
ABSTRACT
The pathogenesis of abdominal aortic aneurysm (AAA) includes inflammatory responses, matrix metalloproteinases (MMPs) degradation, VSMC apoptosis, oxidative stress, and angiogenesis, among which the inflammatory response plays a key role. At present, surgery is the only curing treatment, and no effective drug can delay AAA progression in clinical practice. Therefore, searching for a signaling pathway related to the immune-inflammatory response is an essential direction for developing drugs targeting AAA. Recent studies have confirmed that the PI3K family plays an important role in many inflammatory diseases and is involved in regulating various cellular functions, especially in the immune-inflammatory response. This review focuses on the role of each isoform of PI3K in each stage of AAA immune-inflammatory response, making available explorations for a deeper understanding of the mechanism of inflammation and immune response during the formation and development of AAA.
1. Introduction
Abdominal aortic aneurysm (AAA) is a degenerative vasodilator disease. The incidence of AAA is increasing year by year with the aging of humankind [Citation1]. According to statistics, the mortality rate of AAA is as high as 50% ~ 70%, while the mortality rate of ruptured AAA is already as high as 80% ~ 95% [Citation2]. Aortic aneurysm is permanent, localized dilation of the artery, and commonly defined as an increase of >50% of the expected average diameter. Approximately 80% of AAA occurred in the infrarenal portion of the abdominal aorta [Citation3].
Surgical repair icluding conventional open operation or interventional endovascular stent placement has become the most predominant recommendation for treatment. As for patients who could not afford an operation or have no indication, effective medications are not available to delay or inhibit the progression of AAA. Recently, it has become a hot research field to explore the pathogenic mechanism and new therapeutic strategies of AAA [Citation4].
Relevant studies have confirmed that the innate and acquired immune cells identified in aortic aneurysms, including neutrophils, macrophages, mast cells, etc., can promote the development of aortic aneurysms [Citation5–9]. The MMPs in the aortic wall mainly derive from monocytes, macrophages, and VSMCs [Citation10,Citation11]. MMPs are a class of enzymes most closely related to ECM degradation. Its substrates include collagen and elastin components in the abdominal aortic wall tissue [Citation12–14]. More and more studies have shown that the adaptive and innate immune system plays an important role in the formation and rupture of AAA. The inflammatory response is only a superficial phenomenon [Citation15–17] (see ).
Figure 1. Immune cells in abdominal aortic aneurysm formation. Different immune-inflammatory responses occur at varying levels of AAA wall. Adventitia: Macrophages are mainly polarized to pro-inflammatory M1 in AAA wall. Polarization of CD4+ Th cells and their cytokines can influence macrophage polarization. Neutrophils are recruited early in inflammation and increase over time. Media: Inflammatory cytokines secreted by above cells will promote smooth muscle cell (SMC) apoptosis and ECM degradation. Intima: Most aneurysms possess intraluminal thrombus (ILT), which is related to AAA size, growth, and rupture.
The PI3K signaling is widely involved in modulating cell functions, such as growth, metabolism, differentiation, proliferation, motility, and survival [Citation18]. Thus, many diseases arise as a result of dysregulation or mutation of PI3K signaling, including diabetes, systemic lupus erythematosus, atherosclerosis, cardiovascular disease, and tumor [Citation19].
In this view, we will discuss the different functions played by each isoform of PI3K in immune-inflammatory responses, which could be involved in this pathology, and raise a possibility that the repression of one or more PI3K subtypes may be a potential treatment for AAA.
2. PI3K signaling pathways involved in cell function
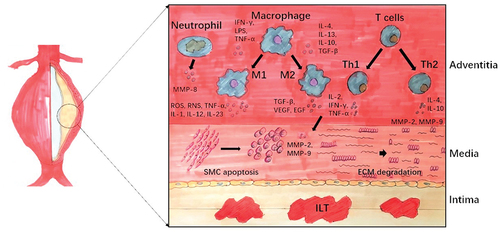
The class I subgroup is most critical when discussing its relationship to the life of cells [Citation20]. Phosphatidylinositol-3,4,5-trisphosphate (PIP3) is a lipid second messenger that activates many downstream molecules by binding to their pleckstrin-homology (PH) domains. The protein serine/threonine kinase AKT (also known as PKB) is a classic target of PIP3. Subsequent phosphorylation by the mTOR (mammalian target of rapamycin)–rictor kinase complex and by PDK1 (3-phosphoinositide-dependent kinase) leads to further activation of AKT. More downstream target proteins are activated to regulate a range of cellular functions. The forkhead (FOXO) family is another important target of AKT [Citation21] ).
2.1 Cell cycle
The effect of AKT on the cell cycle was first discovered in cancer. The initiation and progression of the cell cycle depend on the activation of Cyclin D. After being activated by Cyclin D, CDK4 and CDK6 phosphorylate the tumor suppressor protein Rb to inactivate it, thereby initiating the cell cycle [Citation22,Citation23]. Studies have shown that AKT1 interacts with Cyclin D1 to promote cell proliferation [Citation24]. In addition, AKT indirectly regulates cyclin D1 by inhibiting GSK3 [Citation21]. Cyclin-dependent kinase inhibitors (CKI), of which p21 and p27 can inhibit the CDK2-cyclin complex leading to cell cycle arrest [Citation22,Citation25]. When p21 and p27 are located in the nucleus, they inhibit cell cycle progression. Therefore, AKT1 can phosphorylate p21 to inactivate it or induce its expression in the nucleus to promote cell proliferation [Citation22,Citation26,Citation27]. AKT can also activate GSK3β and downstream Cyclin D1, thereby inhibiting the expression of p21 and p27, and promoting cell proliferation [Citation28,Citation29]. However, p21 also exhibits tumor-promoting effects in some cases, depending on its ability to inhibit cell apoptosis and facilitate the binding of Cyclin D to CDK4 and CDK6 [Citation29].
2.2 Cell survival
How PI3K controls cell apoptosis has been extensively summarized. AKT can directly phosphorylate the pro-apoptotic protein Bad, and at the same time promote its binding to the cytoplasmic 14-3-3 protein to inactivate it, and eliminate the inhibition of anti-apoptotic molecule Bcl-xL by Bad [Citation20,Citation21]. AKT can also directly phosphorylate MDM2, triggering p53 degradation [Citation21,Citation30]. AKT can also phosphorylate the pro-apoptotic protein Bax, the phosphorylated Bax is transferred to the mitochondria, and at the same time heterodimerizes with the anti-apoptotic factor Bcl-2 family members (Mcl-1, Bcl-xL) [Citation31,Citation32]. AKT inhibits cell apoptosis by blocking FoxO-mediated transcription of pro-apoptotic proteins such as FasL and Bim [Citation21,Citation33]. AKT inhibits the anti-apoptotic factor Mcl-1 (Bcl family) by regulating the activity of GSK3 [Citation34]. Together, PI3K signaling serves as an essential anti-apoptotic event.
Figure 2. Cellular functions associated with PI3K/AKT signaling pathway. The black arrows show the direction of signaling, T-bars indicate the suppression of following signaling, and Bold black arrows indicate the direction of protein migration.
3. Innate immunity
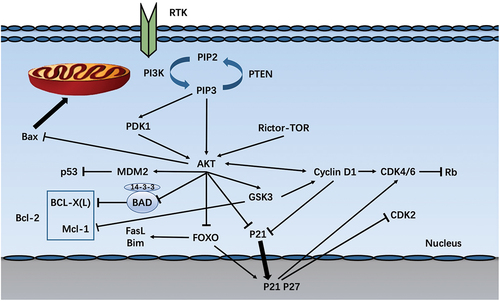
The innate immune system is the first barrier arranged to protect against pathogen invasion in a nonspecific manner. Recent research shows that the expression of multiple receptors controlled by the innate immune system changes in aortic diseases, especially the up-regulation of TLR (Toll-like receptor) and chemokine receptors are involved in the pathological process of aortic aneurysms ().
3.1 PI3K in TLR-induced pro-inflammatory cytokine secretion
Pattern recognition receptors (PRR) can recognize foreign bodies, initiate immune cell responses, and are closely related to the innate immune system [Citation35,Citation36]. At present, the earliest and most studied PPRs are Toll-like receptors, which exist in almost all tissue cells, including APC cells such as monocytes, macrophages, and B cells, as well as endothelial cells, mast cells, fibroblasts, etc. Most of these cells infiltration can be detected in the aneurysm tissue [Citation37]. Generally speaking, TLR signal transduction is classified into MyD88-dependent and MyD88-independent according to the linker molecule. Most TLRs belong to MyD88-dependent signal transduction pathways. But TLR3 is a non-MyD88-dependent signal transduction pathway, which mainly activates the TRIF signal pathway. Only TLR4 works simultaneously in two ways [Citation38]. Since different cells express different kinds of TLR, it has been reported that certain types of TLR perform actively in AAA.
Recent reports prove that TLR4 serves as a vital middle term in AAA progression [Citation39–42]. TLR4 can not only upregulate MMP-2 and MMP-9 expression, but also induce differentiation of immune cells to inflammatory phenotype and secretion of inflammatory chemokines, thereby promoting AAA formation [Citation43,Citation44].
In most cases, PI3K activation is involved in the pro-inflammatory process., However, in some instances, PI3K can also inhibit the secretion of pro-inflammatory factors and play a role in the anti-inflammatory process. PI3K is activated as a downstream of TLR, while feedback regulates TLR activity. They jointly manages the inflammatory response of immune cells [Citation45]. In endotoxin (LPS) -induced mouse spleen cells, MIR-718 expression can directly downregulate PTEN, which promotes the phosphorylation of PI3K/Akt, resulting in reduced production of pro-inflammatory cytokines. It also downregulates TLR4 and further attenuates TLR-4-mediated pro-inflammatory signaling [Citation46]. These results suggest that the negative regulation of TLR signaling proceeds in part through PI3K, thereby limiting inflammatory factors. The following conclusions were obtained by knocking out different PI3K subtypes in mice macrophages. TLR-mediated Akt phosphorylation still occurs commonly in macrophages from p110γ (−/−) mice. TLR-mediated Akt phosphorylation was significantly attenuated in macrophages with p110β knockdown by siRNA. In contrast, in p110α-deficient macrophage cells, the effects of TLR were not impaired, but were enhanced. These results suggest that p110β has a specific function in the negative feedback regulation of TLR signaling [Citation47]. It has been reported that PI3K is a negative regulator of TLR-mediated IL-12 production [Citation48]. Interestingly, knockdown of PI3K p110α suppressed TLR-mediated IL-12 mRNA expression in monocytes and macrophages, but had little effect on monocyte-derived dendritic cells. Knockdown of PI3Kp110β inhibited TLR-mediated expression of IL-12 mRNA in both monocytes and macrophages, and dendritic cells. Knockdown of both PI3Kp110α and PI3Kp110β inhibited the expression of IL-12 protein in both compartments. The role of individual PI3K isoforms in TLR-mediated innate immunity remains to be investigated [Citation49]. Studies have shown that PI3K inhibitors, such as LY294002 and Gambogic acid, can not only decrease TNF‑α, IL-1β, IL-6, and IL-18 contents, but also suppress levels of TGF‑β, MMP‑2, MMP‑9, and NF‑κB in AAA mice, finally repressing the development of aortic aneurysm [Citation50,Citation51]. By the way, Huggins et al. showed that IAXO-102 can negatively regulate non-hematopoietic TLR4 signal transduction and inhibit the occurrence of AAA induced by AngII [Citation52]. Nevertheless, Owens et al. reported that MyD88 deficiency in bone marrow-derived cells could significantly reduce AngII–induced AAA, while lack of TLR4 found no apparent changes [Citation53]. It shows that TLR4 signaling plays a role through non-hematopoietic cell types. These results suggest that TLR4 in AAA composes a complex signaling network with various functional cells and pathways, and exploring this signal crosstalk is the direction of future research.
3.2 PI3K and chemokine receptors
Directed migration of immune cells is an essential condition for the occurrence and completion of immune responses in the body. Chemokines are a class of cytokines that control directed migration of cells [Citation54]. According to the positional relationship of cysteines in the chemokine structure, chemokines are divided into four prominent families: CC, C, CXC, CX3C4 [Citation55]. Correspondingly, multiple chemokine receptors, namely CR, CCR, CXCR, and CX3CR, are widely distributed and have numerous functions in AAA, initiating a series of inflammatory responses [Citation56].
It is believed that CXCR4 contributes to AAA formation. CXCR4 blocking can attenuate the progression of AAA [Citation57,Citation58]. Mo et al. showed that N15P peptide, a CXCR4 antagonist, downregulates the expression of inflammatory factors (TNF-α, IL-6, IL-8, NF-κB, COX-2, TLR4, MyD88, PI3K, and Akt) in the LPS-stimulated peripheral blood mononuclear cell (PBMC) model [Citation59]. It can be concluded that CXCR4 is involved in the progression of AAA by mediating inflammatory response, and PI3K signaling plays a role downstream. SDF1/CXCR4 axis facilitates angiogenesis via activating the PI3K/AKT pathway, inducing recruitment and differentiation of bone mesenchymal stem cells (BMSCs) to vascular endothelial cells and VEGF expression [Citation60]. However, increased expression of CXCR4 might inhibit the progression of AAA with the treatment of BMSCs [Citation61]. In the future, the detailed mechanism needs to be explored.
CXCR3 is highly expressed in AAA tissues and can attract CD45+ cell infiltration [Citation62,Citation63]. Since PI3Kγ and PI3Kδ may regulate the migration and function of immune cells by controlling the expression of chemokine receptors. CXCR3 induces activation of the MAPK and PI3K/Akt pathways in T cells. Interestingly, CXCR3-mediated T-cell migration is dependent on Gi, PLC, and PI-kinase signaling, not the PI3K/Akt or the MAPK pathway [Citation64]. In vitro experiment, activation of T cells from PI3Kγ(-/-) mice express CXCR2 less than wild-type mice. A selective inhibitor or PI3Kγ siRNA suppressed induction of CXCR3 on T cells following activation [Citation65]. Our investigation also showed that inhibition of PI3Kγ by IPI-549 dramatically reduced the mobilization and infiltration of CD45+ cells and CD45+ F4/80+ cells (macrophages) in mice aortas [Citation66]. PI3Kγ signaling may play a significant role in negatively regulating CXCR3 induced infiltration of CD45+ cells in AAA.
In aortic diseases, blockade of CXCR2 effectively delayed the growth of aorta aneurysm by decreasing neutrophil infiltration and down-regulating the expression of inflammatory factors [Citation67]. Brennan et al. showed that C3aR1 engages phosphatase and homolog (PTEN), a negative regulator of the PI3K/AKT pathway, to restrain CXCR2-driven bone marrow neutrophil mobilization following trauma [Citation68]. PI3K signaling may participate in CXCR2-induced neutrophil infiltration in AAA. Nie et al. found that SB265610, a CXCR2 inhibitor, significantly decreased infiltration of CD45+ myelomonocytes, especially F4/80+ macrophages in AAA tissues from AngII–induced ApoE(-/-) mice. Unexpectedly, SB265610 did not change the number of neutrophils and T cells [Citation69]. How CXCR2 crosstalk with PI3K signaling further affect AAA formation remains unknown.
CCR2 is another important chemokine receptor involved in AAA formation. Downregulation of CCR2 has been reported to suppress the expansion of the aorta [Citation70,Citation71]. Rapamycin inhibitor everolimus prevents the growth of bone marrow CCR2 monocytes and reduces levels of these cells in the circulation to limit aortic aneurysm in AngII–induced ApoE(-/-) mice. It is widely accepted that one of its ligands, CCL2 (MCP-1), is the most critical chemokine for macrophages [Citation72]. IL-7 upregulates MCP-1 in endothelial cells and induces recruitment of monocytes/macrophages to endothelium via PI3K/AKT-dependent and -independent activation of NF-κB [Citation73].
Until now, there is no document directly describing how PI3K signaling working in the process of chemokine receptors affecting the progress of AAA. However, we can speculate that the PI3K signaling seems to act downstream of chemokine receptors, regulating the recruitment of various inflammatory cells.
Figure 3. Innate immunity involved in PI3K/AKT signaling pathway. TLR4 initiates the MyD88-dependent way, which promotes IRAK ternary complex assembly. This complex binds to and activates TRAF6. Thus facilitatingTAK1 activation, phosphorylates the IKK complex, and ultimately leads to NF-κB transfer into the nucleus to secrete inflammatory mediators, such as TNF-a, IL1-b, IL6, IL8, IL12, IFN-γ, and MCP1. BCAP is tyrosine phosphorylated upon ligation to TLR, and the TRI domain of BCAP mediates the recruitment and activation of PI3K. Chemokines and their receptors may also be involved in the TLR4 inflammatory pathway and crosstalk with pro-inflammatory factors such as Akt and NF-kB, thereby regulating the distribution of inflammatory cells and the secretion of inflammatory mediators.
4. Immune cell infiltration in AAA
4.1 Neutrophils
Previous studies have summarized the implications of neutrophils in cardiovascular and inflammatory diseases [Citation74,Citation75]. Due to the role of neutrophils in the immune-inflammatory response, they mainly participate in the early inflammatory response and multiple processes of AAA progression, such as oxidative stress, ECM degradation, and intraluminal thrombus (ILT) formation [Citation75,Citation76]. However, the chemotaxis of neutrophils is the basis to help complete the above process. PI3Kγ deficiency affects neutrophil chemotaxis both in vivo and in vitro. For example, during N-formyl-Met-Leu-Phe-induced neutrophil chemotaxis, most PI3Kγ (-/-) neutrophils could not move along the chemotaxis gradient, and abnormal cell morphology partially reduced viability could be observed. In wild-type neutrophils, AKT and F-actin were co-localized at the front of neutrophils, whereas in PI3Kγ (-/-) neutrophils, this alteration was not, as AKT failed to completely overlap with F-actin in the corresponding filled region of the cell membrane [Citation77]. Similarly, intervention with PI3Kγ (-/-) or PI3Kγ inhibitors before and after septic episodes in wild-type mice could reduce the sensitivity of neutrophil migration failure by maintaining the expression of the chemokine receptor CXCR2 on the neutrophil surface to protect vital organ function [Citation78]. Other PI3K isoforms also have some effect on neutrophil migration. Susceptibility to pathogens in the elderly is associated with inaccurate migration of neutrophils, and PI3K signaling activation has been seen in this process. The accuracy of neutrophil migration was restored by blocking PI3K signaling, specifically inhibiting PI3Kγ or PI3Kδ. Consistent with the above results, inhibition of SHIP1 increased migration errors [Citation79]. These lines of evidence suggest that neutrophil migration to inflammatory sites is regulated to some extent by PI3KIA class and PI3KIB class.
4.2 Macrophages
According to phenotype and secreted cytokines, macrophages can be divided into two polarization types: classically activated M1 type and selectively activated M2 type [Citation80–83]. M1 macrophages secrete pro-inflammatory factors and exert pro-inflammatory functions. In response to IFN-γ, LPS, and TNF-α, M1 macrophages secrete ROS, TNF-α, IL-1, IL-12, IL-23, and other chemokines, which are crucial to inflammatory responses against microorganisms, and also impair normal organs and tissues. M2 macrophages reduce inflammatory responses and exert tissue repair functions. Macrophages polarize to M2 type under the action of IL-4, IL-13, IL-10, and TGF-β. M2 macrophages secrete TGF-β, VEGF, EGF, and other factors, synchronously can remove cell debris, promote angiogenesis and wound fibrosis [Citation84–88]. Macrophages are mainly developed from bone marrow stem cells. As generally accepted, the bone marrow stem cells differentiate into mature monocytes in the blood. Monocytes pass through the vascular endothelium, migrate to different tissues, and finally differentiate into tissue-specific macrophages. Not only can untyped monocytes polarize to varying types in response to environmental factors, typed M1 or M2 macrophages can also interconvert with each other [Citation89]. An increased M1/M2 ratio was observed during the AAA development. Many drugs have been shown to inhibit the formation and progression of AAA by preventing the conversion of macrophages to M1 or promoting macrophages to M2.
PI3K translates extracellular stimuli into intracellular signals to control various crucial functions of macrophages [Citation21,Citation48]. In response to extracellular stimulus signals, the PI3K class I accumulate in the plasma membrane. The inhibition of the regulatory subunit p85 releases the catalytic subunit p110. Subsequently, phosphatidylinositol 4,5-bisphosphate (PIP2) is phosphorylated to generate phosphatidylinositol 3,4,5-triphosphate (PIP3). PIP3 serves as a second messenger and other recruits AKT and PDK1 to regulate multiple downstream signaling pathways via activating AKT. Phosphorylated Akt inactivates tuberous sclerosis complex (TSC) 1/2, thereby inhibiting enriched Ras homolog enriched in the brain (Rheb), leading to mTORC1 activation [Citation90–92].
Various lines of evidence suggest that PI3K signaling limits TLR-induced pro-inflammatory responses in macrophages, as shown by attenuation of pro-inflammatory cytokine production (such as IL-12) and enhancement of IL-10 secretion [Citation45,Citation93,Citation94]. As reported, initiating PI3K/AKT/mTOR pathway in macrophages results in increased histone acetylation and enhanced function of a series of genes in favor of the M2 phenotype, proving that PI3Ks attenuate LPS induced inflammation at the genetic level [Citation95]. Whereas TLR-induced bone marrow macrophages (BMM) deficient in p110γ or p85α perform increased levels of pro-inflammatory cytokines (IL-6, IL-12, and TNF) [Citation96], another assay showed that inhibition of PI3K enhances NF-kB activation and inducible NO lyase expression in TLR-induced macrophages, which all contribute to the conversion to M1 phenotype [Citation97–99].
In most studies, PI3K/Akt/mTOR pathway was studied as a whole. Therefore, the specific functions of different isoforms of PI3K or Akt remain to be elucidated. Among the PI3K family, PI3Kδ and PI3Kγ are critical in the immune-inflammatory response of hematopoietic cells [Citation100]. Akts are mainly activated by PIP3 and to a lesser extent by PIP2 [Citation101,Citation102]. The relationship between individual Akt isoforms and specific PI3K subtypes in macrophages is not clear. However, with the application of gene knockout, siRNA technology and, the appearance of specific chemical inhibitors, the particular effects of Akt and PI3K isoforms in macrophages have been reported (see ).
Table 1. Summary of the contribution of each PI3K subtypes and related components in inflammatory responses.
PI3K is shown to mediate M2 macrophage phenotypes [Citation94,Citation97,Citation103]. Accordingly, the dual PI3Kδ/γ inhibitor RP6530 downregulate lactate regulation and shift macrophage activation from an immunosuppressive M2-like phenotype to a pro-inflammatory M1-like phenotype in tumor-induced macrophage [Citation104]. In addition, the expression of M1 (CD86, TNF, IL-12, IL-23) was increased, and the manifestation of M2 (CD206, Arg-1) was decreased in the bone marrow of PI3Kγ knockout compared with wild-type mice. HMGB1 facilitated macrophage reprogramming toward M1-like phenotype dependent on the activation of PI3Κγ [Citation105], thus participating in pro-inflammatory signal transduction. In contrast, the anti-inflammatory effect of naloxone and magnesium sulfate are also related to PI3Kδ and PI3Kγ [Citation106,Citation107]. Furthermore, it has been confirmed that PI3K is involved in mediating the anti-inflammatory effects of vasopressin. Interestingly, the inhibitor of PI3Kβ, PI3Kδ, and PI3Kγ can partially counteract the effects of vasopressin, while the inhibitor of PI3Kα does not [Citation108]. In the human monocytic cell line THP-1, the PI3Kα is involved in phagocytosis, oxidative burst, and secretion of pro-inflammatory mediators, but inversely correlated with the production of TNF-α and IL-10 [Citation109]. Despite the fact that PI3K subunits possess distinct functions, a certain degree of redundancy has been described in several aspects of macrophage activation [Citation110,Citation111].
The primary role of PTEN is to dephosphorylate lipid substrates (e.g. PIP3). Still, PTEN can also dephosphorylate peptides phosphorylated on Tyr, Ser, and Thr in vitro, so PTEN is a dual-specificity protein phosphatase [Citation112,Citation113]. In this case, PTEN prevents activation of the PI3K-Akt signaling pathway [Citation114], and also regulates macrophage polarization via PIP3 acting as an auxiliary messenger [Citation115,Citation116]. SHIP is another negative regulator of PI3K/Akt signaling that dephosphorylates PI3K-generated PIP3 like PTEN. SHIP removes this second messenger from the cell membrane in macrophages [Citation117]. Consistent with the above results, deficiency of SHIP contributes to the generation of M2 phenotype macrophages [Citation118].
4.3 Lymphocytes
T cells are heterogeneous, and possess different developmental stages or immune functions at the same time. According to CD antigen classification, T cells can be divided into two major types: CD4 + and CD8 + Most helper T cells express CD4, whereas most cytotoxic T cells express CD8. In different stages of AAA development, it was detected that the expression of CD4 + T cells in the patient’s serum increased, and it was mainly expressed in AAA tissues. The CD4 + T cell regulates ECM metabolism by recruitment of macrophages and protease synthesis [Citation124].
CD4+ Th cells can be divided into two basic subsets, Th1 and Th2, based on their cytokine secretion profile. Th1 cells promote cellular immunity by secreting lL-2, IFN-γ, and TNF-α, while Th2 cells enhance humoral immunity through secreting IL-4, IL-10, etc [Citation125,Citation126]. Under normal circumstances, Th1/Th2 cells regulate each other and keep in a dynamic balance to maintain the normal immune function of the body [Citation126]. When the Th1/Th2 cell ratio is out of balance, the body’s immune and inflammatory response will follow [Citation127]. Most studies have confirmed that Th1 cells are anti-inflammatory, while Th2 cells are pro-inflammatory in the host [Citation128,Citation129]. Consistent with the observed results, Th2 cells and cytokines were mainly expressed in the AAA wall. IFN-γ receptor-deficient hosts successfully induced severe AAA formation by effectively upregulate levels of MMP-9 and MMP-12. In contrast, deficient in IL-4 negatively develop AAA and likewise exhibited attenuated ability to degrade collagen and elastic fibers [Citation130,Citation131]. However, Galle et al. find that CD4 + T cells generate high levels of IFN-γ instead of IL-4, indicating that Th1 cells may predominate in human AAA lesions [Citation132]. Another study showed the presence of Th1 and Th2 cells activated in situ in the aneurysmal lesions of AAA patients, both of which are involved in the pathogenesis of AAA [Citation133]. Th1 type cytokine IFN-γ may be related to the recruitment and infiltration of early abdominal aortic inflammatory cells, and Th2 type cytokine IL-4 in the local abdominal aortic aggregation environment may induce the increase in the activity of matrix metalloenzymes in the artery, leading to pathological remodeling of the extracellular matrix. The results seem to be contradictory, indicating that IFN-γ and IL-4 may play different roles in different development stages of abdominal aortic aneurysms. The immune response mechanism of AAA pathogenesis shows the characteristics of complexity and diversity. The activation of PI3K may increase Th2 and decrease the number of Th1 and Treg during cell immunity in AAA [Citation134]. Similarly, Kuroda et al. reported that SHIP-/- mice skewed to Th2 accompanied by excessive Th2 cells and its cytokines such as IL-4 in serum and spleen. Neutralizing anti-IL-4 Ab can completely reverse these effects. They conclude that SHIP can negatively regulate IL-4 production from basophils, which indirectly affects Th cell differentiation [Citation135]. Interestingly, whether it is in the context of CD28 co-stimulation, a kinase-inactivating mutation (D910A) in the p110δ isoform of PI3K has no effect on the normal survival of T cells, but reduce division and impair differentiation [Citation136]. The role of PI3K and its different subtypes in the differentiation of Th1 and Th2 cells remains to be elucidated. Inducing Th2 cells switching to Th1 cells may become a new therapeutic target for AAA.
4.4 Vascular smooth muscle cells
The tunica media is mainly composed of vascular smooth muscle cells. As the main site of ECM synthesis, VSMC apoptosis causes extracellular matrix (ECM) loss in the aortic wall, and ECM degradation can also contribute to VSMC loss. This vicious cycle disrupts the integrity of aortic wall structure [Citation137,Citation138]. VSMCs remain quiescent and contractile phenotypes in physiological states, but they switch to proliferative, synthetic, and migratory phenotypes in response to various pathological stimuli [Citation139,Citation140]. It has been demonstrated that VSMC phenotypic switching result in vascular dysfunction [Citation140–142]. Long-term weakening of the structural and functional integrity of the aortic wall leads to aortic aneurysm formation [Citation138].
Many studies have reported the essential role of VSMC phenotype switching in atherosclerosis, and its molecular mechanism has been further elaborated [Citation143,Citation144]. Some subsequent studies show that the transcription factors therein also involved in AAA. KLF-4 acted as a key transcription factor that promotes advanced atherosclerotic plaque formation by regulating multiple VSMC phenotypes and their derived cells through various mechanisms [Citation145,Citation146]. Peroxisome proliferator activated receptor γ (PPAR-γ) takes part in the proliferation, differentiation and apoptosis of cells, including VSMC. PPAR-γ agonists (pyrenone) could activate Akt signaling and reduce KLF4 ubiquitination, finally stabilizing KLF4 protein [Citation147]. P22phox also promotes AngII–induced VSMC phenotype switching via the HO-ERK1/2/AKT-KLF4 signaling pathway [Citation148]. From the above, it can be concluded that the PI3K-AKT-KLF4 axis is involved in VSMC phenotypic switching. In addition, we found that the PI3K-AKT-KLF4 axis is also related to VSMC apoptosis and inflammatory response. Previous studies have proved that curcumin (Cur) attenuate AAA development by suppressing the inflammatory response in ApoE −/− mice [Citation149,Citation150]. CurTn inhibits apoptosis and inflammation in VSMCs by regulating the PVT1/miR-26a/KLF4 axis through PI3K/AKT signaling pathway [Citation151]. Myocardin and serum response factor maintain VSMC quiescent and contractile phenotypes, which may be potential targets for atherosclerosis therapy [Citation152]. In the rat model of subarachnoid hemorrhage(SAH), PPARβ/δ significantly inhibited hemoglobin-induced VSMC phenotype switching, and this inhibition was partially hampered in the presence of LY294002. It is not difficult to speculate that PPARβ/δ-induced PI3K/AKT activation contributes to serum response factor nuclear localization and myocardin expression [Citation153]. FoxO transcription factors act as downstream targets of Akt, which is inactivated by Akt phosphorylation. Spry1 and Spry4 perform opposing roles in VSMC phenotype switching. Spry1 promotes Akt activation in VSMC, while Spry4 antagonized MAPK/ERK and Akt signaling. FoxO3a inhibits the myocardin promoter activity, increases the expression of Spry1, and decreases the expression of Spry4, thereby reducing the myocardin mRNA level [Citation154]. Foxo4 interacts with myocardin and also inhibits its activity [Citation155]. The inhibition of FoxO family on myocardin was relieved by PI3K/AKT signaling pathway, promoting VSMC phenotypic switching. In conclusion, PI3K signaling promotes VSMC phenotype switching leading to vascular damage, ultimately contributing to AAA formation and development.
López-Candales et al. first observed that high p53 expression as well as a clear decrease in VSMC content in AAA tissues. P53 was mainly localized to residual SMC in the arterial wall [Citation156]. This reveals that VSMC apoptosis in the medial layer of the aortic wall occur in early aneurysm development [Citation138]. Except for phenotypic switching, VSMC apoptosis is also associated with various physiological responses, such as inflammatory infiltration, oxidative stress, and autophagy. It has been reported earlier that inflammatory cells infiltrating into the aortic wall secretes cytokines to promote VSMC apoptosis [Citation157]. MCP-1 is a significantly elevated cytokine in arterial inflammation, and the promoting effect of MCP-1 on VSMC apoptosis was observed in different mouse models [Citation158–160]. Elevation of medial MCP-1 accompanied by upregulation of MMP-2 and MMP-9 induces more chemokine secretion or accumulation of macrophages within the aortic wall, causing VSMC apoptosis [Citation161]. AAA patients related studies found that elevated levels of oxidative stress in plasma and aortic tissues [Citation162]. AAA mice model studies reported that elevated concentrations of oxidative stress markers such as 8-hydroxy-2-deoxyguanosine (8-OHdG) [Citation163,Citation164]. NOX generates ROS in cells. NOX subunit p47phox knockdown attenuates Ang II–induced AAA in Apoe −/− mice [Citation165]. Antioxidant was also involved in AAA. Overexpression of catalase, a key H2O2 scavenger, in VSMCs could partially impede Ang II or CaCl2 induced AAA [Citation166,Citation167]. Superoxide dismutase 2 (SOD2) was not only fighting on the forefront of counteracting mitochondrial ROS in VSMC, but also affecting mitophagy, inhibiting PIK/Akt signaling, and delaying vascular aging, thereby protecting blood vessels from calcification [Citation168]. Alternatively, ROS-dependent activation of PI3K/Akt and MAPK/ERK signaling pathways take part in AAA formation. Ang- (1–7) significantly inhibited Ang II–induced VSMCs proliferation, migration, and inflammation. NAC (superoxide scavenger) or apocynin (NOX inhibitor) inhibited Ang II–induced activation of Akt and ERK1/2 signaling [Citation169]. Resveratrol can also inhibit the hyperproliferation of VSMC through ROS-dependent PI3K/Akt and MAPK/ERK signaling pathways [Citation170]. Miquel et al. demonstrated that 7-KC was significantly increased in AAA patients, which correlated with the expression of endoplasmic reticulum stress (ERS) markers. It is accompanied by increased apoptosis, increased reactive oxygen species (ROS) production, and decreased mitochondrial biogenesis in AAA vessel wall cells [Citation171]. Autophagy is an important defense mechanism of cells to clear unrequired proteins and organelles, highly conserved in evolution [Citation172]. Recent evidence suggests that autophagy has a unique role in the development and progression of aging-related cardiovascular diseases, and the function of autophagy declines with aging [Citation173]. Autophagy can be activated by AMPK signaling or inhibited by AKT-mTOR signaling pathway [Citation174]. The release of ROS, cytokines, or growth factors is positively correlated with autophagy levels in aortic aneurysm VSMCs [Citation140,Citation175]. Autophagy has been shown to regulate Ca2+ homeostasis in VSMCs, affecting aortic contractility. Specific deletion of Atg7 leads to an imbalance between Ca2+ uptake and Ca2+ release in VSMC, and increased calcium concentration further contributes to impaired autophagy [Citation175]. Apoptosis can be inhibited by transcription factor EB (TFEB), the master regulator of autophagy. TFEB decrease was observed in both human and mouse aortic aneurysm lesions. It increases VSMC apoptosis and promotes AAA formation in different mouse models [Citation176]. Atg5 insufficiency causes loss of autophagy in VSMCs, activates ERS, promotes vascular inflammation, and induces Ang II–induced dissection [Citation177].
4.5 Endothelial cells
Endothelial cells (ECs) play a critical role in maintaining vascular homeostasis and blood flow via regulating vascular tone, angiogenesis, smooth muscle cell proliferation, and resisting local inflammatory responses and thrombosis [Citation178]. Until now, Multifaceted clinical data confirmed that AAA is closely related to endothelial dysfunction. First, the brachial artery flow-mediated dilation (FMD) is a noninvasive test reflecting endothelial cell function [Citation179–181]. The principle is that the shear stress on endothelial cells can be changed by repeatedly inflating and deflating the cuff of upper arm, in order to induce endothelium-dependent release of NO and dilate the vessels [Citation180]. FMD is the difference in brachial artery diameter before and after induction measured by ultrasound [Citation179]. Sung et al prospectively enrolled 78 patients suspected AAA and found a positive correlation between FMD and circulating endothelial progenitor cells, while a negative correlation between circulating endothelial progenitor cells and AAA. These results suggest that FMD not only effectively predict endothelial function and but also the progression of AAA [Citation182]. In another prospective cohort, Lee et al. directly demonstrated an inverse association between FMD and maximal diameter of AAA in humans. Interestingly, they also found that FMD decreased as AAA diameter increased in the natural course of AAA, which disappeared after surgical repair [Citation183].Moreover, they confirmed the validity of FMD as a predictor of AAA growth by way of machine learning techniques [Citation184]. Secondly, epidemiological investigation shows that AAA and endothelial cell injury possess similar risk factors [Citation185]. For example, smoking is a recognized risk factor for cardiovascular disease, smoking patients AAA incidence increased more than five times [Citation186]. Smoking may damage endothelial cells through oxidative stress and leukocyte adhesion [Citation187]. Smoking contributes to both AAA formation and endothelial dysfunction through a similar mechanism. Consequently, some drugs that inhibit aneurysm growth, such as ACEI, ARBs and statins, also have some protective effect on the endothelium [Citation185,Citation188].
A growing number of evidence suggests that the pathological responses in AAA originate initially in the intima, rather than in the media and adventitia. Endothelial cells convert L-arginine to NO and L-citrulline by the enzyme eNOS. Circulating NO can reach the medial layer through the walls of blood vessels, where it initiating the pathological process by stimulating the production of cyclic GMP membrane (cGMP) [Citation181]. The coupling state of eNOS is determined by its cofactor tetrahydrobiopterin (HB4), whose production is influenced by both the de novo synthesis pathway (the key enzyme is GTP cyclohydroxylase-1(GCH1)) and the remediation pathway (the key enzyme is Dihydrofolate reductase (DHFR)) [Citation185]. Cai et al. observed excessive eNOS uncoupling in both GCH1-and DHFR-knockout Ang II–induced AAA mice, accompanied by higher AAA incidence and rupture rate. Restoration of DHFR expression by oral folic acid protects against AAA initiation and rupture, and attenuates the progressive eNOS uncoupling [Citation189,Citation190]. In Ang II–induced AAA mice with double knockout of GCH1 and four types of NOX (NOX1, NOX2, p47phox, or NOX4), AAA formation was inhibited and eNOS uncoupling activity was significantly reduced due to reduced ROS production [Citation191]. After scavenging mitochondrial ROS with Mito-Tempo in DHFR-knockout Ang II–induced AAA mice, the promotion of DHFR-knockout on hypertension and AAA was completely reversed [Citation190]. It is suggested that oxidative stress plays a regulatory role upstream of the GCH1/DHFR/eNOS axis [Citation192]. In high-glucose-treated cells, tanshinone IIA (Tan IIA) significantly inhibited NOX4 expression and ROS production, improved NO production in a concentration-dependent manner, restored eNOS uncoupling, upregulated GCH1 and DHFR expression, increased the ratio of BH4 to BH2. Furthermore, Tan IIA significantly inhibited PI3K expression. Similar results were obtained by replacing Tan IIA with Wortmannin, a PI3K inhibitor. These results suggest that Tan IIA restores high glucose-induced eNOS uncoupling by targeting NOX, GCH1 and DHFR, as well as PI3K pathways [Citation193].
In addition, vascular remodeling and inflammatory response were also observed in eNOS uncoupled mice [Citation190]. The interaction between oxidative stress and inflammation aggravated arterial tissue damage. Elevated ROS levels are usually observed at sites of chronic inflammation [Citation185]. ROS regulates inflammatory cell recruitment and increases pro-inflammatory cytokine secretion. The pro-inflammatory cytokine also induces ROS production by upregulating NOX activity in endothelial cells [Citation194]. Li et al. proved that Rhubarb inhibits the PI3K/AKT/NF-κB signaling pathway to downregulate the levels of inflammatory factors (e.g. IL-1, IL-6, and TNF-α), ROS, and NO [Citation195].
4.6 Fibroblasts
The major role of fibroblasts in the repair of tissue injury is gradually well-known. As the most abundant cell population in the cardiovascular system [Citation196], proliferation and “outward-inward” migration of adventitial fibroblasts were observed in the injured coronary arteries after balloon expansion, loss of intima was subsequently replenished [Citation197]. Follow-up studies disproved the previous hypothesis that VSMCs are the earliest to respond and dominate neointima formation after intimal injury [Citation198]. The phenotypic switch of fibroblasts to myofibroblasts, resulting in the acquisition of migratory capacity, is considered to be a marker of the adventitia in response to injury [Citation199,Citation200]. Types I and III collagens mainly exist and play a supporting role in blood vessels. Some scholars speculate that they may be associated to the growth and rupture of AAA. TGF-β can induce adventitial fibroblasts to produce collagen types I and III [Citation201]. Complex crosstalk happens between TGF-β and PI3K, even in fibroblasts [Citation202]. Bumdelger et al. observed less aortic dissection and dilatation, more adventitial thickening, and higher collagen I and Trail expressions after osteoprotegerin (Opg) knockout in CaCl2-induced AAA mice. They also showed that the adventitia thickening may be involved in aortic fibrotic remodeling induced by myofibroblast accumulation [Citation203]. When Keulen et al. analyzed the AAA familial inheritance, they found that a small proportion of patients with AAA had type III collagen deficiency [Citation204]. However, with more cases were included, type III collagen deficiency did not appear to affect AAA incidence [Citation205].
Fibroblasts also play an important role in vascular inflammation. During the inward migration, myofibroblasts can induce VSMC phenotypic switching, participate in inflammatory cell infiltration and pro-inflammatory factor secretion [Citation201,Citation206]. Chiang et al found increased levels of Gal-1 (galectin-1) in the serum of AAA patients and tissues of Ang II–induced AAA mice. Gal-1 deletion reduces the incidence and severity of AAA by ameliorating medial degeneration and adventitial inflammation. Gal-1 induces the expression of MMP9 and inflammatory cytokines to varying degrees in macrophages, VSMCs, and fibroblasts by activating MAPK signaling [Citation207]. Gal-1 is closely related to tissue repair. Continuous Gal-1 over-expression enhances angiogenesis and ECM secretion via PI3K/Akt pathway [Citation208]. Binding of Gal-1 to platelets triggers PI3K phosphorylation, which further activates platelets to control primary bleeding [Citation209]. In conclusion, Gal-1 appears to mediate injury repair and inflammatory response in fibroblast, including AAA tissue. PI3K may be involved as a downstream signaling pathway of Gal-1.
4.7 Mast cells
A series of studies on the association between AAA and mast cells (MC) was carried out as early as more than a decade ago. First Tsuruda et al. observed that the number of mast cells in the adventitia of AAA was higher than in atherosclerotic aortas without any aneurysmal changes [Citation210]. It can be explained that mast cell activation followed by adrenomedullin (AM) release exerts an anti-fibrotic effect leading to adventitial dysfunction, consequently contributing to AAA development [Citation210]. Interestingly, real-world data suggest a positive correlation between adventitial mast cell number and AAA diameter [Citation211]. Tsuruda et al. successfully replicated the above results in CaCl2 and Ang II–induced AAA models. Compared to controls, mast cell-deficient mutant (Ws/Ws) rats showed reduced inflammatory cell infiltration (macrophages, T cells, mast cells), MMP-9 accumulation, elastin degradation, and angiogenesis [Citation211]. Inhibiting mast cell viability by asparagus, an inhibitor of mast cell degranulation, the repeated experiment come to similar conclusions [Citation211]. However, how mast cells contribute to AAA progression is not clear.
As reported, MC-derived renin, chymase, and cathepsin G promote the conversion of Ang I to Ang II [Citation212–214]. In addition, Mast cells can cause the death of VSMCs through both direct and indirect methods. TNF-α secreted from MCs can bind to surrounding VSMCs surface receptors, directly triggering caspase-mediated apoptosis [Citation215,Citation216]. MCs derived chymases and cathepsins degrade the ECM. VSMCs lose the support of adjacent cells and surroundings, indirectly causing anoikis [Citation215,Citation217]. Many articles have shown that PI3K/AKT/mTOR pathway activates in Ang II-induced mice, accompanied by a series of pathological events such as inflammatory cell infiltration, VSMC phenotypic conversion and apoptosis, and MMP accumulation [Citation50,Citation218–220].
5. ILT in AAA
Thrombosis is a ubiquitous feature of AAA and appears to correspond with AAA size, growth, and rupture [Citation221,Citation222]. Thrombus-covered aneurysm wall demonstrated a thinner underlying AAA vessel wall and a higher level of immune-inflammation response [Citation223]. Previous studies indicate that proteases in the thrombus develop local hypoxia and may lead to the generation of reactive oxygen species to intensify aortic wall weakening [Citation224,Citation225]. It has also been suggested that the accumulation of inflammatory cells in ILT may perturb the structural integrity and stability of the aortic wall by promoting VSMC differentiation and apoptosis [Citation226]. In the rat aortic aneurysm model, AZD6140, a P2Y(12) receptor antagonist, inhibits platelet activation and intraluminal thrombosis, reduces inflammatory infiltration and MMP expression, thereby impairing experimental abdominal aortic aneurysm development [Citation227]. Therefore, antiplatelet or anticoagulant drugs may inhibit the progression of AAA by reducing the number or volume of ILT.
Platelets play critical roles in thrombosis by adhering to and aggregating at sites of vascular injury to prevent blood loss. Growing evidence suggesting PI3K pathways which metabolism is highly active in platelets play a major role in the above two processes [Citation228]. All PI3K isoforms are expressed in platelets. However, the PI3Kβ and PI3Kγ are enriched in platelet activation. Platelet aggregation was initiated in response to collagen and Glycoprotein (GP) VI, PI3K plays an important role in GPVI-mediated platelet activation [Citation229]. Consistent with these findings, PI3Kβ-deficiency significantly inhibits GPVI-mediated platelet aggregation, secretion, and intracellular Ca(2+) mobilization, PI3Kα-deficiency has a weak inhibitory effect, when PI3Kγ- and PI3Kδ-deficiency have no effect [Citation230,Citation231]. Although both PI3Kα and PI3Kβ are involved in GPVI-mediated platelet activation in a non-redundant way, PI3Kβ is the main isoform involved following a potent activation of GPVI [Citation231,Citation232]. The PI3Kδ plays a tiny role in platelet activation mediated by the GPVI and integrin αIIbβ3 [Citation233]. PI3Kγ-deficiency defects aggregation response to ADP and threshold concentrations of collagen, strongly associated with impaired ADP-induced integrin αIIbβ3 activation [Citation234,Citation235]. Interestingly, both PI3Kγ- and PI3Kβ- deficiency are accompanied by impaired P2Y(12) signaling [Citation234,Citation236]. Deficiency of PI3Kγ and PI3Kβ can significantly decrease arterial thrombosis without a corresponding increase in bleeding time, indicating that selective inhibition of one or more PI3K isoforms for antithrombotic therapy may reduce the risk of bleeding, with clear advantages compared to existing anticoagulant drugs.
6. Sex difference and estrogen in AAA
It is well known that the male is independent risk factors for AAA formation. A more in-depth study has shown that estrogen levels inversely correlated with AAA in men [Citation237]. Associations between estrogen or estrogen receptors and the PI3K signaling pathway have been reported in different diseases, although the molecular mechanisms vary or remain unclear. Recent studies have shown that the PI3K/AKT/mTOR pathway can counteract resistance to endocrine therapy in estrogen receptor-positive breast cancer [Citation238]. The BOLERO-2 trial profoundly illustrated that the mTOR inhibitor works in the treatment of estrogen receptor-positive breast cancer [Citation239]. Normally, estrogen receptor (ER) activates the PI3K/AKT/mTOR pathway to directly or indirectly regulate cyclin-dependent kinases 4 and 6 (CDK4/6) in the nucleus [Citation240,Citation241]. When PIK3CA mutations in breast cancers, ERs could still drive tumor cell proliferation by activating alternative pathways such as the RAS/MEK/ERK pathway, the ER pathway, or the HER2 pathway [Citation242]. Activation of the PI3K signaling pathway by estrogen has also been reported to be beneficial in cardiovascular disease. Estrogen binding to ER induces endothelial progenitor cell (EPCs) migration, proliferation, and apoptosis by PI3K pathway [Citation243,Citation244]. ER is involved in endothelial eNOS activation through rapid non-genomic effects with downstream signaling of the PI3K pathway [Citation245]. Unfortunately, no original studies have explored whether sex differences in AAA formation result from activation of the PI3K pathway by estrogen or ERs.
7. Current PI3K-mediated pharmacological treatments of AAA
Many drugs that inhibit AAA by modulating the PI3K signaling pathway have been demonstrated in murine models( and ). However, most of the drugs lack clinical data support. Metformin, a first-line drug for type 2 diabetes mellitus (T2DM), has been widely used in clinical practice. Recent basic and clinical studies have shown that metformin inhibits AAA development and reduces the risk of aneurysm rupture. Wang et al. showed that metformin inhibited AAA progression by modulating the function of VSMCs through the PI3K/AKT/mTOR pathway. In addition, PI3K inhibitors and agonists enhanced and attenuated the protective effect of metformin on AAA, respectively [Citation218]. A pool of clinical studies reported that metformin reduced the annual rate of aneurysm growth and the incidence of fatal AAA events, including aneurysm rupture or death, in patients with T2DM. The effect of metformin on AAA in patients without T2DM remains unknown [Citation246]. A multicenter cohort study involving 1.9 million subjects demonstrated that T2DM was also associated with a lower incidence of AAA [Citation246,Citation247]. Whether diabetes itself or hypoglycemic drugs have priority in the protective effect on AAA is not conclusive yet. Nevertheless, metformin deserves more research to explore its protective mechanism against AAA and can to be a breakthrough in the pharmacological prevention and treatment of AAA.
Table 2. Pharmacological treatment of AAA via PI3K signaling.
Table 3. PI3K-related microRNA in AAA.
There are three main murine models of AAA reported, induced by angiotensin II, elastase, and calcium chloride (CaCl2) [Citation248]. Murine models induced by angiotensin II are the most common. Besides easy to prepare, it has similar pathological features to humans, such as inflammatory infiltration, extracellular matrix degradation, arterial wall plaque, and thrombosis [Citation249]. The model also has some limitations. Local inflammation induced by angiotensin II was mainly dominated by macrophage infiltration [Citation250]. Angiotensin II can tear the intima of the artery to form a dissection. Hence angiotensin II-induced AAA is essentially a dissecting aneurysm, not a true aneurysm commonly seen in patients, and the thrombus is subintimal rather than intramural [Citation251]. In addition, angiotensin II induces aneurysm formation throughout the entire length of the aorta, while aneurysms are localized below the renal arteries in most patients [Citation250]. The elastase-induced murine model, also known as the PPE model, is the second most common model. Compared with the angiotensin II model, the PPE model requires exposure and incision of the murine aorta for intraluminal drug perfusion, which is more challenging to operate [Citation252]. Compared with the angiotensin II model, the arterial wall of the PPE model was evenly dilated and the intima was intact, which was more in line with patient characteristics [Citation253,Citation254]. Pathological features showed inflammation, elastic fibers destruction in arterial tissue, but adnexal thrombosis was rare [Citation253].In addition, the PPE model has a limited ability to sustain growth, the aneurysm does not rupture, suggesting that it cannot be used to study the natural course of AAA or late ruptured AAA [Citation248]. The mouse model induced by CaCl2 is commonly used to complement the results. The aortic diameter in the CaCl2 model is smaller than the above two models [Citation255,Citation256]. Inflammation, more serious degradation of elastic fiber and intimal calcifications are observed in arterial walls [Citation256–258]. Arterial rupture and intraluminal thrombosis also did not occur with the CaCl2 model [Citation256]. Mechanistically, inflammatory infiltration and elastic fiber degradation were present in all three models. Significantly, arterial plaques and intraluminal thrombi are only observed in the angiotensin II model. In terms of the degree of arterial dilatation, the angiotensin II model and PPE model are more obvious. Ni, X., et al. found that IMD could protect AAA by inhibiting inflammation mediated by the Notch1 signaling pathway in both the angiotensin II model and the CaCl2 model, whereas PI3K inhibitors could reverse this protection [Citation249]. These evidences indicate that despite PI3K is activated in three murine models to regulate the inflammatory response and promote AAA formation, its expression level, activation degree and focus differ.
Conclusions
The present study suggests that chronic inflammation of the aortic wall is involved in the pathology of AAA, leading to thickening of the aortic wall, adhesion to surrounding tissues and massive inflammatory cell infiltration. However, a vital role in AAA formation is the immune response, and the inflammatory response is just a surface phenomenon. In recent years, PI3K has been reported as a potential therapeutic target for many chronic inflammatory diseases. The use of PI3K pan-inhibitors in the treatment of related diseases is often accompanied by non-negligible side effects. The emergence of specific PI3K isotype inhibitors has dramatically increased the controllability of drugs. The impact of single isotype PI3K on the immune inflammatory response in AAA and its role in the formation and expansion of AAA may become the focus of future research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Li G, Qin L, Wang L, et al. Inhibition of the mTOR pathway in abdominal aortic aneurysm: implications of smooth muscle cell contractile phenotype, inflammation, and aneurysm expansion. Am J Physiol Heart Circ Physiol. 2017;312(6):H1110–H1119.
- Luebke T, Brunkwall J. Risk-adjusted meta-analysis of 30-day mortality of endovascular versus open repair for ruptured abdominal aortic aneurysms. Ann Vasc Surg. 2015;29(4):845–863.
- Daugherty A, Cassis L. Mechanisms of abdominal aortic aneurysm formation. Curr Atheroscler Rep. 2002;4(3):222–227.
- Zhang S, Du X, Chen Y-Q, et al. Potential medication treatment according to pathological mechanisms in abdominal aortic aneurysm. J Cardiovasc Pharmacol. 2018;71(1):46–57.
- Yan Y, Fan J, Bai S-L, et al. Zinc prevents abdominal aortic aneurysm formation by induction of a20-mediated suppression of NF-κB pathway. PloS one. 2016;11(2):e0148536.
- Rateri D, Howatt DA, Moorleghen JJ, et al. Prolonged infusion of angiotensin II in apoE(-/-) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am J Pathol. 2011;179(3):1542–1548.
- Wang J, Lindholt JS, Sukhova GK, et al. IgE actions on CD 4 + T cells, mast cells, and macrophages participate in the pathogenesis of experimental abdominal aortic aneurysms. EMBO Mol Med. 2014;6(7):952–969.
- Forester N, Cruickshank SM, Scott DJA, et al. Increased natural killer cell activity in patients with an abdominal aortic aneurysm. Br J Surg. 2006;93(1):46–54.
- Eliason J, Hannawa KK, Ailawadi G, et al. Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation. 2005;112(2):232–240.
- Longo G, Xiong W, Greiner TC, et al. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110(5):625–632.
- Longo G, Buda SJ, Fiotta N, et al. MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery. 2005;137(4):457–462.
- Rizzo R, McCarthy WJ, Dixit SN, et al. Collagen types and matrix protein content in human abdominal aortic aneurysms. J Vasc Surg. 1989;10(4):365–373.
- Keeling W, Armstrong PA, Stone PA, et al. An overview of matrix metalloproteinases in the pathogenesis and treatment of abdominal aortic aneurysms. Vasc Endovascular Surg. 2005;39(6):457–464.
- Culav E, Clark C, Merrilees M. Connective tissues: matrix composition and its relevance to physical therapy. Phys Ther. 1999;79(3):308–319.
- Yamaguchi T, Yokokawa M, Suzuki M, et al. The effect of immunosuppression on aortic dilatation in a rat aneurysm model. Surg Today. 2000;30(12):1093–1099.
- Liu Y, Liao J, Zhao M, et al. Increased expression of TLR2 in CD4 + T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur J Immunol. 2015;45(9):2683–2693.
- Jagadesham V, Scott D, Carding S. Abdominal aortic aneurysms: an autoimmune disease? Trends Mol Med. 2008;14(12):522–529.
- Nunes-Santos C, Uzel G, Rosenzweig S. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol. 2019;143(5):1676–1687.
- Elmenier F, Lasheen D, Abouzid K. Phosphatidylinositol 3 kinase (PI3K) inhibitors as new weapon to combat cancer. Eur J Med Chem. 2019;183:111718.
- Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415(3):333–344.
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619.
- Basu A, Lambring C. Akt isoforms: a family affair in breast cancer. Cancers (Basel). 2021;13(14):3445.
- Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220–5227.
- Chen K, Jiao X, Di Rocco A, et al. Endogenous cyclin D1 promotes the rate of onset and magnitude of mitogenic signaling via Akt1 Ser473 phosphorylation. Cell Rep. 2020;32(11):108151.
- Abukhdeir A, Park B. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19.
- Li Y, Dowbenko D, Lasky L. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem. 2002;277(13):11352–11361.
- Héron-Milhavet L, Franckhauser C, Rana V, et al. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol Cell Biol. 2006;26(22):8267–8280.
- Yang X, Feng Y, Liu Y, et al. Fuzheng Jiedu Xiaoji formulation inhibits hepatocellular carcinoma progression in patients by targeting the AKT/CyclinD1/p21/p27 pathway. Phytomedicine. 2021;87:153575.
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414.
- Mayo L, Donner D. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98(20):11598–11603.
- Linton M, Moslehi J, Babaev V. Akt signaling in macrophage polarization, survival, and atherosclerosis. Int J Mol Sci. 2019;20(11):2703.
- Gardai S, Hildeman DA, Frankel SK, et al. Phosphorylation of bax ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem. 2004;279(20):21085–21095.
- Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868.
- Maurer U, Charvet C, Wagman AS, et al. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21(6):749–760.
- Sellge G, Kufer T. PRR-signaling pathways: learning from microbial tactics. Semin Immunol. 2015;27(2):75–84.
- Hontelez S, Sanecka A, Netea MG, et al. Molecular view on PRR cross-talk in antifungal immunity. Cell Microbiol. 2012;14(4):467–474.
- Pryshchep O, Ma-Krupa W, Younge BR, et al. Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation. 2008;118(12):1276–1284.
- Richards M, Black AS, Bonnet DJ, et al. The LPS2 mutation in TRIF is atheroprotective in hyperlipidemic low density lipoprotein receptor knockout mice. Innate Immun. 2013;19(1):20–29.
- Yang Y, Ma Z, Yang G, et al. Alginate oligosaccharide indirectly affects toll-like receptor signaling via the inhibition of microRNA-29b in aneurysm patients after endovascular aortic repair. Drug Des Devel Ther. 2017;11:2565–2579.
- Shang T, Ran F, Qiao Q, et al. Tanshinone IIA attenuates elastase-induced AAA in rats via inhibition of myd88-dependent TLR-4 signaling. VASA Z Gefasskrankheiten. 2014;43(1):39–46.
- Pirianov G, Torsney E, Howe F, et al. Rosiglitazone negatively regulates c-Jun N-terminal kinase and toll-like receptor 4 proinflammatory signalling during initiation of experimental aortic aneurysms. Atherosclerosis. 2012;225(1):69–75.
- Balistreri C. Genetic contribution in sporadic thoracic aortic aneurysm? Emerging evidence of genetic variants related to TLR-4-mediated signaling pathway as risk determinants. Vascul Pharmacol. 2015;74:1–10.
- Lai C, Wang K-C, Lee F-T, et al. Toll-like receptor 4 is essential in the development of abdominal aortic aneurysm. PloS one. 2016;11(1):e0146565.
- Qin Z, Bagley J, Sukhova G, et al. Angiotensin II-induced TLR4 mediated abdominal aortic aneurysm in apolipoprotein E knockout mice is dependent on STAT3. J Mol Cell Cardiol. 2015;87:160–170.
- Fukao T, Tanabe M, Terauchi Y, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3(9):875–881.
- Zhang X, Li N, Shao H, et al. Methane limit LPS-induced NF-κB/MAPKs signal in macrophages and suppress immune response in mice by enhancing PI3K/AKT/GSK-3β-mediated IL-10 expression. Sci Rep. 2016;6(1):29359.
- Hazeki K, Kinoshita S, Matsumura T, et al. Opposite effects of wortmannin and 2-(4-morpholinyl)-8-phenyl-1(4 H)-benzopyran-4-one hydrochloride on toll-like receptor-mediated nitric oxide production: negative regulation of nuclear Factor-κB by phosphoinositide 3-kinase. Mol Pharmacol. 2006;69(5):1717–1724.
- Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24(7):358–363.
- (Baltimore, Md.: 1950), Utsugi M, Dobashi K, Ono A, et al. PI3K p110beta positively regulates lipopolysaccharide-induced IL-12 production in human macrophages and dendritic cells and JNK1 plays a novel role. J Iimmunol. 2009;182(9):5225–5231.
- Liu Q, Shan P, Li H. Gambogic acid prevents angiotensin II‑induced abdominal aortic aneurysm through inflammatory and oxidative stress dependent targeting the PI3K/Akt/mTOR and NF‑κB signaling pathways. Mol Med Rep. 2019;19(2):1396–1402.
- Ma X, Yao H, Yang Y, et al. miR-195 suppresses abdominal aortic aneurysm through the TNF-α/NF-κB and VEGF/PI3K/Akt pathway. Int J Mol Med. 2018;41(4):2350–2358.
- Huggins C, Pearce S, Peri F, et al. A novel small molecule TLR4 antagonist (IAXO-102) negatively regulates non-hematopoietic toll like receptor 4 signalling and inhibits aortic aneurysms development. Atherosclerosis. 2015;242(2):563–570.
- Owens A, Rateri DL, Howatt DA, et al. MyD88 deficiency attenuates angiotensin II-induced abdominal aortic aneurysm formation independent of signaling through toll-like receptors 2 and 4. Arterioscler Thromb Vasc Biol. 2011;31(12):2813–2819.
- Nagarsheth N, Wicha M, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–572.
- Marchese A. Endocytic trafficking of chemokine receptors. Curr Opin Cell Biol. 2014;27:72–77.
- van der Vorst E, Döring Y, Weber C. Chemokines. Arterioscler Thromb Vasc Biol. 2015;35(11):e52–6.
- Michineau S, Franck G, Wagner-Ballon O, et al. Chemokine (C-X-C motif) receptor 4 blockade by AMD3100 inhibits experimental abdominal aortic aneurysm expansion through anti-inflammatory effects. Arterioscler Thromb Vasc Biol. 2014;34(8):1747–1755.
- Tanios F, Pelisek J, Lutz B, et al. CXCR4: a potential marker for inflammatory activity in abdominal aortic aneurysm wall. Eur J Vasc Endovasc Surg. 2015;50(6):745–753.
- Mo X, Sun H. The anti-inflammatory effect of the CXCR4 antagonist-N15P peptide and its modulation on inflammation-associated mediators in LPS-induced PBMC. Inflammation. 2015;38(3):1374–1383.
- Zhang H, Wang P, Zhang X, et al. SDF1/CXCR4 axis facilitates the angiogenesis via activating the PI3K/AKT pathway in degenerated discs. Mol Med Rep. 2020;22(5):4163–4172.
- Long M, Li -H-H, Pen X-Z, et al. Expression of chemokine receptor-4 in bone marrow mesenchymal stem cells on experimental rat abdominal aortic aneurysms and the migration of bone marrow mesenchymal stem cells with stromal-derived factor-1. Kaohsiung J Med Sci. 2014;30(5):224–228.
- van den Borne P, Quax PHA, Hoefer IE, et al. The multifaceted functions of CXCL10 in cardiovascular disease. Biomed Res Int. 2014;2014:893106.
- Gallo A, Saad A, Ali R, et al. Circulating interferon-γ-inducible Cys-X-Cys chemokine receptor 3 ligands are elevated in humans with aortic aneurysms and Cys-X-Cys chemokine receptor 3 is necessary for aneurysm formation in mice. J Thorac Cardiovasc Surg. 2012;143(3):704–710.
- Smit M, Verdijk P, van der Raaij-helmer EMH, et al. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood. 2003;102(6):1959–1965.
- Barbi J, Cummings HE, Lu B, et al. PI3Kgamma (PI3Kgamma) is essential for efficient induction of CXCR3 on activated T cells. Blood. 2008;112(8):3048–3051.
- Liu R, Huang J, Ge Y, et al. Inhibition of phosphatidylinositol 3-kinase γ by IPI-549 attenuates abdominal aortic aneurysm formation in mice. Eur J Vasc Endovasc Surg. 2020;60(2):254–263.
- Anzai A, Shimoda M, Endo J, et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ Res. 2015;116(4):612–623.
- Kim N, Woo D-C, Joo SJ, et al. Reduction in renal ischemia-reperfusion injury in mice by a phosphoinositide 3-kinase p110gamma-specific inhibitor. Transplantation. 2015;99(10):2070–2076.
- Nie H, Wang H-X, Tian C, et al. Chemokine (C-X-C motif) receptor 2 blockade by SB265610 inhibited angiotensin II-induced abdominal aortic aneurysm in apo E mice. Heart Vessels. 2019;34(5):875–882.
- (London, England: 1979), Daugherty A, Rateri D, Charo I, et al. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE-/- mice. Clin Sci. 2010;118(11):681–689.
- MacTaggart J, Xiong W, Knispel R, et al. Deletion of CCR2 but not CCR5 or CXCR3 inhibits aortic aneurysm formation. Surgery. 2007;142(2):284–288.
- Zhang J, Chen H, Liu L, et al. Chemokine (C-C motif) receptor 2 mediates mast cell migration to abdominal aortic aneurysm lesions in mice. Cardiovasc Res. 2012;96(3):543–551.
- Li R, Paul A, Ko KWS, et al. Interleukin-7 induces recruitment of monocytes/macrophages to endothelium. Eur Heart J. 2012;33(24):3114–3123.
- Klopf J, Brostjan C, Eilenberg W, et al. Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease. Int J Mol Sci. 2021;22(2):559.
- (London England: 1979), Fougerat A, Gayral S, Malet N, et al. Phosphoinositide 3-kinases and their role in inflammation: potential clinical targets in atherosclerosis? Clin Sci. 2009;116(11):791–804.
- Michel J, Martin-Ventura J-L, Egido J, et al. Novel aspects of the pathogenesis of aneurysms of the abdominal aorta in humans. Cardiovasc Res. 2011;90(1):18–27.
- Hannigan M, Zhan L, Li Z, et al. Neutrophils lacking phosphoinositide 3-kinase γ show loss of directionality during N -formyl-Met-Leu-Phe-induced chemotaxis. Proc Natl Acad Sci U S A. 2002;99(6):3603–3608.
- Sasaki T, Sasaki J, Watanabe K, et al. Non-invasive visualization of the lipid product of class I PI3K in transgenic mouse models. Biochem Soc Trans. 2007;35(2):215–218.
- Sapey E, Greenwood H, Walton G, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123(2):239–248.
- Manning B, Cantley L. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274.
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35.
- Gordon S, Martinez F. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604.
- Liu X, Shi Y, Birnbaum MJ, et al. Quantitative analysis of anti-apoptotic function of Akt in Akt1 and Akt2 double knock-out mouse embryonic fibroblast cells under normal and stressed conditions. J Biol Chem. 2006;281(42):31380–31388.
- Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686.
- (Baltimore, Md.: 1950), Dale M, Xiong W, Carson JS, et al. Elastin-derived peptides promote abdominal aortic aneurysm formation by modulating M1/M2 macrophage polarization. J Iimmunol. 2016;196(11):4536–4543.
- Pope N, Salmon M, Davis JP, et al. D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J. 2016;30(12):4192–4201.
- Rao J, Brown BN, Weinbaum JS, et al. Distinct macrophage phenotype and collagen organization within the intraluminal thrombus of abdominal aortic aneurysm. J Vasc Surg. 2015;62(3):585–593.
- Raffort J, Lareyre F, Clément M, et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14(8):457–471.
- (Baltimore, Md.: 1950), Stout R, Jiang C, Matta B, et al. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Iimmunol. 2005;175(1):342–349.
- Covarrubias A, Aksoylar H, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015; 27(4):286–296.
- Troutman T, Bazan J, Pasare C, (GeorgetownTex.). Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle. 2012;11(19):3559–3567.
- Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71.
- Aksoy E, Vanden Berghe W, Detienne S, et al. Inhibition of phosphoinositide 3-kinase enhances TRIF-dependent NF-kappa B activation and IFN-beta synthesis downstream of toll-like receptor 3 and 4. Eur J Immunol. 2005;35(7):2200–2209.
- Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277(35):32124–32132.
- Covarrubias A, Aksoylar HI, Yu J, et al. Akt-mTORC1 signaling regulates acly to integrate metabolic input to control of macrophage activation. eLife. 2016;5.
- Luo L, Wall AA, Yeo JC, et al. Rab8a interacts directly with PI3Kγ to modulate TLR4-driven PI3K and mTOR signalling. Nat Commun. 2014;5(1):4407.
- (Baltimore, Md.: 1950), Luyendyk J, Schabbauer GA, Tencati M, et al. Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J Iimmunol. 2008;180(6):4218–4226.
- (Baltimore, Md.: 1950)Díaz-Guerra M, Castrillo A, Martín-Sanz, P, et al. Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J Iimmunol 1999;162(10):6184–6190.
- Weichhart T, Säemann M. The multiple facets of mTOR in immunity. Trends Immunol. 2009;30(5):218–226.
- Fung-Leung W. Phosphoinositide 3-kinase delta (PI3Kδ) in leukocyte signaling and function. Cell Signal. 2011;23(4):603–608.
- Franke T, Yang S-I, Chan TO, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81(5):727–736.
- Jean S, Kiger A. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci. 2014;127(5):923–928.
- (Baltimore, Md.: 1950), Beharka A, Crowther JE, McCormack FX, et al. Pulmonary surfactant protein A activates a phosphatidylinositol 3-kinase/calcium signal transduction pathway in human macrophages: participation in the up-regulation of mannose receptor activity. J Iimmunol. 2005;175(4):2227–2236.
- Locatelli S, Careddu G, Serio S, et al. Targeting cancer cells and tumor microenvironment in preclinical and clinical models of Hodgkin lymphoma using the dual PI3Kδ/γ inhibitor RP6530. Clin Cancer Res off J Am Assoc Cancer Res. 2019;25(3):1098–1112.
- Su Z, Zhang P, Yu Y, et al. HMGB1 facilitated macrophage reprogramming towards a proinflammatory M1-like phenotype in experimental autoimmune myocarditis development. Sci Rep. 2016;6(1):21884.
- Lee P, Yang C-H, Kao M-C, et al. Phosphoinositide 3-kinase β, phosphoinositide 3-kinase δ, and phosphoinositide 3-kinase γ mediate the anti-inflammatory effects of magnesium sulfate. J Surg Res. 2015;197(2):390–397.
- Wang T, Su N-Y, Shih P-C, et al. Anti-inflammation effects of naloxone involve phosphoinositide 3-kinase delta and gamma. J Surg Res. 2014;192(2):599–606.
- Jan W, Kao M-C, Yang C-H, et al. Phosphoinositide 3-kinase is involved in mediating the anti-inflammation effects of vasopressin. Inflammation. 2017;40(2):435–441.
- Lee J, Nauseef WM, Moeenrezakhanlou A, et al. Monocyte p110alpha phosphatidylinositol 3-kinase regulates phagocytosis, the phagocyte oxidase and cytokine production. J Leukoc Biol. 2007;81(6):1548–1561.
- Guillermet-Guibert J, Bjorklof K, Salpekar A, et al. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A. 2008;105(24):8292–8297.
- Bourdonnay E, Serezani CH, Aronoff DM, et al. Regulation of alveolar macrophage p40phox: hierarchy of activating kinases and their inhibition by PGE 2. J Leukoc Biol. 2012;92(1):219–231.
- Li D, Sun H. TEP1, encoded by a candidate tumor suppressor locus is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57(11):2124–2129.
- Myers M, Stolarov JP, Eng C, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94(17):9052–9057.
- Yehia L, Ngeow J, Eng C. PTEN-opathies: from biological insights to evidence-based precision medicine. J Clin Invest. 2019;129(2):452–464.
- Funamoto S, Meili R, Lee S, et al. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 2002;109(5):611–623.
- (Baltimore, Md.: 1950), Sahin E, Haubenwallner S, Kuttke M, et al. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol. 2014;193(4):1717–1727.
- Park M, Sheng R, Silkov A, et al. SH2 domains serve as lipid-binding modules for pTyr-signaling proteins. Mol Cell. 2016;62(1):7–20.
- Sly L, Ho V, Antignano F, et al. The role of SHIP in macrophages. Front Biosci. 2007;12(8–12):2836–2848.
- Peng K, Gu J-F, Su S-L, et al. Salvia miltiorrhiza stems and leaves total phenolic acids combination with tanshinone protect against DSS-induced ulcerative colitis through inhibiting TLR4/PI3K/AKT/mTOR signaling pathway in mice. J Ethnopharmacol. 2021;264:113052.
- Huang J, Ding Y, Huang D-S, et al. RNA interference targeting p110β reduces tumor necrosis factor-alpha production in cellular response to wear particles in vitro and osteolysis in vivo. Inflammation. 2013;36(5):1041–1054.
- Molnarfi N, Brandt KJ, Gruaz L, et al. Differential regulation of cytokine production by PI3Kdelta in human monocytes upon acute and chronic inflammatory conditions. Mol Immunol. 2008;45(12):3419–3427.
- Takeda A, Maher TJ, Zhang Y, et al. Human PI3Kγ deficiency and its microbiota-dependent mouse model reveal immunodeficiency and tissue immunopathology. Nat Commun. 2019;10(1):4364.
- Luo L, Wall AA, Tong SJ, et al. TLR crosstalk activates LRP1 to recruit Rab8a and PI3Kγ for suppression of inflammatory responses. Cell Rep. 2018;24(11):3033–3044.
- Dale M, Ruhlman M, Baxter B. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol. 2015;35(8):1746–1755.
- Abbas A, Murphy K, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383(6603):787–793.
- discussion 154-8: Mosmann T, Li L, Hengartner H, et al. Differentiation and functions of T cell subsets. Ciba Found Symp. 1997;204:148–154.
- (Hoboken N.J.), Saadoun D, Garrido M, Comarmond C, et al. Th1 and Th17 cytokines drive inflammation in Takayasu arteritis. Arthritis Rheumatol. 2015;67(5):1353–1360.
- Wang L, Gao S, Xu W, et al. Allergic asthma accelerates atherosclerosis dependent on Th2 and Th17 in apolipoprotein E deficient mice. J Mol Cell Cardiol. 2014;72:20–27.
- Yu W, Du H, Fu Q, et al. The influence of Th1/Th2 and CD4+ regulatory t cells of mesenteric lymph nodes on systemic lipopolysaccharide. POL J PATHOL. 2014;65(2):125–129.
- Shimizu K, Shichiri M, Libby P, et al. Th2-predominant inflammation and blockade of IFN-gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114(2):300–308.
- Schönbeck U, Sukhova GK, Gerdes N, et al. T(H)2 predominant immune responses prevail in human abdominal aortic aneurysm. Am J Pathol. 2002;161(2):499–506.
- Galle C, Schandené L, Stordeur P, et al. Predominance of type 1 CD4+ T cells in human abdominal aortic aneurysm. Clin Exp Immunol. 2005;142(3):519–527.
- Téo F, de Oliveira RTD, Villarejos L, et al. Characterization of CD4 + T cell subsets in patients with abdominal aortic aneurysms. Mediators Inflamm. 2018;2018:6967310.
- Huang L, Wang M, Yan Y, et al. OX40L induces helper T cell differentiation during cell immunity of asthma through PI3K/AKT and P38 MAPK signaling pathway. J Transl Med. 2018;16(1):74.
- (Baltimore, Md.: 1950), Kuroda E, Antignano F, Ho VW, et al. SHIP represses Th2 skewing by inhibiting IL-4 production from basophils. J Iimmunol. 2011;186(1):323–332.
- (Baltimore, Md.: 1950), Okkenhaug K, Patton DT, Bilancio A, et al. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Iimmunol. 2006;177(8):5122–5128.
- Ailawadi G, Moehle CW, Pei H, et al. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. 2009;138(6):1392–1399.
- Rombouts K, van Merrienboer TAR, Ket JCF, et al. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest. 2021;52(4):e13697.
- Gurung R, Choong AM, Woo CC, et al. Genetic and epigenetic mechanisms underlying vascular smooth muscle cell phenotypic modulation in abdominal aortic aneurysm. Int J Mol Sci. 2020;21(17):6334.
- Lu H, Du W, Ren L, et al. Vascular smooth muscle cells in aortic aneurysm: from genetics to mechanisms. J Am Heart Assoc. 2021;10(24):e023601.
- Spin J, Maegdefessel L, Tsao P. Vascular smooth muscle cell phenotypic plasticity: focus on chromatin remodelling. Cardiovasc Res. 2012;95(2):147–155.
- Petsophonsakul P, Furmanik M, Forsythe R, et al. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2019;39(7):1351–1368.
- Basatemur G, Jørgensen HF, Clarke MCH, et al. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. 2019;16(12):727–744.
- Sorokin V, Vickneson K, Kofidis T, et al. Role of vascular smooth muscle cell plasticity and interactions in vessel wall inflammation. Front Immunol. 2020;11:599415.
- Alencar G, Owsiany KM, Karnewar S, et al. Stem cell pluripotency genes Klf4 and oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation. 2020;142(21):2045–2059.
- Shankman L, Gomez D, Cherepanova OA, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21(6):628–637.
- Sun Y, Zheng B, Zhang X-H, et al. PPAR-γ agonist stabilizes KLF4 protein via activating Akt signaling and reducing KLF4 ubiquitination. Biochem Biophys Res Commun. 2014;443(2):382–388.
- Tang Y, Huang Q, Liu C, et al. p22phox promotes Ang-II-induced vascular smooth muscle cell phenotypic switch by regulating KLF4 expression. Biochem Biophys Res Commun. 2019;514(1):280–286.
- Garg A, Devereaux PJ, Hill A, et al. Oral curcumin in elective abdominal aortic aneurysm repair: a multicentre randomized controlled trial. CMAJ = Journal de l’Association Medicale Canadienne. 2018;190(43):E1273–E1280.
- Hao Q, Chen X, Wang X, et al. Curcumin attenuates angiotensin II-induced abdominal aortic aneurysm by inhibition of inflammatory response and ERK signaling pathways. Evid Based Complement Alternat Med. 2014;2014:270930.
- Xiong J, Liu H, Chen J, et al. Curcumin nicotinate suppresses abdominal aortic aneurysm pyroptosis via lncRNA PVT1/miR-26a/KLF4 axis through regulating the PI3K/AKT signaling pathway. Toxicol Res (Camb). 2021;10(3):651–661.
- Kansakar U, Jankauskas SS, Gambardella J, et al. Targeting the phenotypic switch of vascular smooth muscle cells to tackle atherosclerosis. Atherosclerosis. 2021;324:117–120.
- Zhang H, Jiang L, Guo Z, et al. PPARβ/δ, a novel regulator for vascular smooth muscle cells phenotypic modulation and vascular remodeling after subarachnoid hemorrhage in rats. Sci Rep. 2017;7(1):45234.
- Yang X, Gong Y, Tang Y, et al. Spry1 and Spry4 differentially regulate human aortic smooth muscle cell phenotype via Akt/FoxO/myocardin signaling. PloS one. 2013;8(3):e58746.
- Liu Z, Wang Z, Yanagisawa H, et al. Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev Cell. 2005;9(2):261–270.
- López-Candales A, Holmes DR, Liao S, et al. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150(3):993–1007.
- Henderson E, Geng Y-J, Sukhova GK, et al. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99(1):96–104.
- Wang Q, Shu C, Su J, et al. A crosstalk triggered by hypoxia and maintained by MCP-1/miR-98/IL-6/p38 regulatory loop between human aortic smooth muscle cells and macrophages leads to aortic smooth muscle cells apoptosis via Stat1 activation. Int J Clin Exp Pathol. 2015;8(3):2670–2679.
- Yamanouchi D, Morgan S, Kato K, et al. Effects of caspase inhibitor on angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2010;30(4):702–707.
- Morgan S, Yamanouchi D, Harberg C, et al. Elevated protein kinase C-δ contributes to aneurysm pathogenesis through stimulation of apoptosis and inflammatory signaling. Arterioscler Thromb Vasc Biol. 2012;32(10):2493–2502.
- Xue M, Li G, Li D, et al. Up-regulated MCPIP1 in abdominal aortic aneurysm is associated with vascular smooth muscle cell apoptosis and MMPs production. Biosci Rep. 2019;39(11).
- (London, England: 1979), Emeto T, Moxon J, Au M, et al. Oxidative stress and abdominal aortic aneurysm: potential treatment targets. Clin Sci. 2016;130(5):301–315.
- Sawada H, Hao H, Naito Y, et al. Aortic iron overload with oxidative stress and inflammation in human and murine abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35(6):1507–1514.
- Kaneko H, Anzai T, Morisawa M, et al. Resveratrol prevents the development of abdominal aortic aneurysm through attenuation of inflammation, oxidative stress, and neovascularization. Atherosclerosis. 2011;217(2):350–357.
- Thomas M, Gavrila D, McCormick ML, et al. Deletion of p47 phox attenuates angiotensin II–Induced abdominal aortic aneurysm formation in apolipoprotein E–Deficient mice. Circulation. 2006;114(5):404–413.
- Maiellaro-Rafferty K, Weiss D, Joseph G, et al. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am J Physiol Heart Circ Physiol. 2011;301(2):H355–62.
- Parastatidis I, Weiss D, Joseph G, et al. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2013;33(10):2389–2396.
- Tsai Y, Yeh H, Chao C, et al. Superoxide dismutase 2 (SOD2) in vascular calcification: a focus on vascular smooth muscle cells, calcification pathogenesis. And Therapeutic Strategies. Oxidative Medicine and Cellular Longevity. 2021;2021:6675548.
- Zhang F, Ren X, Zhao M, et al. Angiotensin-(1-7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci Rep. 2016;6(1):34621.
- Almajdoob S, Hossain E, Anand-Srivastava MB. Resveratrol attenuates hyperproliferation of vascular smooth muscle cells from spontaneously hypertensive rats: role of ROS and ROS-mediated cell signaling. Vascul Pharmacol. 2018;101:48–56.
- (London, England: 1979), Navas-Madroñal M, Rodriguez C, Kassan M, et al. Enhanced endoplasmic reticulum and mitochondrial stress in abdominal aortic aneurysm. Clin Sci. 2019;133(13):1421–1438.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075.
- Ren J, Zhang Y. Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol Sci. 2018;39(12):1064–1076.
- Shirakabe A, Ikeda Y, Sciarretta S, et al. Aging and autophagy in the heart. Circ Res. 2016;118(10):1563–1576.
- Phadwal K, Feng D, Zhu D, et al. Autophagy as a novel therapeutic target in vascular calcification. Pharmacol Ther. 2020;206:107430.
- Lu H, Sun J, Liang W, et al. Cyclodextrin prevents abdominal aortic aneurysm via activation of vascular smooth muscle cell transcription factor EB. Circulation. 2020;142(5):483–498.
- Clément M, Chappell J, Raffort J, et al. Vascular smooth muscle cell plasticity and autophagy in dissecting aortic aneurysms. Arterioscler Thromb Vasc Biol. 2019;39(6):1149–1159.
- Widmer R, Lerman A. Endothelial dysfunction and cardiovascular disease. Glob Cardiol Sci Pract. 2014;2014(3):291–308.
- Murad F. Shattuck lecture. nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006;355(19):2003–2011.
- Furchgott R. The 1996 Albert Lasker medical research awards. the discovery of endothelium-derived relaxing factor and its importance in the identification of nitric oxide. JAMA. 1996;276(14):1186–1188.
- DeRoo E, Stranz A, Yang H, et al. Endothelial dysfunction in the pathogenesis of abdominal aortic aneurysm. Biomolecules. 2022;12(4):509.
- Sung S, Wu T-C, Chen J-S, et al. Reduced number and impaired function of circulating endothelial progenitor cells in patients with abdominal aortic aneurysm. Int J Cardiol. 2013;168(2):1070–1077.
- Lee R, Bellamkonda K, Jones A, et al. Flow mediated dilatation and progression of abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 2017;53(6):820–829.
- Lee R, Jarchi D, Perera R, et al. Applied machine learning for the prediction of growth of abdominal aortic aneurysm in humans. EJVES Short Rep. 2018;39:24–28.
- Sun J, Deng H, Zhou Z, et al. Endothelium as a potential target for treatment of abdominal aortic aneurysm. Oxid Med Cell Longev. 2018;2018:6306542.
- Ahmed R, Ghoorah K, Kunadian V. Abdominal aortic aneurysms and risk factors for adverse events. Cardiol Rev. 2016;24(2):88–93.
- Barua R, Ambrose JA, Srivastava S, et al. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: an in vitro demonstration in human coronary artery endothelial cells. Circulation. 2003;107(18):2342–2347.
- Baxter B, Terrin M, Dalman R. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117(14):1883–1889.
- (Dallas, Tex.: 1979), Gao L, Siu KL, Chalupsky K, et al. Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: treatment with folic acid. Hypertension. 2012;59(1):158–166.
- Li Q, Youn JY, Siu KL, et al. Knockout of dihydrofolate reductase in mice induces hypertension and abdominal aortic aneurysm via mitochondrial dysfunction. Redox Biol. 2019;24:101185.
- Siu K, Li Q, Zhang Y, et al. NOX isoforms in the development of abdominal aortic aneurysm. Redox Biol. 2017;11:118–125.
- Huang K, Wang Y, Siu KL, et al. Targeting feed-forward signaling of TGFβ/NOX4/DHFR/eNOS uncoupling/TGFβ axis with anti-TGFβ and folic acid attenuates formation of aortic aneurysms: novel mechanisms and therapeutics. Redox Biol. 2021;38:101757.
- Zhou Z, Xie X-L, Zhou S-F, et al. Mechanism of reversal of high glucose-induced endothelial nitric oxide synthase uncoupling by tanshinone IIA in human endothelial cell line EA.hy926. Eur J Pharmacol. 2012;697(1–3):97–105.
- McCormick M, Gavrila D, Weintraub N. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2007;27(3):461–469.
- Li X, Huang S, Zhuo B, et al. κComparison of three species of rhubarb in inhibiting vascular endothelial injury via regulation of PI3K/AKT/NF-B signaling pathway. Oxid Med Cell Longev. 2022;2022:8979329.
- Hu C, Zhang X, Teng T, et al. Cellular senescence in cardiovascular diseases: a systematic review. Aging Dis. 2022;13(1):103–128.
- Wilcox J, Waksman R, King SB, et al. The role of the adventitia in the arterial response to angioplasty: the effect of intravascular radiation. Int J Radiat Oncol Biol Phys. 1996;36(4):789–796.
- Shi Y, O’Brien JE, Fard A, et al. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation. 1996;94(7):1655–1664.
- Scott N, Cipolla GD, Ross CE, et al. Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries. Circulation. 1996;93(12):2178–2187.
- Maiellaro K, Taylor W. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75(4):640–648.
- Gao J, Guo W. Mechanisms of abdominal aortic aneurysm progression: a review. Vasc Med. 2022;27(1):88–96.
- Feng E, Wang J, Wang X, et al. Inhibition of HMGB1 might enhance the protective effect of taxifolin in cardiomyocytes via PI3K/AKT signaling pathway. Iran J Pharm Res. 2021;20(2):316–332.
- Bumdelger B, Otani M, Karasaki K, et al. Disruption of osteoprotegerin has complex effects on medial destruction and adventitial fibrosis during mouse abdominal aortic aneurysm formation. PloS one. 2020;15(7):e0235553.
- van Keulen C, van de Akker E, Pals G, et al. The role of type III collagen in the development of familial abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 1999;18(1):65–70.
- van Keulen C, van den Akker E, van den Berg FG, et al. The role of type III collagen in family members of patients with abdominal aortic aneurysms. Eur J Vasc Endovasc Surg. 2000;20(4):379–385.
- Haurani M, Pagano P. Adventitial fibroblast reactive oxygen species as autacrine and paracrine mediators of remodeling: bellwether for vascular disease? Cardiovasc Res. 2007;75(4):679–689.
- Chiang M, Chen I-M, Hsu -F-F, et al. Gal-1 (Galectin-1) upregulation contributes to abdominal aortic aneurysm progression by enhancing vascular inflammation. Arterioscler Thromb Vasc Biol. 2021;41(1):331–345.
- Hermenean A, Oatis D, Herman H, et al. Galectin 1-A key player between tissue repair and fibrosis. Int J Mol Sci. 2022;23(10):5548.
- Romaniuk M, Croci D, Lapponi M, et al. Binding of galectin-1 to αIIbβ₃ integrin triggers “outside-in” signals, stimulates platelet activation. And Controls Primary Hemostasis. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2012;26(7):2788–2798.
- Tsuruda T, Kato J, Hatakeyama K, et al. Adrenomedullin in mast cells of abdominal aortic aneurysm. Cardiovasc Res. 2006;70(1):158–164.
- Tsuruda T, Kato J, Hatakeyama K, et al. Adventitial mast cells contribute to pathogenesis in the progression of abdominal aortic aneurysm. Circ Res. 2008;102(11):1368–1377.
- Veerappan A, Reid AC, Estephan R, et al. Mast cell renin and a local renin-angiotensin system in the airway: role in bronchoconstriction. Proc Natl Acad Sci U S A. 2008;105(4):1315–1320.
- Miyazaki M, Takai S, Jin D, et al. Pathological roles of angiotensin II produced by mast cell chymase and the effects of chymase inhibition in animal models. Pharmacol Ther. 2006;112(3):668–676.
- Owen CA, Campbell EJ. Angiotensin II generation at the cell surface of activated neutrophils: novel cathepsin G-mediated catalytic activity that is resistant to inhibition. J Immunol. 1998;160(3):1436–1443.
- Swedenborg J, Mäyränpää MI, Kovanen PT. Mast cells: important players in the orchestrated pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2011;31(4):734–740.
- Kovanen PT. Mast cells: multipotent local effector cells in atherothrombosis. Immunol Rev. 2007;217(1):105–122.
- Leskinen MJ, Heikkila H, Speer M, et al. Mast cell chymase induces smooth muscle cell apoptosis by disrupting NF-kappaB-mediated survival signaling. Exp Cell Res. 2006;312(8):1289–1298.
- Wang Z, Guo J, Han X, et al. Metformin represses the pathophysiology of AAA by suppressing the activation of PI3K/AKT/mTOR/autophagy pathway in ApoE(-/-) mice. Cell Biosci. 2019;9(1):68.
- Wang J, Zhou Y, Wu S, et al. Astragaloside IV attenuated 3,4-benzopyrene-induced abdominal aortic aneurysm by ameliorating macrophage-mediated inflammation. Front Pharmacol. 2018;9:496.
- Takahara Y, Tokunou T, Ichiki T. Suppression of abdominal aortic aneurysm formation in mice by teneligliptin, a dipeptidyl peptidase-4 inhibitor. J Atheroscler Thromb. 2018;25(8):698–708.
- Satta J, Läärä E, Juvonen T. Intraluminal thrombus predicts rupture of an abdominal aortic aneurysm. J Vasc Surg. 1996;23(4):737–739.
- Parr A, McCann M, Bradshaw B, et al. Thrombus volume is associated with cardiovascular events and aneurysm growth in patients who have abdominal aortic aneurysms. J Vasc Surg. 2011;53(1):28–35.
- Sagan A, Mrowiecki W, Mikolajczyk T, et al. Local inflammation is associated with aortic thrombus formation in abdominal aortic aneurysms. relationship to clinical risk factors. Thromb Haemost. 2012;108(5):812–823.
- Vorp D, Lee PC, Wang DHJ, et al. Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening. J Vasc Surg. 2001;34(2):291–299.
- Swedenborg J, Eriksson P. The intraluminal thrombus as a source of proteolytic activity. Ann N Y Acad Sci. 2006;1085(1):133–138.
- Kazi M, Thyberg J, Religa P, et al. Influence of intraluminal thrombus on structural and cellular composition of abdominal aortic aneurysm wall. J Vasc Surg. 2003;38(6):1283–1292.
- Dai J, Louedec L, Philippe M, et al. Effect of blocking platelet activation with AZD6140 on development of abdominal aortic aneurysm in a rat aneurysmal model. J Vasc Surg. 2009;49(3):719–727.
- Min S, Abrams C. Regulation of platelet plug formation by phosphoinositide metabolism. Blood. 2013;122(8):1358–1365.
- Watanabe N, Nakajima H, Suzuki H, et al. Functional phenotype of phosphoinositide 3-kinase p85alpha-null platelets characterized by an impaired response to GP VI stimulation. Blood. 2003;102(2):541–548.
- Kim S, Mangin P, Dangelmaier C, et al. Role of phosphoinositide 3-kinase beta in glycoprotein VI-mediated Akt activation in platelets. J Biol Chem. 2009;284(49):33763–33772.
- Gilio K, Munnix ICA, Mangin P, et al. Non-redundant roles of phosphoinositide 3-kinase isoforms alpha and beta in glycoprotein VI-induced platelet signaling and thrombus formation. J Biol Chem. 2009;284(49):33750–33762.
- Laurent P, Hechler B, Solinhac R, et al. Impact of PI3Kα (phosphoinositide 3-kinase alpha) inhibition on hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2018;38(9):2041–2053.
- Senis Y, Atkinson BT, Pearce AC, et al. Role of the p110delta PI 3-kinase in integrin and ITAM receptor signalling in platelets. Platelets. 2005;16(3–4):191–202.
- Hirsch E, Bosco O, Tropel P, et al. Resistance to thromboembolism in PI3Kgamma-deficient mice. FASEB J. 2001;15(11):2019–2021.
- Lian L, Wang Y, Draznin J, et al. The relative role of PLCbeta and PI3Kgamma in platelet activation. Blood. 2005;106(1):110–117.
- Jackson S, Schoenwaelder SM, Goncalves I, et al. PI 3-kinase p110beta: a new target for antithrombotic therapy. Nat Med. 2005;11(5):507–514.
- Ohlsson C, Langenskiöld M, Smidfelt K, et al. Low progesterone and low estradiol levels associate with abdominal aortic aneurysms in men. J Clin Endocrinol Metab. 2022;107(4):e1413–e1425.
- Schiff R, Massarweh SA, Shou J, et al. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004;10(1 Pt 2):331s–6s.
- Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529.
- Thangavel C, Dean JL, Ertel A, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. 2011;18(3):45–333.
- du Rusquec P, Blonz C, Frenel JS, et al. Targeting the PI3K/Akt/mTOR pathway in estrogen-receptor positive HER2 negative advanced breast cancer. Ther Adv Med Oncol. 2020;12:1758835920940939.
- Brandão M, Caparica R, Eiger D, et al. Biomarkers of response and resistance to PI3K inhibitors in estrogen receptor-positive breast cancer patients and combination therapies involving PI3K inhibitors. Ann Oncol. 2019;30(Suppl_10):x27–x42.
- Zhao X, Huang L, Yin Y, et al. Estrogen induces endothelial progenitor cells proliferation and migration by estrogen receptors and PI3K-dependent pathways. Microvasc Res. 2008;75(1):45–52.
- Zhang T, Liang X, Shi L, et al. Estrogen receptor and PI3K/Akt signaling pathway involvement in S-(-)equol-induced activation of Nrf2/ARE in endothelial cells. PLoS One. 2013;8(11):e79075.
- Hohmann N, Xia N, Steinkamp-Fenske K, et al. Estrogen receptor signaling and the PI3K/Akt pathway are involved in betulinic acid-induced eNOS activation. Molecules. 2016;21(8):973.
- Yuan Z, Heng Z, Lu Y, et al. The protective effect of metformin on abdominal aortic aneurysm: a systematic review and meta-analysis. Front Endocrinol (Lausanne). 2021;12:721213.
- Shah A, Langenberg C, Rapsomaniki E, et al. Type 2 diabetes and incidence of cardiovascular diseases: a cohort study in 1·9 million people. The Lancet Diabetes & Endocrinology. 2015;3(2):105–113.
- Golledge J, Krishna S, and Wang Y. Mouse models for abdominal aortic aneurysm. Br J Pharmacol. 2022;179(5):792–810.
- Ni X, Zhang Y-R, Jia L-X, et al. Inhibition of notch1-mediated inflammation by intermedin protects against abdominal aortic aneurysm via PI3K/Akt signaling pathway. Aging (Albany NY). 2021;13(4):5164–5184.
- Daugherty A, Manning M, Cassis L. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105(11):1605–1612.
- Saraff K, Babamusta F, Cassis LA, et al. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. arteriosclerosis, thrombosis. And Vascular Biology. 2003;23(9):1621–1626.
- Thompson R, Curci JA, Ennis TL, et al. Pathophysiology of abdominal aortic aneurysms: insights from the elastase-induced model in mice with different genetic backgrounds. Ann N Y Acad Sci. 2006;1085(1):59–73.
- Busch A, Holm A, Wagner N, et al. Extra- and intraluminal elastase induce morphologically distinct abdominal aortic aneurysms in mice and thus represent specific subtypes of human disease. J Vasc Res. 2016;53(1–2):49–57.
- Lu G, Su G, Davis JP, et al. A novel chronic advanced stage abdominal aortic aneurysm murine model. J Vasc Surg. 2017;66(1):232–242.e4.
- Phillips E, Yrineo AA, Schroeder HD, et al. Morphological and biomechanical differences in the elastase and AngII apoE −/− rodent models of abdominal aortic aneurysms. Biomed Res Int. 2015;2015:413189.
- Wang Y, Krishna S, Golledge J. The calcium chloride-induced rodent model of abdominal aortic aneurysm. Atherosclerosis. 2013;226(1):29–39.
- Chen H, Wang F, Gao P, et al. Age-associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res. 2016;119(10):1076–1088.
- Liu C, Wang Y, Liao M, et al. Allergic lung inflammation aggravates angiotensin II-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2016;36(1):69–77.
- Li G, Qin L, Wang L, et al. Inhibition of the mTOR pathway in abdominal aortic aneurysm: implications of smooth muscle cell contractile phenotype, inflammation, and aneurysm expansion. Am J Physiol Heart Circ Physiol. 2017;312(6):H1110–H1119.
- Yu J, Liu R, Huang J, et al. Inhibition of phosphatidylinositol 3-kinease suppresses formation and progression of experimental abdominal aortic aneurysms. Sci Rep. 2017;7(1):15208.
- Escudero P, Navarro A, Ferrando C, et al. Combined treatment with bexarotene and rosuvastatin reduces angiotensin-II-induced abdominal aortic aneurysm in apoE −/− mice and angiogenesis. Br J Pharmacol. 2015;172(12):2946–2960.
- Zhang S, Kan X, Li Y, et al. Deficiency of γδT cells protects against abdominal aortic aneurysms by regulating phosphoinositide 3-kinase/AKT signaling. J Vasc Surg. 2018;67(3):899–908.e1.
- Rabkin S. The effect of nicotine and tobacco on aortic matrix metalloproteinases in the production of aortic aneurysm. Curr Vasc Pharmacol. 2016;14(6):514–522.
- Yu M, Chen C, Cao Y, et al. Inhibitory effects of doxycycline on the onset and progression of abdominal aortic aneurysm and its related mechanisms. Eur J Pharmacol. 2017;811:101–109.
- (Georgetown, Tex.), Tian Y, Li X, Bai C, et al. MiR-17-5p promotes the endothelialization of endothelial progenitor cells to facilitate the vascular repair of aneurysm by regulating PTEN-mediated PI3K/AKT/VEGFA pathway. Cell Cycle. 2020;19(24):3608–3621.
- Zhou Y, Wang M, Zhang J, et al. MicroRNA-29a-3p regulates abdominal aortic aneurysm development and progression via direct interaction with PTEN. J Cell Physiol. 2020;235(12):9414–9423.
- Li K, Cui M-Z, Zhang K-W, et al. Effect of miR-21 on rat thoracic aortic aneurysm model by regulating the expressions of MMP-2 and MMP-9. Eur Rev Med Pharmacol Sci. 2020;24(2):878–884.
- Maegdefessel L, Azuma J, Toh R, et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci Transl Med. 2012;4(122):122ra22.
- Zhao L, Huang J, Zhu Y, et al. miR-33-5p knockdown attenuates abdominal aortic aneurysm progression via promoting target adenosine triphosphate-binding cassette transporter A1 expression and activating the PI3K/Akt signaling pathway. Perfusion2019;35(1):57–65.
- Chen S, Chen H, Yu C, et al. Long noncoding RNA myocardial infarction associated transcript promotes the development of thoracic aortic by targeting microRNA-145 via the PI3K/Akt signaling pathway. J Cell Biochem. 2019;120(9):14405–14413.
- Ma X, Yao H, Yang Y, et al. miR-195 suppresses abdominal aortic aneurysm through the TNF-alpha/NF-kappaB and VEGF/PI3K/Akt pathway. Int J Mol Med. 2018;41(4):2350–2358.
- J P, He X, Zhang L, et al. MicroRNA‑26a protects vascular smooth muscle cells against H2O2‑induced injury through activation of the PTEN/AKT/mTOR pathway. Int J Mol Med. 2018;42(3):1367–1378.
- Licholai S, Szczeklik W, Sanak M. miR-29c-3p is an effective biomarker of abdominal aortic aneurysm in patients undergoing elective surgery. MicroRNA (Shariqah United Arab Emir). 2016;5(2):124–131.
- Shen G, Sun Q, Yao Y, et al. Role of ADAM9 and miR-126 in the development of abdominal aortic aneurysm. Atherosclerosis. 2020;297:47–54.
- Li B, Wang Z, Chen R, et al. Up regulation of isoleucyl-tRNA synthetase promotes vascular smooth muscle cells dysfunction via p38 MAPK/PI3K signaling pathways. Life Sci. 2019;224:51–57.