
ABSTRACT
The cGAS/STING signaling pathway is an important part of the cytoplasmic DNA sensor, which can trigger a type I interferon response to microbial infection when pathogenic DNA is detected. However, continuous inhibition of cGAS/STING signaling by viral infection may be an important cause of tumorigenesis. At the same time, recent studies have shown that although the cGAS/STING signaling pathway also plays a core role in anti-tumor immunity and cell senescence, the inflammatory response induced by cGAS/STING signaling will also promote tumorigenesis in different backgrounds. Here, we discuss the role of cGAS/STING in the context of infection, senescence, and tumors, especially with respect to progression, to facilitate a better understanding of the mechanism of the cGAS/STING pathway.
KEYWORDS:
Introduction
Quickly identifying and removing the invading pathogenic microorganisms are important steps for maintaining homeostasis. PRRs are a key part of the innate immune system and play an important role in detecting viral nucleic acid in the body [Citation1,Citation2]. PRRs s are expressed in cell membranes, organelle membranes, and even serum and are widespread in mammals [Citation3]. Its function is to initiate innate immunity by recognizing viral PAMPs and host-derived DAMP [Citation4]. When pathogens enter the cell to replicate, cytoplasmic biosensors participate in the immune response according to the properties of pathogen-related molecular patterns, one of which is cGAS. As a cytosolic DNA sensor, it can activate the innate immune response [Citation5,Citation6].
Because cGAS binds to DNA using positively charged residues, cGAS can be activated by any dsDNA in a sequence-independent manner [Citation7], leading to DNA accumulation caused by senescence or a tumor, which also activates the cGAS/STING signaling pathway [Citation8]. Through the induction of this abnormal DNA, cGAS can induce senescent cell death or activate the body’s strong immune response to inhibit tumor growth [Citation9]. Therefore, a variety of tumor treatments based on the cGAS/STING signaling pathway have emerged. However, according to in-depth research, cGAS/STING signaling may not only have a protective function in the process of tumorigenesis but also promote tumorigenesis in special circumstances, indicating that cGAS/STING signaling has dual roles in the regulation of the body. Therefore, in this paper, I review research progress and the application of the cGAS/STING signaling pathway in infection, senescence, and tumors in recent years to facilitate a better understanding of the role of cGAS/STING signaling.
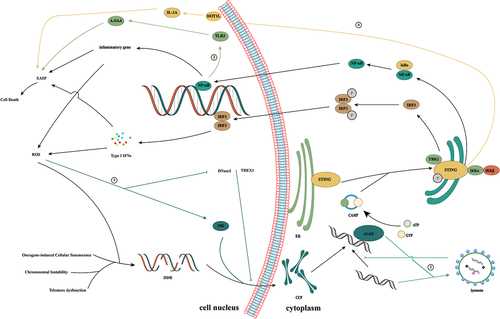
Activation of the cGAS/STING signaling ()
The cGAS contains approximately 520 amino acids and consists of a catalytic domain and an extended amino-terminal domain. The catalytic domain has a double-leaf folded structure and contains three DNA binding sites [Citation5]. It has been reported that cGAS is enriched in centromeric satellite DNA and linear DNA repeats [Citation10]. In the nucleosome, cGAS is immobilized by acidic plaques composed of H2A-H2B, and through this interaction, the cGAS-DNA binding site is buried, and cGAS is simultaneously locked into a monomeric state, thereby preventing the formation of active cGAS dimers [Citation11–15]. This explains why cGAS is not abnormally activated in the nucleus when cGAS can not specifically recognize DNA sequences. During mitosis, with the rupture of the nuclear membrane, cGAS is rapidly transferred to the chromosome. During mitosis, the CDK1/cyclin B complex inhibits the synthesis of cGAMP by phosphorylating human cGAS at S305.In addition, cGAS is dephosphorylated by protein phosphatase-1 after mitosis, which restores its DNA-sensing capacity [Citation16].
Figure 1. Activation of the cGAS/STING signaling pathway. cGAS is a cytoplasmic nucleic acid sensor that recognizes double-stranded DNA from tumor cells, bacteria, and viruses in the cytoplasm. By binding to heterologous DNA to form a dimer, it induces a conformational change in itself, thereby promoting the conversion of ATP and GTP to cGAMP. As a second messenger, cGAMP binds to STING anchored in the endoplasmic reticulum, induces its conformational change, and promotes the transfer of STING from the endoplasmic reticulum to the Golgi. Subsequent recruitment of TBK1 induces IRF3 phosphorylation, which elicits type 1 IFN-mediated immune responses. In addition, STING can also activate IκB kinase, phosphorylate IκB, release NF-κB into the nucleus, and induce the expression of IFN and inflammatory cytokines. ①: Ribonucleoprotein A2B1 promotes the nucleocytoplasmic transport of cGAS, IFI16, and STING through N6-methyladenosine modification, thereby forming a positive feedback loop for TANK/ TBK1/ IRF3 activation. ② the Yip family and STING ER exit protein recruit STING to COPII. STEEP promotes VPS34 complex-mediated phosphatidylinositol-3-phosphate production and ER membrane bending by binding to STING and recruits the curvature-binding protein Sar1 to promote COPII-mediated ER transport of STING to the Golgi apparatus. ③: TBK1 enhances the affinity of p62 for ubiquitinated STING through the phosphorylation of p62 and promotes its transport to autophagosomes for degradation. ④: the interferon gamma-inducible protein 16 acts on STING through the pyrin domain and cooperates with cGAMP to promote the phosphorylation and translocation of STING.
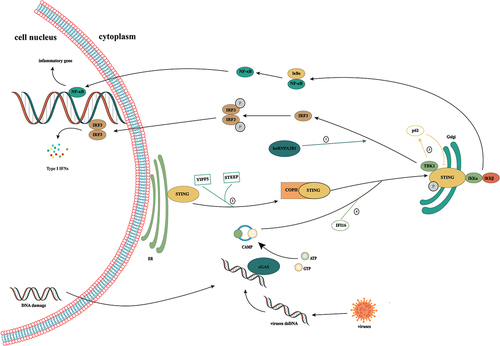
The process of DNA recognition by cGAS causes conformational changes to promote the binding of cGAS to its substrate and catalyze the formation of a dimer complex between cGAS and DNA at a 2:2 ratio [Citation17]. DNA greater than 45 bp can promote the formation of a stable ladder network of cGAS dimers, thereby promoting the production of cGAMP [Citation18]. Recent studies have shown that K187 and L195 in human cGAS play important roles in the regulation of cGAS activity [Citation19]. They can specifically inhibit the formation of minimal cGAS/DNA complexes. When murine cGAS was replaced with human cGAS, the substitution of K187 and L195 blocks total cGAMP production, thereby inhibiting immune activation. Additionally, these residues significantly enhance the ability of cGAS to differentiate DNA in a length-dependent manner, which explains why the level of cGAMP synthesis by human cGAS is severely reduced compared to that of murine cGAS as well as the difference in DNA length between human and mouse cGAS.
cGAS dimers can catalyze the synthesis of the cyclic dinucleotide cGAMP [Citation20,Citation21]. Interestingly, the interaction between cGAS and DNA forms micron-sized droplets, which undergo a liquid-solid transformation to transition into a mature gel-like state, allowing the droplets to function as microreactors, thereby markedly increasing the yield of cGAMP [Citation22,Citation23]. Zinc ions promote the occurrence of this reaction [Citation22]. Additionally, this change also promotes cGAS resistance to negative regulation and effective DNA sensing [Citation24].
cGAMP binds to the STING protein at the ER. STING (also known as MITA, MPYS, ERIS and TMEM173) is a stimulator of interferon genes and is a natural immune sensor of cyclic dinucleotides [Citation25]. It consists of 379 amino acids, and the entire protein is composed of three structural domains, namely, the N-terminal transmembrane domain, cyclic dinucleotide binding domain, and C-terminal tail [Citation26,Citation27]. In the resting state, the STING protein exists in the form of homodimers and is anchored in the ER through the N-terminal transmembrane domain. cGAMP binding promotes conformational changes in STING, promoting the formation of STING dimers and ultimately tetramers and polymer complexes [Citation28,Citation29]. In addition, STING exits the ER in COPII vesicles from the ER-Golgi intermediate compartment [Citation30,Citation31] The Yip family and STING ER exit protein recruit STING to COPII [Citation32,Citation33] STEEP promotes VPS34 complex-mediated phosphatidylinositol-3-phosphate production and ER membrane bending by binding to STING and recruits the curvature-binding protein Sar1 to promote COPII-mediated ER transport of STING to the Golgi apparatus [Citation33].
In keratinocytes, the interferon gamma-inducible protein 16 acts on STING through the pyrin domain and cooperates with cGAMP to promote the phosphorylation and translocation of STING [Citation34]. Additionally, IFN-γ promotes the attachment of STAT1 to the cGAS sequence in the human STING promoter region through the IFN-γ/Janus kinase/STAT1 pathway, thereby promoting upregulation of STING expression [Citation35]. Ribonucleoprotein A2B1 promotes the nucleocytoplasmic transport of cGAS, IFI16, and STING through N6-methyladenosine modification, thereby forming a positive feedback loop for TBK1/IRF3 activation [Citation36]. In addition, studies have shown that changes in autophagy and calcium ions also regulate STING activity [Citation37,Citation38]. These results suggest that STING activity is regulated at multiple levels in the body.
After STING is transferred to the Golgi apparatus, it activates TBK1. Subsequently, TBK1 induces the phosphorylation of STING at Ser366 in a highly conserved sequence in the CTT region, thereby promoting the binding of STING and IRF3; it also induces IRF3 phosphorylation and dimer formation [Citation39,Citation40]. TBK1 can also play a negative feedback role in regulating STING activation. TBK1 enhances the affinity of p62 for ubiquitinated STING through the phosphorylation of p62 and promotes its transport to autophagosomes for degradation [Citation41].
After IRF3 dimers are formed, they are transported into the nucleus to trigger the production of type I interferon [Citation40]. Moreover, activated STING induces activation of the inhibitor of nuclear factor kappa-B kinase, thereby promoting NF-κB expression. As IRF3 and NF-κB enter the nucleus, they induce the expression of interferon-stimulated genes and inflammatory cytokines to jointly coordinate antiviral defense mechanisms [Citation42].
cGAS/STING promotes anti-tumor immunity by activating immune cells ()
Effective anti-tumor immunity depends on the cross-presentation of tumor antigens by APCs to CD8+T lymphocytes, and APC activation requires type I IFN signals activated by innate immune sensors [Citation43]. In anti-tumor immunity, cGAMP produced by tumor cells can be transferred to tumor-associated DCs through gap junctions and activate intracellular cGAS/STING signaling pathways, thus promoting the cross-presentation of tumor antigens and tumor-specific cell responses by APCs to CD8+T lymphocytes [Citation44,Citation45]. In addition, ROS produced by oxidative stress in the tumor microenvironment TME can promote SENP3 accumulation in DCs, and induce the interaction between SENP3 and IFI204 and IFI204 desulfonylation and thus promote activation of the STING signaling pathway [Citation46].
Figure 2. Activation and inhibition of cGAS/STING signaling in tumor immunity. In tumor immunity, dsDNA generated by gene replication of tumor cells can activate the cGAS/STING pathway, in which dsDNA can be delivered to APC cells through extracellular vesicles or gap junctions, and GAMP can be delivered to APC cells through cGAMP exporters. APC activates its own cGAS/STING by recognizing this dsDNA and cGAMP to induce a type I IFN-mediated immune response, thereby recruiting CD8+ cells and NK cells to mediate tumor cell apoptosis. ①: STING expression is epigenetically inhibited by histone H3K4 lysine demethylase KDM5B and KDM5C, which inhibit cGAS/STING signaling by maintaining low H3K4me3 levels in the STING promoter region. ②: high expression of DNMT1 and EZH2 was found to lead to significant silencing of STING expression and inhibition of cGAS/STING signaling. ③: receptor tyrosine kinase HER2 binds to the C-terminal domain of STING through its domain, recruits AKT1 to STING and phosphorylates TBK1S510 through AKT1 to prevent the TBK1/STING and TBK1K63-ubiquitin associations. ④: Tumor cells inhibit STING activation by upregulating exonuclease ENPP1 expression and hydrolyzing the immune transmitter cGAMP. ⑤: Tumor cells promote STING degradation in a SOX2 autophagy-dependent manner.
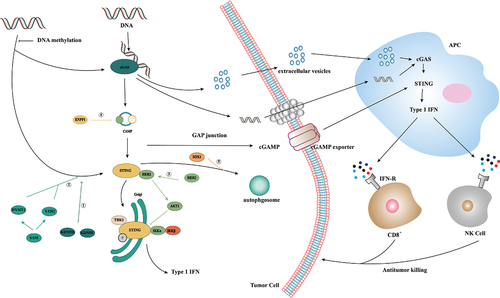
NK cells are involved in monitoring and eliminating cancer [Citation47]. The tumor rejection of NK cells largely depends on STING activation in non-tumor cells, and cGAS activation in tumor cells is a prerequisite for NK cells to produce anti-tumor immunity [Citation48]. In B16D8 tumors, STING in host immune cells and tumor cells can induce different chemokines, including CXCL10, CCL5, and IL-33, which can promote the infiltration and activation of NK cells in B16D8 tumors [Citation49]. Studies show that cGAS/STING induces activation of immune cells through the network of tumor and host cells, which contributes to the natural monitoring and inhibition of tumors in vivo.
Inhibition of cGAS/STING signaling is an important means for tumor cells to escape
Although cGAS/STING activation can effectively inhibit tumor growth, many reports have pointed out that this signaling pathway is inhibited in tumors, preventing the body from effectively activating the anti-tumor response [Citation45,Citation50]. Therefore, elucidation of the tumor escape mechanism can deepen our understanding of cGAS/STING.
Epigenetic modification of the cGAS/STING gene is an important mechanism for tumor escape. Loss-of-function mutation or epigenetic silencing in the promoter region of cGAS or STING has been observed in many cancers [Citation51]. For example, in melanoma and colon cancer, tumor cells’ epigenetic hypermethylation-induced silencing of STING and cGAS expression is an important means for tumor cells to escape from immune system recognition [Citation50,Citation52]. In breast cancer, STING expression is epigenetically inhibited by histone H3K4 lysine demethylase KDM5B and KDM5C, which inhibit cGAS/STING signaling by maintaining low H3K4me3 levels in the STING promoter region [Citation53]. In KRAS/LKB1 mutant lung cancer, high expression of DNMT1 and EZH2 was found to lead to significant silencing of STING expression and inhibition of cGAS/STING signaling [Citation54].
Abnormally activated cGAS in tumor cells can induce cGAMP production. Through paracrine signaling, cGAMP can be secreted into the TME and then recognized by immune cells to activate anti-tumor immunity. When this pathway is damaged, the body will not be able to produce an effective immune response; thus, inhibiting activation of cGAS/STING signaling is an important means for tumor cells to escape. Receptor tyrosine kinase HER2 binds to the C-terminal domain of STING through its domain, recruits AKT1 to STING, and phosphorylates TBK1 S510 through AKT1 to prevent the TBK1/STING and TBK1 K63-ubiquitin associations, thus weakening DNA induction and preventing tumor cell senescence and apoptosis [Citation55]. In the ALT+ cancer cell line, inhibiting STING expression is an important means of immune escape. ECTR is an important mechanism for ALT+ cancer cells to maintain telomere length. ECTR can effectively activate the cGAS/STING/TBK/IRF3 signaling axis in normal human fibroblasts, which triggers IFN-β production and the type I interferon response and leads to the emergence of the SASP, which leads to cell proliferation defects. However, this pathway is inhibited due to the inhibition of STING expression in ALT cancer cell lines [Citation56]. In the presence of CIN, tumor cells inhibit STING activation by upregulating exonuclease ENPP1 expression and hydrolyzing the immune transmitter cGAMP [Citation57]. In head and neck squamous cell carcinoma, tumor cells promote STING degradation in a SOX2 autophagy-dependent manner, thus inhibiting tumor immunity [Citation58].
These results suggest that inhibition of cGAS/STING signaling is an important means for tumor cells to escape the immune response, thus restoring the expression of cGAS/STING signaling as an important strategy for tumor treatment.
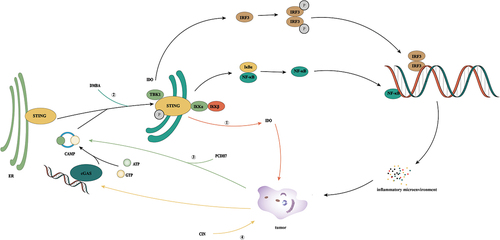
cGAS/STING signal activation promotes tumor immunity ()
Due to the widespread inhibition of cGAS/STING signaling in tumor cells, [Citation45,Citation50] activating the cGAS/STING signaling pathway through a variety of pathways is an important strategy for tumor therapy. One such pathway is an activation of the cGAS/STING signaling pathway by increasing DNA damage in tumor cells and inducing abnormal accumulation of DNA in the cytoplasm. For example, the telomere-targeting drug 6-thio-20-deoxyguanosine induces telomere dysfunction in tumor cells, resulting in telomere-associated DNA damage in tumor cells. When this DNA damage is absorbed by DCs, it can effectively activate the internal cGAS/STING pathway of host cells and enhance the cross-initiation ability of DCs and subsequent tumor-specific T cell activation [Citation59].
Figure 3. cGAS/STING signal activation promotes tumor immunity. Since cGAS-STING signaling is generally inhibited in tumor cells, activation of the cGAS-STING signaling pathway through multiple pathways is an important idea for tumor therapy. ① cdiGMP, c-diAmp,cgamp, and 2′, 3′-cGAMP are human natural cyclic dinucleotides (CDN), which can effectively induce inflammation and tumor necrosis by intratumoral injection. ② DSDP and BNBC are small-molecule STING agonists that can induce the activation of the cGAS/STING signaling pathway. ③ RR-S2 CDA and MLRR-S2 CDA are CDN derivatives, which have the same effect as natural CDA, but their stability in vivo is better. ④ It activates the cGAS-STING signaling pathway by increasing the DNA damage of tumor cells and inducing abnormal DNA aggregation in the cytoplasm. For example, telomere targeting drugs 6-thio-20-deoxyguanosine and doxorubicin are used.
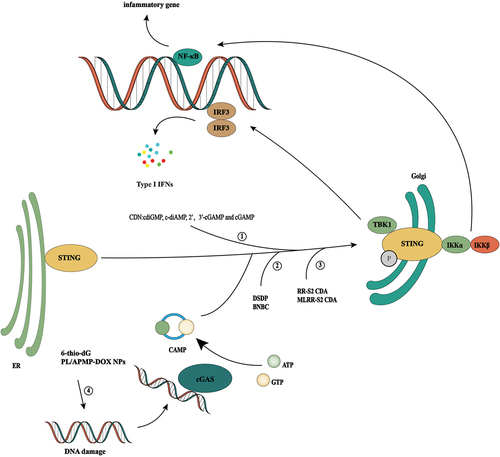
STING activation is an important step for the body to produce an interferon response; therefore, direct activation of STING is also an effective treatment. Four known natural agonists of human STING exist, which correspond to four known natural CDNs: cdiGMP, c-diAMP, 2′,3′-cGAMP, and cGAMP. Intratumoral injection of CDNs can effectively induce inflammation and tumor necrosis and can produce long-lasting tumor-specific adaptability [Citation60]. Intratumoral injection of CDNs can effectively induce inflammation and tumor necrosis and can produce long-lasting tumor-specific adaptability [Citation61,Citation62]. Intramuscular injections of cGAMP can stimulate the activation of DCs and their presentation of tumor antigens to CD8+T cells in a STING-dependent manner, thus significantly enhancing the anti-tumor effect of PD-L1 blockers [Citation63]. At the same time, activating intra-tumor STING can strongly induce the expression of type I and type II IFN in the TME in a CD8+T-dependent manner and promote activation of vascular stability genes (such as Cdh5, Angpt1, Pdgfrb, Mcam, and Col4a) and upregulation of adhesion molecules. Combining VEGFR2 and immune checkpoint inhibitors can effectively delay tumor growth and inhibit distant metastasis in a spontaneous breast cancer model [Citation64].
In addition to natural CDNs, a variety of CDN derivatives have also shown efficient STING activation. CDN derivatives with Rp, Rp dithiol diastereomer and a noncanonical c[A(2′,5′)pA(3′,5′)p] phosphate bridge structure can induce not only lymphocyte tumor infiltration by promoting the binding of human STING but also tumor regression in combination with PD-1 blockers. More importantly, compared with natural CDNs, these derivatives are more stable in vivo [Citation65]. MLRR-S2CDA is a CDN-derived molecule and compared with endogenous and pathogen-derived CDN, it shows lipophilicity and high stability. At the same time, it can activate all known human STING allele variants and produce effective anti-tumor effects on established B16 melanoma [Citation66].
In addition to CDNs and their derivatives, small molecular STING agonists can also exert an effective anti-tumor effect. DSDP, as an activator of the cGAS/STING pathway, induces the mRNA expression of type I and III IFN in human fibroblasts and peripheral blood mononuclear cells in a dose-dependent manner and inhibits the replication of various flaviviruses. More importantly, DSDP induces an IFN-based pro-inflammatory cytokine response in a human STING-dependent manner [Citation67]. 6-Bromo-N-(naphthalen-1-yl)benzo[d][1,3] dioxole-5-carboxamide (BNBC) is another human STING agonist similar to DSDP that promotes the establishment of pro-inflammatory cytokine response and an antiviral state in a STING-dependent manner and effectively inhibits the replication of dengue virus, yellow fever virus and Zika virus in fibroblasts. In addition, BNBC can effectively induce an inflammatory cytokine response and promote the maturation of peripheral blood mononuclear cell PBMC-derived dendritic cells [Citation68]. These results show that activating cGAS/STING signaling in tumor cells is a promising treatment.
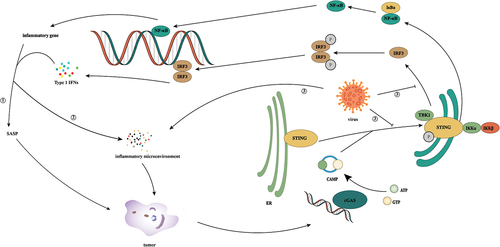
Inhibition of cGAS/STING signaling in viral infection ()
Innate immunity plays an important role in early defense against pathogens including viruses. cGAS activates innate immune response by identifying virus-derived DNA in the cytoplasm during primary infection and virus production. When cGAS/STING is absent in macrophages and DCs, the fatal infection can occur because an effective immune response cannot be produced [Citation69].
Figure 4. Activation and inhibition of cGAS/STING signaling in viral infection. During viral infection, cells recognize viral DNA through cGAS and activate the cGas/sting pathway, thereby exerting antiviral immunity. However, viruses have evolved a variety of mechanisms to evade host immunity in the confrontation with humans. ① hbx interferes with cGas’s recognition of DNA by interacting with cGAS. But HBx was found to upregulate TBK1 expression at the transcriptional level, thus promoting the occurrence of HCC by activating NF-κB. ② HPV16 E7 can mediate transcriptional activation of chromatin repressor SUV39H1, induce the formation of heterochromatin in the promoter region of the STING gene and effectively inhibit the intracellular cGAS/STING signaling pathway. ③: BFRF1 can evade the innate immunity of the host by inhibiting IRF3 activation by interacting with the downstream signal IKKi of the cGAS/STING pathway. ④: in KSHV infection, the cytoplasmic LANA variant inhibits the production of cGAS/STING-dependent interferon through direct interaction with cGAS. Meanwhile, Kaposi’s sarcoma virus ORF52 directly inhibits the activity of the cGAS enzyme by binding to cGAS and DNA. ⑤: vIRF1 targets STING by blocking the interaction between STING and TBK1, thus inhibiting the phosphorylation and concomitant activation of STING and ultimately inhibiting the DNA induction pathway. ⑥: by binding to STING, CMV protein M152 slows its transport to the Golgi chamber and inhibits STING from initiating type I IFN signaling in the Golgi chamber. ⑦: HTLV-1 interacts with STING through the protein Tax and reduces ubiquitination of its K63 connection, resulting in a decrease in the STING/TBK1 association and IFN-I production. Meanwhile, Tax-1 directly targets the autophagy complex containing Beclin1, which relieves the regulation of autophagy by stimulating the IKK complex, thus leading to continuous activation of NF-kB.
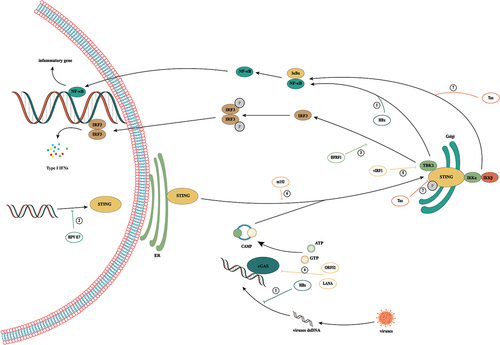
In the long-term struggle with human beings, many viruses have evolved a variety of mechanisms to inhibit the activation of cGAS/STING signals and maintain the long-term survival of viruses in cells. Tumor-associated virus, also known as carcinogenic virus or cancer virus, mainly includes HBV, HCV, EBV, KSHV, HPVs, and HTLV-1 [Citation70]. However, the cGAS/STING pathway and senescence mechanisms are generally damaged in the infection of these viruses. Given the close relationship between cGAS/STING and the mechanism of senescence, we have reason to suspect that inhibition of cGAS/STING signaling may be an important cause of precancerous lesions.
HBV infection is an important risk factor for HCC. Activation of cGAS/STING signaling in hepatocytes can effectively inhibit HBV replication [Citation71]. However, when hepatocytes are faced with HBV infection, the intracellular cGAS/STING signaling pathway is not significantly activated [Citation72,Citation73], which indicates that HBV has a mechanism of immune evasion. On the one hand, the expression of cGAS/STING protein in hepatocytes may be low, resulting in an inability to effectively perceive replication of the virus [Citation72]. On the other hand, rapid degradation of viral DNA by cytoplasmic nuclease may result in the inability of cGAS to sense infection [Citation74]. A recent study has shown that HBx plays an important role in promoting immune escape by HBV. HBx is a unique non-structural protein of HBV encoded by one of the four open reading frames of HBV. It is an important coactivator of HBV replication and HBV-related HCC. It interferes with cGAS’s recognition of DNA by interacting with cGAS [Citation75]. Inhibition of cGAS/STING signaling may be an important cause of HBV infection.
Interestingly, in early studies, HBx was found to upregulate TBK1 expression at the transcriptional level, thus promoting the occurrence of HCC by activating NF-κB [Citation76]. The lack of STING in HCC can accelerate the progression of HCC. Activation of cGAS/STING signaling can effectively inhibit HCC progression [Citation77]. At the same time, an important relationship exists between senescence induced by HBV and the occurrence of liver cancer. Therefore, we speculate that cGAS/STING plays an important role in the transformation from hepatitis to liver cancer. On the one hand, HBV cannot effectively clear senescent cells by inhibiting activation of the cGAS/STING signaling pathway, while on the other hand, inflammatory factors secreted by senescent cells eventually induce the emergence of inflammation-related tumors.
HPV is a small, unenveloped double-stranded DNA virus, which is an important cause of cervical cancer [Citation78]. HPV16 E7 can mediate transcriptional activation of chromatin repressor SUV39H1, induce the formation of heterochromatin in the promoter region of the STING gene and effectively inhibit the intracellular cGAS/STING signaling pathway [Citation79,Citation80]. At the same time, HPV’s unique L2-dependent vesicular membrane transport can also effectively shield CGA/STING monitoring in the cytoplasm, thus promoting the persistence of the virus [Citation81]. Recent studies have shown that a dysfunctional cGAS/STING pathway is an important cause of a poor prognosis in patients with cervical cancer [Citation82]. Hypermethylation of the cGAS gene promoter was found in cervical precancerous lesions [Citation83], and activation of STING can effectively increase the number of tumor-infiltrating CD8+T cells and CD103+ DCs and inhibit tumor growth in vivo [Citation84]. These studies suggest that persistent inhibition of the cGAS/STING signaling pathway by HPVs may be another important cause of cell carcinogenesis.
EBV is the pathogen of infectious mononucleosis and is related to the development of various B-cell lymphoproliferative diseases, including Hodgkin’s lymphoma, non-Hodgkin’s lymphoma and Burkitt’s lymphoma. The early cleavage protein BFRF1 encoded by EBV can evade the innate immunity of the host by inhibiting IRF3 activation by interacting with the downstream signal IKK of the cGAS/STING pathway [Citation85].
KSHV (also known as HHV-8) is the pathogen of Kaposi’s sarcoma, multicentre Castleman’s disease and primary exudative lymphoma. The LANA of KSHV is mainly located in the nuclei of latently infected cells and plays a key role in the replication and maintenance of DNA in the outer body of the latent virus. Through direct interaction with cGAS, the cytoplasmic LANA variant inhibits the production of cGAS/STING-dependent interferon, thus promoting reactivation of KSHV from the latent period [Citation86]. Kaposi’s sarcoma virus ORF52 is a specific protein that directly inhibits the activity of the cGAS enzyme by binding to cGAS and DNA, thus destroying cell solute DNA sensing [Citation87]. On the other hand, vIRF1 targets STING by blocking the interaction between STING and TBK1, thus inhibiting the phosphorylation and concomitant activation of STING and ultimately inhibiting the DNA induction pathway [Citation88].
CMV, a kind of beta herpes virus, is one of the most common pathogens in chronic viral infection in humans. CMV infection is associated with various forms of chronic inflammation, including cancer, and the cGAS/STING/TBK1/IRF3 pathway is an important means to limit early CMV infection [Citation89]. By binding to STING, CMV protein M152 slows its transport to the Golgi chamber and inhibits STING from initiating type I IFN signaling in the Golgi chamber. Interestingly, this process does not affect NF-κB signaling mediated by STING, and activation of NF- κB signaling can promote early CMV transcription [Citation90]. EBV, cytomegalovirus and KSHV are all herpesviruses. When they infect a host, they not only inhibit activation of cGAS/STING signaling in various manners but also have a common feature: activation of the DDR [Citation91]. Whether this will lead to the occurrence of tumors deserves more in-depth study.
HTLV-1 is associated with a variety of diseases, including invasive blood cancer and adult T-cell leukaemia/lymphoma [Citation92]. HTLV-1 virus can not only inhibit activation of the cGAS/STING signaling pathway but also activate NF-kB and induce the senescence response [Citation93,Citation94]. On the one hand, HTLV-1 interacts with STING through the protein Tax and reduces ubiquitination of its K63 connection, resulting in a decrease in the STING/TBK1 association and IFN-I production [Citation95,Citation96]. On the other hand, Tax-1 protein can also damage the DNA repair pathway of host cells, thus inducing gene instability. Tax-1 directly targets the autophagy complex containing Beclin1, which relieves the regulation of autophagy by stimulating the IKK complex, thus leading to continuous activation of NF-kB [Citation92]. However, NF-kB plays an important role in promoting cell senescence.
Tumor-associated viruses show a long lag between virus-driven carcinogenesis and initial infection. Tumor-associated viruses’ insertion of mutagenic and viral oncogenes into cellular DNA is an important cause of tumorigenesis, while the role of chronic inflammation and an immunosuppressive state in tumorigenesis cannot be ignored [Citation70]. Here, we emphasize the importance of tumor-associated virus damage to host cells’ cGAS/STING pathway in inducing tumorigenesis, and inhibition of the cGAS/STING pathway may lead to an inability of the immune system to effectively respond to tumor-associated virus-induced senescence, leading to tumorigenesis. Sustained activation of NF-κB may be another reason for virus-induced tumorigenesis. Activation of NF-κB in liver cancer is an important factor in inducing tumor development, and activation of NF-κB was also found in cytomegalovirus and human T-lymphotropic virus type 1 infection, but this hypothesis requires more experiments verification.
cGAS/STING and senescence ()
Cell senescence is an irreversible state of cell cycle stagnation. Various types of cellular and environmental stress can cause senescence, such as gene instability, oncogene-induced senescence OIS and telomere dysfunction [Citation5,Citation43,Citation97,Citation98]. Senescent cells can secrete a variety of cytokines, chemokines and growth factors. This unique phenotype is called the SASP [Citation99,Citation100] In the presence of the SASP, the body can remove senescence cells by recruiting immune cells and stimulate a local immune response to eliminate cells that express oncogenes, which is called “senescence monitoring” [Citation101]. The cGAS/STING pathway is a key regulatory factor induced by the SASP [Citation102]. Under normal circumstances, cells can induce the removal of pro-inflammatory DNA through exocrine and autophagy to prevent the activation of cytoplasmic cGAS/STING [Citation103]. Interestingly, cGAS can promote not only inflammation by sensing micronuclei but also the elimination of micronuclei by regulating micronucleus autophagy. cGAS, as an autophagy receptor, binds to LC3B through its LC3 interaction region and promotes the transfer of autophagy micronuclei to lysosomes in an ATG14-and ATG7-dependent manner, which inhibits micronucleus-driven cGAS activation and subsequent cGAMP production [Citation104].
Figure 5. cGAS/STING signaling pathway and cellular senescence. CCFs can be induced by dsDNA generated in senescent cells due to oncogene activation, telomere dysfunction, and chromosomal instability. cGAS activates the cGAS/STING pathway by recognizing CCFs and triggers the production of SASP factors, thereby promoting the recruitment of immune cells to senescent cells, and inducing the clearance of senescent cells through immune cell action. ①: ROS drives the formation of CCFs through ROS/JNK retrograde signaling pathways. ②: cGAS, as an autophagy receptor, binds to LC3B through its LC3 interaction region and promotes the transfer of autophagy micronuclei to lysosomes in an ATG14 and ATG7 dependent manner, which inhibits micronucleus-driven cGAS activation and subsequent cGAMP production. ③: TLR2 promotes cell cycle arrest by regulating the tumor inhibitor and regulates the SASP by inducing serum A-SAA in the acute phase. ④: STING can regulate the senescence microenvironment through DOT1L-mediated H3K79 methylation at the IL1A site in OIS cells.
When senescence is inevitable, nuclear-encoded mitochondrial oxidative phosphorylation genes will be downregulated, resulting in mitochondrial dysfunction, and the resulting ROS drive the formation of cytoplasmic chromatin fragments through ROS/JNK retrograde signaling pathways [Citation105]. Moreover, due to the downregulation of the expression of DNase2 and TREX1 and the damage from autophagy in senescent cells, the cells could not effectively clear the CCFs, which led to abnormal activation of cGAS/STING and induced the release of inflammatory factors and the appearance of the SASP through NF-κB pathway [Citation106,Citation107]. These molecules can induce cell death, senescence or the immune responses to clear infected or damaged cells. When the cGAS/STING signaling pathway was damaged, it could promote the spontaneous immortalization of mouse embryonic fibroblasts and inhibit the SASP induced by DNA damage [Citation108,Citation109]. At the same time, it will lead to the weakening of the infiltration of anti-tumor immune cells, a decrease in the clearance rate of senescence cells and the growth of tumor cells.[Citation110].
However, inflammatory processes induced by long-standing senescent cells may be harmful to the body by, for example, creating a microenvironment that promotes tumor growth, metastasis or immunosuppression [Citation111]. In the study of B-cell lymphoma and T-cell acute lymphoblastic leukemia, therapeutically induced senescence allows cancer cells to acquire new stem cell characteristics when they enter into cell senescence [Citation112]. At the same time, in the study of thyroid papillary carcinoma, senescent cells produced a gradually increasing CXC motif ligand 12 chemokine gradient from the tumor center to the tumor edge through the SASP, which led to increased invasiveness of thyroid papillary carcinoma characterized by enhanced lymphatic vascular invasion and apoptosis resistance [Citation113].
cGAS/STING inhibits tumorigenesis by promoting tumor senescence
Telomere dysfunction is an important cause of senescence [Citation114]. Telomeres are special structures at the ends of linear chromosomes, which are essential for maintaining genomic stability. Telomere shortening leads to telomere capping, which triggers a DDR similar to double-strand breaks, thus initiating the senescence process [Citation115,Citation116]. However, during telomere dysfunction, G2/M checkpoints cannot prevent cells with fused chromosomes from mitosis, resulting in micronucleus production. After nuclear membrane rupture, cGAS binds to the CCF to initiate innate immune signals, resulting in a premature senescence phenotype caused by non-telomere shortening [Citation117], which shows that the cGAS/STING signaling pathway plays an important role in initiating the senescence process in patients with telomere dysfunction. When the cGAS/STING signaling pathway is damaged, it is unable to respond effectively to telomere abnormalities, which leads to tumorigenesis [Citation56].
OIS is a cellular response that limits the replication of cells expressing oncogenes. Therefore, OIS is an effective tumor inhibition mechanism that can limit cancer progression [Citation118]. OIS markers include persistent DDR induced by DNA replication stress and upregulation of the p53/p21 and p16/pRb pathways [Citation119]. Increased DNA damage and decreased laminin expression induced by OIS can cause CCFs to activate cGAS/STING and downstream effectors IRF3 and NF-κB [Citation120,Citation121]. Among them, NF-κB plays an important role in regulating the process of TLR2 induced by OIS. TLR2 is the key mediator of senescence. It promotes cell cycle arrest by regulating the tumor inhibitor p53-p21CIP1, p16INK4a and p15INK4b and regulates the SASP by inducing A-SAA in the acute phase [Citation122]. In addition, STING can also regulate the senescence microenvironment through DOT1 L-mediated H3K79 methylation at the IL1A site in OIS cells [Citation123].
CIN refers to the increase in the rate of chromosome segregation defects during iterations of mitosis, resulting in heterogeneous cell populations with chromosome layouts that are no longer multiples of haploid complement. The resulting karyotype imbalance is called aneuploidy and is usually harmful at both the organic and cellular levels [Citation124]. CIN can promote the production of solute DNA, increase the level of STING and promote the formation of micronuclei [Citation125]. Due to the error in chromosome segregation, the whole chromosome or chromosome fragments cannot reach the cell poles, and the abnormal nuclear chamber is formed [Citation126–128]. cGAS is located in a large amount of chromatin during mitosis but dissociates when mitosis is completed. cGAS is concentrated in micronuclei after a short delay, and micronuclei containing cGAS initiate inflammatory signals [Citation129].
A subgroup of UM revealed that PRC1 deletion leads to transcriptional disinhibition of PRC1- target genes and mitotic chromosome segregation errors and drives tumorigenesis. Chromosome segregation errors lead to micronucleus formation and exposure of dsDNA to the cytoplasm to activate cGAS/STING signals [Citation130]. BLM deficiency can lead to CIN and accumulation of micronuclei in the cytoplasm and constitutively upregulate the expression of inflammatory the interferon-stimulating gene mediated by the cGAS/STING/IRF3 cytoplasmic DNA sensing pathway [Citation131]. Inflammation induced by these two pathways can effectively inhibit the occurrence of tumors. These results indicate that cGAS/STING signaling plays an important role in driving the inflammatory response induced by CIN.
The cGAS/STING signaling pathway promotes tumor progression ()
Activation of cGAS/STING is related to efficient anti-tumor immunity. However, many studies have shown that the expression of cGAS/STING signaling is upregulated in some cancer tissues [Citation132,Citation133]. The highly upregulated cGAS/STING signal is negatively correlated with immune cell infiltration and indicates a poor prognosis for some cancer patients [Citation133]. In low-antigenicity tumors, STING can promote the growth of Lewis lung cancer by inducing indoleamine 2 and 3 dioxygenase responses in the TME [Citation134]. Dimethylbenzene (a) anthracene is a carcinogen that can activate STING/dependent signal transduction to produce inherent inflammation, thus inducing tumor formation in mice [Citation135]. In a study of breast cancer and lung cancer, tumor cells were found to selectively establish gap junctions with astrocytes by expressing protocadherin 7. These gap junctions allow tumor cells to transfer cGAMP to astrocytes, activate the STING pathway and produce the inflammatory cytokines IFN α and TNF α. These inflammatory cytokines secreted by astrocytes can be used as paracrine signals to activate the STAT1 and NF-κB pathways in brain metastatic tumor cells, thus supporting tumor growth and chemotherapy resistance [Citation136].
Figure 6. The cGAS/STING signaling pathway promotes tumor progression. Tumor cells are often filled with cytosolic dsDNA. Normally, tumor growth can be inhibited by inducing the cGAS/STING signaling pathway. However, studies have shown that in some cases, activation of the cGAS/STING signaling pathway can induce the generation of an inflammatory tumor microenvironment, thereby inducing tumorigenesis. ① STING can promote tumor growth by inducing IDO responses in the tumor microenvironment. ② DMBA activates STING-dependent signaling with intrinsic inflammatory effects that induce tumorigenesis. ③ Tumor cells selectively establish gap junctions with astrocytes through PCDH7, transfer tumor cells to cGAMP to astrocytes, activate the STING pathway, and produce inflammatory cytokines IFNα and TNFα. These inflammatory factors secreted by astrocytes can activate STAT1 and NF-κB pathways in tumor cells, thereby supporting tumor growth and chemotherapy resistance. ④ CIN can activate the cGAS/STING signaling pathway in tumor cells, thereby activating the inflammation-related tumor microenvironment to promote tumor progression.
The relationship between cGAS/STING, CIN and senescence has been described previously, and cGAS/STING activation induced by CIN and senescence may be an important cause of tumorigenesis. In a study of colorectal cancer, the expression of N-acetyltransferase 10 has been found to mediate the formation of micronuclei, which induces cGAS to activate SASP and promotes the progression of colorectal cancer [Citation137]. In uveal melanoma, a subset of tumor cells are driven by deletion of polycomb repressive complex 1. polycomb repressive complex 1 deletion triggers chromosome segregation errors, thus promoting the signal transduction of chronic inflammation before metastasis. In the presence of small molecular inhibitors of CIN or STING, the migration phenotype induced by PRC1 inhibition was inhibited [Citation130]. The expression of EMT and metastasis-related genes increased in CIN-high cells, with an increase in migration and invasion in vitro. Interestingly, when the STING in the cells decreased, the malignant phenotype of the cells was inhibited [Citation125]. These findings suggest that activation of the cGAS/STING signaling pathway may induce inflammation-dependent tumorigenesis, while activation of cGAS/STING and downstream inflammatory factors induced by CIN may be an important reason for tumor metastasis promotion. However, the mechanism leading to the transition from anti-tumor effects to tumorigenesis is unknown.
In tumor immunity, the cGAS/STING pathway shows diversity. On the one hand, the cGAS/STING pathway can associate DNA damage with immune surveillance, cell senescence and other anti-tumor mechanisms and induce the SASP by sensing DNA damage and oncogene activation to coordinate the immune response and promote the clearance function of the immune system. On the other hand, the inflammatory response caused by cGAS/STING in the induction of DNA damage and senescence will induce changes in tumor cells and the local microenvironment to promote tumor progression.
Conclusion and future directions ()
The study of the cGAS/STING signaling pathway provides a new understanding of the mechanism of intracellular DNA detection. In normal innate immunity, abnormal DNA accumulation in the cytoplasm can effectively activate the cGAS/STING signaling pathway, promote cell senescence and maintain the stability of the internal environment by inducing an interferon response. However, with a deepening understanding of senescence, senescence is an important means to resist tumor progression, but when senescent cells cannot be effectively cleared, senescence will also promote tumor production [Citation111]. Given the important regulatory role of the cGAS/STING signaling pathway in innate immunity and senescence, the damage from cGAS/STING signaling may lead to an inability of the body to effectively remove senescence cells, leading to persistent inflammation and thus resulting in the emergence of tumors. When we extend this hypothesis to various tumor-related viruses, we can find that inhibition of the cGAS/STING signaling pathway is an important means for tumor-related viruses to evade immunity, and the cGAS/STING signaling pathway is an important mechanism for senescence regulation; therefore, we have reason to suspect that the senescence mechanism may be impaired when tumor-related viruses are present, but senescence cannot be eliminated effectively. The secretion of various inflammatory factors may be the cause of tumors induced by tumor-associated viruses.
Figure 7. The crosstalk of cGAS/STING pathway in infection, senescence, and tumor. ① In the face of tumor formation, cells induce tumor cell senescence through the cGAS/STING signaling pathway. ② Under specific circumstances, the cGAS/STING signaling pathway can induce the generation of an inflammatory microenvironment, which can lead to the occurrence of inflammation-related tumors. ③ During virus infection, on the hand, it inhibits the cGAS/STING activity of host cells by inhibiting the activities of cGAS, STING, and IRF3, on the other hand, it promotes the release of inflammatory factors by activating NF-κB, and at the same time induces the production of an inflammatory microenvironment through self-reproduction, which induces tumorigenesis.
Senescence is an alarm response of cells to harmful stimuli or abnormal proliferation, which can induce irreversible cell cycle arrest [Citation138]. As mentioned earlier, cGAS/STING signaling plays an important role in the regulation of senescence, and promoting tumor cell senescence by relying on the cGAS/STING signaling pathway is an important strategy for tumor treatment at present. In some cases, however, the emergence of senescence-related secretory phenotypes promotes tumorigenesis, which is a concern with this treatment method. Therefore, studying the mechanism of the cGAS/STING signaling pathway in different backgrounds is important, which may be a challenge for future research on the cGAS/STING signaling pathway.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing does not apply to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Roers A, Hiller B, Hornung V. Recognition of endogenous nucleic acids by the innate immune system. Immunity. 2016;44(4):739–754.
- Evans MJ, Ni G, Ma Z, et al. cGAS and STING: At the intersection of DNA and RNA virus-sensing networks. PLoS Pathog. 2018;14(8):e1007148
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820.
- Couto D, Zipfel C. Regulation of pattern recognition receptor signalling in plants. Nat Rev Immunol. 2016;16(9):537–552.
- Zhang X, Bai XC, Chen ZJ. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. 2020;53(1):43–53.
- Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20(11):657–674.
- Ablasser A, Chen ZJ. cGAS in action: expanding roles in immunity and inflammation. Science. 2019;363:6431.
- Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142–1149.
- Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 2020;10(1):26–39.
- Gentili M, Lahaye X, Nadalin F, et al. The N-terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep. 2019;26(9):2377–2393.e2313. DOI:10.1016/j.celrep.2019.01.105
- Michalski S, de Oliveira Mann CC, Stafford CA, et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature. 2020;587(7835):678–682. DOI:10.1038/s41586-020-2748-0
- Pathare GR, Decout A, Gluck S, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature. 2020;587(7835):668–672. DOI:10.1038/s41586-020-2750-6
- Boyer JA, Spangler CJ, Strauss JD, et al. Structural basis of nucleosome-dependent cGAS inhibition. Science. 2020;370(6515):450–454. DOI:10.1126/science.abd0609
- Kujirai T, Zierhut C, Takizawa Y, et al. Structural basis for the inhibition of cGAS by nucleosomes. Science. 2020;370(6515):455–458. DOI:10.1126/science.abd0237
- Zhao B, Xu P, Rowlett CM, et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature. 2020;587(7835):673–677. DOI:10.1038/s41586-020-2749-z
- Zhong L, Hu M-M, Bian L-J, et al. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discov. 2020;6(1). DOI:10.1038/s41421-020-0162-2
- Zhang X, Wu J, Du F, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014;6(3):421–430. DOI:10.1016/j.celrep.2014.01.003
- Andreeva L, Hiller B, Kostrewa D, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature. 2017;549(7672):394–398. DOI:10.1038/nature23890
- Zhou W, Whiteley AT, de Oliveira Mann CC, et al. Structure of the human cGAS–DNA complex reveals enhanced control of immune surveillance. Cell. 2018;174(2):300–311 e311. DOI:10.1016/j.cell.2018.06.026
- Ablasser A, Goldeck M, Cavlar T, et al. cGAS produces a 2‘-5’-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380–384. DOI:10.1038/nature12306
- Zhang X, Shi H, Wu J, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51(2):226–235. DOI:10.1016/j.molcel.2013.05.022
- Du M, Chen ZJ. DNA-Induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361(6403):704–709.
- Gao D, Li T, Li XD, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci USA. 2015;112(42):E5699–5705. DOI:10.1073/pnas.1516465112
- Zhou W, Mohr L, Maciejowski J, et al. cGAS phase separation inhibits TREX1-mediated DNA degradation and enhances cytosolic DNA sensing. Mol Cell. 2021;81(4):739–755 e737.
- Cheng Z, Dai T, He X, et al. The interactions between cGAS-STING pathway and pathogens. Signal Transduct Target Ther. 2020;5(1):91. DOI:10.1038/s41392-020-0198-7
- Shang G, Zhu D, Li N, et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol. 2012;19(7):725–727. DOI:10.1038/nsmb.2332
- Shu C, Yi G, Watts T, et al. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol. 2012;19(7):722–724.
- Civril F, Deimling T, de Oliveira Mann CC, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498(7454):332–337. DOI:10.1038/nature12305
- Shang G, Zhang C, Chen ZJ, et al. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. 2019;567(7748):389–393.
- Dobbs N, Burnaevskiy N, Chen D, et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe. 2015;18(2):157–168.
- Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567(7747):262–266. DOI:10.1038/s41586-019-1006-9
- Ran Y, Xiong MG, Xu ZS, et al. YIPF5 is essential for innate immunity to DNA virus and facilitates COPII-dependent STING trafficking. J Immunol. 2019;203(6):1560–1570.
- B-C Z, Nandakumar R, Reinert LS, et al. STEEP mediates STING ER exit and activation of signaling. Nat Immunol. 2020;21(8):868–879. DOI:10.1038/s41590-020-0730-5
- Almine JF, O’Hare CA, Dunphy G, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun. 2017;8(1):14392. DOI:10.1038/ncomms14392
- Nishikawa Y, Matsuzaki Y, Kimura K, et al. Modulation of stimulator of interferon genes (STING) expression by interferon-gamma in human keratinocytes. Biochem Genet. 2018;56(1–2):93–102.
- Wang L, Wen M, Cao X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science. 2019;365:6454.
- Kwon D, Sesaki H, Kang SJ. Intracellular calcium is a rheostat for the STING signaling pathway. Biochem Biophys Res Commun. 2018;500(2):497–503.
- Hou P, Lin Y, Li Z, et al. Autophagy receptor CCDC50 tunes the STING-mediated interferon response in viral infections and autoimmune diseases. Cell Mol Immunol. 2021;18(10):2358–2371. DOI:10.1038/s41423-021-00758-w
- Li J, Bakhoum SF. Expanding the role of STING in cellular homeostasis and transformation. Trends Cancer. 2019;5(4):195–197.
- Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791.
- Prabakaran T, Bodda C, Krapp C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/sqstm1-dependent autophagy pathway activated by TBK1. Embo J. 2018;37(8). DOI:10.15252/embj.201797858
- Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826–830. DOI:10.1126/science.1229963
- Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–1299.
- Schadt L, Sparano C, Schweiger NA, et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019;29(5):1236–1248 e1237. DOI:10.1016/j.celrep.2019.09.065
- Woo SR, Fuertes MB, Corrales L, et al. STING-Dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830–842. DOI:10.1016/j.immuni.2014.10.017
- Hu Z, Teng XL, Zhang T, et al. SENP3 senses oxidative stress to facilitate STING-dependent dendritic cell antitumor function. Mol Cell. 2021;81(5):940–952 e945. DOI:10.1016/j.molcel.2020.12.024
- Hodgins JJ, Khan ST, Park MM, et al. Killers 2.0: NK cell therapies at the forefront of cancer control. J Clin Invest. 2019;129(9):3499–3510.
- Marcus A, Mao AJ, Lensink-Vasan M, et al. Tumor-Derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity. 2018;49(4):754–763.e754.
- Takashima K, Takeda Y, Oshiumi H, et al. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem Biophys Res Commun. 2016;478(4):1764–1771. DOI:10.1016/j.bbrc.2016.09.021
- Xia T, Konno H, Ahn J, et al. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 2016;14(2):282–297.
- Konno H, Yamauchi S, Berglund A, et al. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene. 2018;37(15):2037–2051.
- Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76(22):6747–6759.
- Wu L, Cao J, Cai WL, et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018;16(8):e2006134. DOI:10.1371/journal.pbio.2006134
- Kitajima S, Ivanova E, Guo S, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019;9(1):34–45. DOI:10.1158/2159-8290.CD-18-0689
- Wu S, Zhang Q, Zhang F, et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat Cell Biol. 2019;21(8):1027–1040. DOI:10.1038/s41556-019-0352-z
- Chen YA, Shen YL, Hsia HY, et al. Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat Struct Mol Biol. 2017;24(12):1124–1131.
- Li J, Duran MA, Dhanota N, et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov. 2021;11(5):1212–1227. DOI:10.1158/2159-8290.CD-20-0387
- Tan YS, Sansanaphongpricha K, Xie Y, et al. Mitigating SOX2-potentiated immune escape of head and neck squamous cell carcinoma with a STING-inducing nanosatellite vaccine. Clin Cancer Res. 2018;24(17):4242–4255. DOI:10.1158/1078-0432.CCR-17-2807
- Mender I, Zhang A, Ren Z, et al. Telomere stress potentiates STING-dependent anti-tumor immunity. Cancer Cell. 2020;38(3):400–411 e406. DOI:10.1016/j.ccell.2020.05.020
- Lioux T, Mauny MA, Lamoureux A, et al. Design, synthesis, and biological evaluation of novel cyclic adenosine-inosine monophosphate (cAIMP) analogs that activate stimulator of interferon genes (STING). J Med Chem. 2016;59(22):10253–10267. DOI:10.1021/acs.jmedchem.6b01300
- Francica BJ, Ghasemzadeh A, Desbien AL, et al. TNFalpha and radioresistant stromal cells are essential for therapeutic efficacy of cyclic dinucleotide STING agonists in nonimmunogenic tumors. Cancer Immunol Res. 2018;6(4):422–433. DOI:10.1158/2326-6066.CIR-17-0263
- Li T, Cheng H, Yuan H, et al. Antitumor activity of cGAMP via stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci Rep. 2016;6(1):19049. DOI:10.1038/srep19049
- Wang H, Hu S, Chen X, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA. 2017;114(7):1637–1642. DOI:10.1073/pnas.1621363114
- Yang H, Lee WS, Kong SJ, et al. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest. 2019;129(10):4350–4364. DOI:10.1172/JCI125413
- Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7(283):283ra252. DOI:10.1126/scitranslmed.aaa4306
- Corrales L, Glickman LH, McWhirter SM, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11(7):1018–1030. DOI:10.1016/j.celrep.2015.04.031
- Liu B, Tang L, Zhang X, et al. A cell-based high throughput screening assay for the discovery of cGAS-STING pathway agonists. Antiviral Res. 2017;147:37–46.
- Zhang X, Liu B, Tang L, et al. Discovery and mechanistic study of a novel human-stimulator-of-interferon-genes agonist. ACS Infect Dis. 2019;5(7):1139–1149. DOI:10.1021/acsinfecdis.9b00010
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792.
- Hatano Y, Ideta T, Hirata A, et al. Virus-driven carcinogenesis. Cancers (Basel). 2021;13(11):2625. DOI:10.3390/cancers13112625
- Guo F, Tang L, Shu S, et al. Activation of stimulator of interferon genes in hepatocytes suppresses the replication of hepatitis B virus. Antimicrob Agents Chemother. 2017;61(10). DOI:10.1128/AAC.00771-17
- Lauterbach-Riviere L, Bergez M, Monch S, et al. Hepatitis B virus DNA is a substrate for the cGAS/STING pathway but is not sensed in infected hepatocytes. Viruses. 2020;12(6):592. DOI:10.3390/v12060592
- Verrier ER, Yim SA, Heydmann L, et al. Hepatitis B virus evasion from cyclic guanosine monophosphate-adenosine monophosphate synthase sensing in human hepatocytes. Hepatology. 2018;68(5):1695–1709. DOI:10.1002/hep.30054
- Hu J, Tang L, Cheng J, et al. Hepatitis B virus nucleocapsid uncoating: biological consequences and regulation by cellular nucleases. Emerg Microbes Infect. 2021;10(1):852–864. DOI:10.1080/22221751.2021.1919034
- Chen H, Jiang L, Chen S, et al. Hbx inhibits DNA sensing signaling pathway via ubiquitination and autophagy of cGAS. Virol J. 2022;19(1):55. DOI:10.1186/s12985-022-01785-3
- Kim HR, Lee SH, Jung G. The hepatitis B viral X protein activates NF-kappaB signaling pathway through the up-regulation of TBK1. FEBS Lett. 2010;584(3):525–530.
- Karakousis ND, Papatheodoridi A, Chatzigeorgiou A, et al. Cellular senescence and hepatitis B-related hepatocellular carcinoma: An intriguing link. Liver Int. 2020;40(12):2917–2927.
- Lo Cigno I, Calati F, Albertini S, et al. Subversion of host innate immunity by human papillomavirus oncoproteins. Pathogens. 2020;9(4):292.
- Bortnik V, Wu M, Julcher B, et al. Loss of HPV type 16 E7 restores cGAS-STING responses in human papilloma virus-positive oropharyngeal squamous cell carcinomas cells. J Microbiol Immunol Infect. 2021;54(4):733–739. DOI:10.1016/j.jmii.2020.07.010
- Lo Cigno I, Calati F, Borgogna C, et al. Human papillomavirus E7 oncoprotein subverts host innate immunity via SUV39H1-mediated epigenetic silencing of immune sensor genes. J Virol. 2020;94(4). DOI:10.1128/JVI.01812-19
- Uhlorn BL, Jackson R, Li S, et al. Vesicular trafficking permits evasion of cGAS/STING surveillance during initial human papillomavirus infection. PLoS Pathog. 2020;16(11):e1009028.
- Lubbers JM, Koopman B, de Klerk-Sluis JM, et al. Association of homozygous variants of STING1 with outcome in human cervical cancer. Cancer Sci. 2021;112(1):61–71. DOI:10.1111/cas.14680
- Huang S, Li R, Huang X, et al. Association study between methylation in the promoter regions of cGAS, MAVS, and TRAF3 genes and the risk of cervical precancerous lesions and cervical cancer in a southern Chinese population. Front Genet. 2019;10:1123.
- Shi F, Su J, Wang J, et al. Activation of STING inhibits cervical cancer tumor growth through enhancing the anti-tumor immune response. Mol Cell Biochem. 2021;476(2):1015–1024.
- Wang P, Deng Y, Guo Y, et al. Epstein-barr virus early protein BFRF1 suppresses IFN-beta activity by inhibiting the activation of IRF3. Front Immunol. 2020;11:513383.
- Zhang G, Chan B, Samarina N, et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc Natl Acad Sci USA. 2016;113(8):E1034–1043. DOI:10.1073/pnas.1516812113
- Wu JJ, Li W, Shao Y, et al. Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe. 2015;18(3):333–344. DOI:10.1016/j.chom.2015.07.015
- Ma Z, Jacobs SR, West JA, et al. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci USA. 2015;112(31):E4306–4315. DOI:10.1073/pnas.1503831112
- Lio CW, McDonald B, Takahashi M, et al. cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J Virol. 2016;90(17):7789–7797. DOI:10.1128/JVI.01040-16
- Stempel M, Chan B, Juranic Lisnic V, et al. The herpesviral antagonist m152 reveals differential activation of STING-dependent IRF and NF-kappaB signaling and STING’s dual role during MCMV infection. Embo J. 2019;38(5). DOI:10.15252/embj.2018100983
- Leidal AM, Pringle ES, McCormick C. Evasion of oncogene-induced senescence by gammaherpesviruses. Curr Opin Virol. 2012;2(6):748–754.
- Zhang LL, Wei JY, Wang L, et al. Human T-cell lymphotropic virus type 1 and its oncogenesis. Acta Pharmacol Sin. 2017;38(8):1093–1103.
- Shudofsky AMD, Giam CZ. Cells of adult T-cell leukemia evade HTLV-1 Tax/NF-kappaB hyperactivation-induced senescence. Blood Adv. 2019;3(4):564–569.
- Giam CZ, Semmes OJ. HTLV-1 infection and adult T-cell leukemia/lymphoma—a tale of two proteins: tax and HBZ. Viruses. 2016;8(6):161.
- Wang J, Yang S, Liu L, et al. HTLV-1 tax impairs K63-linked ubiquitination of STING to evade host innate immunity. Virus Res. 2017;232:13–21.
- Yuen CK, Chan CP, Fung SY, et al. Suppression of type I interferon production by human T-Cell leukemia virus type 1 oncoprotein tax through inhibition of IRF3 phosphorylation. J Virol. 2016;90(8):3902–3912. DOI:10.1128/JVI.00129-16
- Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972.
- Coppe JP, Desprez PY, Krtolica A, et al. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol. 2010;5(1):99–118.
- Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-Induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. DOI:10.1016/j.cell.2008.03.039
- Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. DOI:10.1371/journal.pbio.0060301
- Loo TM, Miyata K, Tanaka Y, et al. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020;111(2):304–311.
- Birch J, Gil J. Senescence and the SASP: many therapeutic avenues. Genes Dev. 2020;34(23–24):1565–1576.
- Takahashi A, Okada R, Nagao K, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. 2017;8(1):15287. DOI:10.1038/ncomms15287
- Zhao M, Wang F, Wu J, et al. CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy. 2021;17(12):3976–3991.
- Vizioli MG, Liu T, Miller KN, et al. Mitochondria-To-Nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020;34(5–6):428–445. DOI:10.1101/gad.331272.119
- Takahashi A, Loo TM, Okada R, et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun. 2018;9(1):1249. DOI:10.1038/s41467-018-03555-8
- Han X, Chen H, Gong H, et al. Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress-induced senescence. J Biol Chem. 2020;295(14):4451–4463. DOI:10.1074/jbc.RA119.010734
- Yang H, Wang H, Ren J, et al. cGAS is essential for cellular senescence. Proc Natl Acad Sci USA. 2017;114(23):E4612–E4620.
- Gluck S, Guey B, Gulen MF, et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 2017;19(9):1061–1070. DOI:10.1038/ncb3586
- Dou Z, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–406. DOI:10.1038/nature24050
- Sieben CJ, Sturmlechner I, van de Sluis B, et al. Two-step senescence-focused cancer therapies. Trends Cell Biol. 2018;28(9):723–737.
- Milanovic M, Fan DNY, Belenki D, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553(7686):96–100. DOI:10.1038/nature25167
- Kim YH, Choi YW, Lee J, et al. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun. 2017;8(1):15208.
- Cleal K, Baird DM. Catastrophic endgames: emerging mechanisms of telomere-driven genomic instability. Trends Genet. 2020;36(5):347–359.
- Victorelli S, Lagnado A, Halim J, et al. Senescent human melanocytes drive skin ageing via paracrine telomere dysfunction. Embo J. 2019;38(23):e101982. DOI:10.15252/embj.2019101982
- Freund A, Orjalo AV, Desprez PY, et al. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med. 2010;16(5):238–246.
- Abdisalaam S, Bhattacharya S, Mukherjee S, et al. Dysfunctional telomeres trigger cellular senescence mediated by cyclic GMP-AMP synthase. J Biol Chem. 2020;295(32):11144–11160. DOI:10.1074/jbc.RA120.012962
- Innes AJ, Gil J. IMR90 ER:RAS: a cell model of oncogene-induced senescence. Methods Mol Biol. 2019;1896:83–92.
- Aird KM, Zhang R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. 2015;356(2 Pt A):204–210.
- Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-Induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–642. DOI:10.1038/nature05327
- Dunphy G, Flannery SM, Almine JF, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol Cell. 2018;71(5):745–760.e745. DOI:10.1016/j.molcel.2018.07.034
- Hari P, Millar FR, Tarrats N, et al. The innate immune sensor Toll-like receptor 2 controls the senescence-associated secretory phenotype. Sci Adv. 2019;5(6):eaaw0254. DOI:10.1126/sciadv.aaw0254
- Leon KE, Buj R, Lesko E, et al. DOT1L modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. J Cell Biol. 2021;220(8). DOI:10.1083/jcb.202008101
- Barroso-Vilares M, Logarinho E. Chromosomal instability and pro-inflammatory response in aging. Mech Ageing Dev. 2019;182:111118.
- Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553(7689):467–472. DOI:10.1038/nature25432
- Fenech M. Cytokinesis-block micronucleus cytome assay evolution into a more comprehensive method to measure chromosomal instability. Genes (Basel). 2020;11(10):1203.
- Lewis CW, Golsteyn RM. Cancer cells that survive checkpoint adaptation contain micronuclei that harbor damaged DNA. Cell Cycle. 2016;15(22):3131–3145.
- Kalsbeek D, Golsteyn RM. G2/M-phase checkpoint adaptation and micronuclei formation as mechanisms that contribute to genomic instability in human cells. Int J Mol Sci. 2017;18(11):2344.
- Harding SM, Benci JL, Irianto J, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–470.
- Bakhoum MF, Francis JH, Agustinus A, et al. Loss of polycomb repressive complex 1 activity and chromosomal instability drive uveal melanoma progression. Nat Commun. 2021;12(1):5402. DOI:10.1038/s41467-021-25529-z
- Gratia M, Rodero MP, Conrad C, et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J Exp Med. 2019;216(5):1199–1213. DOI:10.1084/jem.20181329
- Yan-Fei H, Han Y, Yan-Ting Z, et al. Dysregulation in nucleic acid-sensing pathway genes is associated with cancer patients’ prognosis. Cancer Sci. 2020;111(7):2212–2222. DOI:10.1111/cas.14450
- An X, Zhu Y, Zheng T, et al. An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol Ther Nucleic Acids. 2019;14:80–89.
- Lemos H, Mohamed E, Huang L, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76(8):2076–2081. DOI:10.1158/0008-5472.CAN-15-1456
- Ahn J, Xia T, Konno H, et al. Inflammation-driven carcinogenesis is mediated through STING. Nat Commun. 2014;5(1):5166.
- Chen Q, Boire A, Jin X, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533(7604):493–498. DOI:10.1038/nature18268
- Cao Y, Yao M, Wu Y, et al. N-acetyltransferase 10 promotes micronuclei formation to activate the senescence-associated secretory phenotype machinery in colorectal cancer cells. Transl Oncol. 2020;13(8):100783.
- Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813–827. DOI:10.1016/j.cell.2019.10.005