settings, some common challenges that are limitations to some studies remain to be solved. First, although in vitro experiments have greatly increased the understanding of EC ferroptosis, further in vivo confirmation is still required. Second, although the physiological significance of ferroptosis has attracted the attention of researchers, the effect of ferroptosis on healthy ECs requires further investigation. Additionally, although PCD has different genetic, morphological, and biochemical characteristics, it can jointly participate in the dysfunction of ECs when stimulated by the same injury. Whether there is an intersection between ferroptosis and other PCD forms in ECs remains unknown. Finally, ECs are a heterogeneous group and the phenotypes of vascular ECs in different species, diameters, and tissues and tumor vascular ECs differ in structure, function, and surface molecules. Under different pathological conditions, whether the threshold of ferroptosis in these ECs is different or the same.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability Statement
All data generated or used during the study appear in the submitted article.
Additional information
Funding
References
- Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282.
- Abdul Y, Li W, Ward R, et al. Deferoxamine treatment prevents post-stroke vasoregression and neurovascular unit remodeling leading to improved functional outcomes in type 2 male diabetic rats: role of endothelial ferroptosis. Transl Stroke Res. 2021;12(4):615–630.
- Bai T, Li M, Liu Y, et al. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. 2020;160:92–102.
- Lopes-Coelho F, Martins F, Hipólito A, et al. The activation of endothelial cells relies on a ferroptosis-like mechanism: novel perspectives in management of angiogenesis and cancer therapy. Front Oncol. 2021;11:656229.
- Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–636.
- Zhang Z, Lu M, Chen C, et al. Holo-lactoferrin: the link between ferroptosis and radiotherapy in triple-negative breast cancer. Theranostics. 2021;11(7):3167–3182.
- Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32(6):920–937.
- Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. 2017;69(6):423–434.
- Kappler A, Bryce C, Mansor M, et al. An evolving view on biogeochemical cycling of iron. Nat Rev Microbiol. 2021;19(6):360–374.
- Brown CW, Amante JJ, Chhoy P, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575–586.e4.
- Roemhild K, von Maltzahn F, Weiskirchen R, et al. Iron metabolism: pathophysiology and pharmacology. Trends Pharmacol Sci. 2021;42(8):640–656.
- Kapralov AA, Yang Q, Dar HH, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16(3):278–290.
- Magtanong L, Ko PJ, To M, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26(3):420–432.e9.
- Lei G, Zhang Y, Koppula P, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30(2):146–162.
- Viswanathan VS, Ryan MJ, Dhruv HD, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–457.
- Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–185.
- Friedmann Angeli JP, Conrad M. Selenium and GPX4, a vital symbiosis. Free Radic Biol Med. 2018;127:153–159.
- Mandal PK, Seiler A, Perisic T, et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285(29):22244–22253.
- Liu T, Jiang L, Tavana O, et al. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019;79(8):1913–1924.
- Hayano M, Yang WS, Corn CK, et al. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–278.
- Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308.
- Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234.
- Shin D, Lee J, You JH, et al. Dihydrolipoamide dehydrogenase regulates cystine deprivation-induced ferroptosis in head and neck cancer. Redox Biol. 2020;30:101418.
- Li D, Yang S, Xing Y, et al. Novel insights and current evidence for mechanisms of atherosclerosis: mitochondrial dynamics as a potential therapeutic target. Front Cell Dev Biol. 2021;9:673839.
- Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590.
- Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698.
- Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41–53.
- Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125.
- Shimada K, Hayano M, Pagano NC, et al. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol. 2016;23(2):225–235.
- Chen X, Comish PB, Tang D, et al. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021;9:637162.
- Doonan F, Cotter TG. Morphological assessment of apoptosis. Methods. 2008;44(3):200–204.
- Jan R, Chaudhry GE. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv Pharm Bull. 2019;9(2):205–218.
- Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417.
- Xie B, Guo Y. Molecular mechanism of cell ferroptosis and research progress in regulation of ferroptosis by noncoding RNAs in tumor cells. Cell Death Discov. 2021;7(1):101.
- Yang J, Hu S, Bian Y, et al. Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol. 2022;9:789948.
- Eelen G, de Zeeuw P, Simons M, et al. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116(7):1231–1244.
- Xu S, Ilyas I, Little PJ, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73(3):924–967.
- Zhang Y, Guallar E, Qiao Y, et al. Is carotid intima-media thickness as predictive as other noninvasive techniques for the detection of coronary artery disease? Arterioscler Thromb Vasc Biol. 2014;34(7):1341–1345.
- Prats-Puig A, Moreno M, Carreras-Badosa G, et al. Serum ferritin relates to carotid intima-media thickness in offspring of fathers with higher serum ferritin levels. Arterioscler Thromb Vasc Biol. 2016;36(1):174–180.
- Vinchi F, Porto G, Simmelbauer A, et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J. 2020;41(28):2681–2695.
- Zhang WJ, Wei H, Frei B. The iron chelator, desferrioxamine, reduces inflammation and atherosclerotic lesion development in experimental mice. Exp Biol Med (Maywood). 2010;235(5):633–641.
- Kautz L, Gabayan V, Wang X, et al. Testing the iron hypothesis in a mouse model of atherosclerosis. Cell Rep. 2013;5(5):1436–1442.
- Marques VB, Leal MAS, Mageski JGA, et al. Chronic iron overload intensifies atherosclerosis in apolipoprotein E deficient mice: role of oxidative stress and endothelial dysfunction. Life Sci. 2019;233:116702.
- Guo L, Harari E, Virmani R, et al. Linking hemorrhage, angiogenesis, macrophages, and iron metabolism in atherosclerotic vascular diseases. Arterioscler Thromb Vasc Biol. 2017;37(4):e33–e39.
- Yang K, Song H, Yin D. PDSS2 inhibits the ferroptosis of vascular endothelial cells in atherosclerosis by activating Nrf2. J Cardiovasc Pharmacol. 2021;77(6):767–776.
- Liang S, Zhang J, Ning R, et al. The critical role of endothelial function in fine particulate matter-induced atherosclerosis. Part Fibre Toxicol. 2020;17(1):61.
- Wang Y, Tang M. PM2.5 induces ferroptosis in human endothelial cells through iron overload and redox imbalance. Environ Pollut. 2019;254(Pt A):112937.
- Sampilvanjil A, Karasawa T, Yamada N, et al. Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2020;318(3):H508–H518.
- Miller MR, Poland CA. Nanotoxicology: the need for a human touch? Small. 2020;16(36):e2001516.
- Qin X, Zhang J, Wang B, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. 2021;17(12):4266–4285.
- Liu Z, Xia X, Lv X, et al. Iron-bearing nanoparticles trigger human umbilical vein endothelial cells ferroptotic responses by promoting intracellular iron level. Environ Pollut. 2021;287:117345.
- Xiao FJ, Zhang D, Wu Y, et al. miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem Biophys Res Commun. 2019;515(3):448–454.
- Li L, Wang H, Zhang J, et al. Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discov. 2021;7(1):235.
- Xia J, Song X, Meng J, et al. Endothelial progenitor cells-derived exosomes transfer microRNA-30e-5p to regulate Erastin-induced ferroptosis in human umbilical vein endothelial cells via the specificity protein 1/adenosine monophosphate-activated protein kinase axis. Bioengineered. 2022;13(2):3566–3580.
- Schober A, Weber C. Mechanisms of MicroRNAs in atherosclerosis. Annu Rev Pathol. 2016;11(1):583–616.
- Yifan C, Jianfeng S, Jun P. Development and validation of a random forest diagnostic model of acute myocardial infarction based on ferroptosis-related genes in circulating endothelial cells. Front Cardiovasc Med. 2021;8:663509.
- Zheng K, Dong Y, Yang R, et al. Regulation of ferroptosis by bioactive phytochemicals: implications for medical nutritional therapy. Pharmacol Res. 2021;168:105580.
- Sakai O, Yasuzawa T, Sumikawa Y, et al. Role of GPx4 in human vascular endothelial cells, and the compensatory activity of brown rice on GPx4 ablation condition. Pathophysiology. 2017;24(1):9–15.
- Muntwyler J, Hennekens CH, Manson JE, et al. Vitamin supplement use in a low-risk population of US male physicians and subsequent cardiovascular mortality. Arch Intern Med. 2002;162(13):1472–1476.
- Myung SK, Ju W, Cho B, et al. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;346(jan18 1):f10.
- Sozen E, Demirel T, Ozer NK. Vitamin E: regulatory role in the cardiovascular system. IUBMB Life. 2019;71(4):507–515.
- He L, Liu YY, Wang K, et al. Tanshinone IIA protects human coronary artery endothelial cells from ferroptosis by activating the NRF2 pathway. Biochem Biophys Res Commun. 2021;575:1–7.
- Sheng S, Xu J, Liang Q, et al. Astragaloside IV inhibits bleomycin-induced ferroptosis in human umbilical vein endothelial cells by mediating LPC. Oxid Med Cell Longev. 2021;2021:6241242.
- Wang Y, Kuang X, Yin Y, et al. Tongxinluo prevents chronic obstructive pulmonary disease complicated with atherosclerosis by inhibiting ferroptosis and protecting against pulmonary microvascular barrier dysfunction. Biomed Pharmacother. 2021;145:112367.
- Ouyang S, You J, Zhi C, et al. Ferroptosis: the potential value target in atherosclerosis. Cell Death Dis. 2021;12(8):782.
- Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. 2011;15(5):1405–1414.
- Fink MP. Animal models of sepsis. Virulence. 2014;5(1):143–153.
- Huerta LE, Rice TW. Pathologic difference between sepsis and bloodstream infections. J Appl Lab Med. 2019;3(4):654–663.
- Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318.
- Yao W, Liao H, Pang M, et al. Inhibition of the NADPH oxidase pathway reduces ferroptosis during septic renal injury in diabetic mice. Oxid Med Cell Longev. 2022;2022:1193734.
- Li J, Li M, Li L, et al. Hydrogen sulfide attenuates ferroptosis and stimulates autophagy by blocking mTOR signaling in sepsis-induced acute lung injury. Mol Immunol. 2022;141:318–327.
- Wei XB, Jiang WQ, Zeng JH, et al. Exosome-derived lncRNA NEAT1 exacerbates sepsis-associated encephalopathy by promoting ferroptosis through regulating miR-9-5p/TFRC and GOT1 axis. Mol Neurobiol. 2022;59(3):1954–1969.
- Joffre J, Hellman J, Ince C, et al. Endothelial responses in sepsis. Am J Respir Crit Care Med. 2020;202(3):361–370.
- She H, Hu Y, Zhou Y, et al. Protective effects of dexmedetomidine on sepsis-induced vascular leakage by alleviating ferroptosis via regulating metabolic reprogramming. J Inflamm Res. 2021;14:6765–6782.
- Wang C, Yuan W, Hu A, et al. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22(1):175–184.
- Khan KA, McMurray JL, Mohammed F, et al. C-type lectin domain group 14 proteins in vascular biology, cancer and inflammation. FEBS J. 2019;286(17):3299–3332.
- Klein D. The tumor vascular endothelium as decision maker in cancer therapy. Front Oncol. 2018;8:367.
- Rohlenova K, Veys K, Miranda-Santos I, et al. Endothelial cell metabolism in health and disease. Trends Cell Biol. 2018;28(3):224–236.
- He YC, Halford MM, Achen MG, et al. Exploring the role of endothelium in the tumour response to anti-angiogenic therapy. Biochem Soc Trans. 2014;42(6):1569–1575.
- Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7(10):803–815.
- Fasolo F, Di Gregoli K, Maegdefessel L, et al. Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc Res. 2019;115(12):1732–1756.
- Ma Q, Dai X, Lu W, et al. Silencing long non-coding RNA MEG8 inhibits the proliferation and induces the ferroptosis of hemangioma endothelial cells by regulating miR-497-5p/NOTCH2 axis. Biochem Biophys Res Commun. 2021;556:72–78.
- Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–296.
- Dumas SJ, García-Caballero M, Carmeliet P. Metabolic signatures of distinct endothelial phenotypes. Trends Endocrinol Metab. 2020;31(8):580–595.
- Maruhashi T, Higashi Y. Pathophysiological association between diabetes mellitus and endothelial dysfunction. Antioxidants (Basel). 2021;10(8):1306.
- Meng Z, Liang H, Zhao J, et al. HMOX1 upregulation promotes ferroptosis in diabetic atherosclerosis. Life Sci. 2021;284:119935.
- Luo EF, Li HX, Qin YH, et al. Role of ferroptosis in the process of diabetes-induced endothelial dysfunction. World J Diabetes. 2021;12(2):124–137.
- Rodríguez ML, Pérez S, Mena-Mollá S, et al. Oxidative stress and microvascular alterations in diabetic retinopathy: future therapies. Oxid Med Cell Longev. 2019;2019:4940825.
- Zhang J, Qiu Q, Wang H, et al. TRIM46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating GPX4 ubiquitination. Exp Cell Res. 2021;407(2):112800.
- Deng L, Du C, Song P, et al. The role of oxidative stress and antioxidants in diabetic wound healing. Oxid Med Cell Longev. 2021:8852759.
- Li S, Li Y, Wu Z, et al. Diabetic ferroptosis plays an important role in triggering on inflammation in diabetic wound. Am J Physiol Endocrinol Metab. 2021;321(4):E509–E520.
- Tabit CE, Chung WB, Hamburg NM, et al. Endothelial dysfunction in diabetes mellitus: molecular mechanisms and clinical implications. Rev Endocr Metab Disord. 2010;11(1):61–74.
- Zhao Y, Liu Y, Xu Y, et al. The role of ferroptosis in blood-brain barrier injury. Cell Mol Neurobiol. 2022. DOI:10.1007/s10571-022-01197-5.
- Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–150.
- Pandit R, Chen L, Götz J. The blood-brain barrier: physiology and strategies for drug delivery. Adv Drug Deliv Rev. 2020;165-166:1–14.
- Enciu AM, Gherghiceanu M, Popescu BO. Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxid Med Cell Longev. 2013;2013:297512.
- Jiang X, Andjelkovic AV, Zhu L, et al. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2018;163-164:144–171.
- Chen C, Huang Y, Xia P, et al. Long noncoding RNA Meg3 mediates ferroptosis induced by oxygen and glucose deprivation combined with hyperglycemia in rat brain microvascular endothelial cells, through modulating the p53/GPX4 axis. Eur J Histochem. 2021;65(3):3224.
- Jiang Y, Liu N, Han J, et al. Diabetes mellitus/poststroke hyperglycemia: a detrimental factor for tPA thrombolytic stroke therapy. Transl Stroke Res. 2021;12(3):416–427.
- Chen B, Wang H, Lv C, et al. Long non-coding RNA H19 protects against intracerebral hemorrhage injuries via regulating microRNA-106b-5p/acyl-CoA synthetase long chain family member 4 axis. Bioengineered. 2021;12(1):4004–4015.
- Keep RF, Andjelkovic AV, Xiang J, et al. Brain endothelial cell junctions after cerebral hemorrhage: changes, mechanisms and therapeutic targets. J Cereb Blood Flow Metab. 2018;38(8):1255–1275.
- Gao S, Zhou L, Lu J, et al. Cepharanthine attenuates early brain injury after subarachnoid hemorrhage in mice via inhibiting 15-lipoxygenase-1-mediated microglia and endothelial cell ferroptosis. Oxid Med Cell Longev. 2022;2022:4295208.
- Dos Santos CC. Advances in mechanisms of repair and remodeling in acute lung injury. Intensive Care Med. 2008;34(4):619–630.
- Ma TL, Zhou Y, Wang C, et al. Targeting ferroptosis for lung diseases: exploring novel strategies in ferroptosis-associated mechanisms. Oxid Med Cell Longev. 2021;2021:1098970.
- Wijerathne H, Langston JC, Yang Q, et al. Mechanisms of radiation-induced endothelium damage: emerging models and technologies. Radiother Oncol. 2021;158:21–32.
- Guo XW, Zhang H, Huang JQ, et al. Via PIEZO1 ion channel mediates ionizing radiation-induced pulmonary endothelial cell ferroptosis Ca/Calpain/VE-cadherin signaling. Front Mol Biosci. 2021;8:725274.
- Qu M, Zhang H, Chen Z, et al. The role of ferroptosis in acute respiratory distress syndrome. Front Med (Lausanne). 2021;8:651552.
- Zhou H, Li F, Niu JY, et al. Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng Li Xue Bao. 2019;71(5):689–697.
- He R, Liu B, Xiong R, et al. Itaconate inhibits ferroptosis of macrophage via Nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. 2022;8(1):43.
- Lovatt M, Kocaba V, Hui Neo DJ, et al. Nrf2: a unifying transcription factor in the pathogenesis of Fuchs’ endothelial corneal dystrophy. Redox Biol. 2020;37:101763.
- Lovatt M, Adnan K, Kocaba V, et al. Peroxiredoxin-1 regulates lipid peroxidation, in corneal endothelial cells. Redox Biol. 2020;30:101417.
- Zhao T, Guo X, Sun Y. Iron accumulation and lipid peroxidation in the aging retina: implication of ferroptosis in age-related macular degeneration. Aging Dis. 2021;12(2):529–551.
- Sun Y, Zheng Y, Wang C, et al. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018;9(7):753.
- Totsuka K, Ueta T, Uchida T, et al. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp Eye Res. 2019;181:316–324.
- Vonk Noordegraaf A, Groeneveldt JA, Bogaard HJ. Pulmonary hypertension. Eur Respir Rev. 2016;25(139):4–11.
- Bianchi ME, Crippa MP, Manfredi AA, et al. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol Rev. 2017;280(1):74–82.
- Xie SS, Deng Y, Guo SL, et al. Endothelial cell ferroptosis mediates monocrotaline-induced pulmonary hypertension in rats by modulating NLRP3 inflammasome activation. Sci Rep. 2022;12(1):3056.