
ABSTRACT
Osteoarthritis (OA) is a common chronic and frequently occurring orthopedic disease in the middle-aged and elderly individuals. Numerous studies have shown that long noncoding RNAs (lncRNAs) play major roles in various diseases. However, the potential molecular mechanism of action of NAV2-AS5 in OA remains unclear. The present study was designed to explore the influence of NAV2-AS5 on the progression of chondrocyte inflammation and its underlying molecular mechanisms. To simulate the inflammatory environment in OA, the human chondrocyte cell line was treated with LPS. Cell proliferation, cell cycle progression, and apoptosis were assessed using Cell Counting Kit-8, 5-ethynyl-2’-deoxyuridine analysis, and flow cytometry. Proliferation- and cycle-related proteins and the release of inflammatory factors were examined by western blot analysis and enzyme-linked immunosorbent assay. The downstream targets of NAV2-AS5 were determined using bioinformatics and confirmed by a luciferase reporter assay. In our study, patients with OA showed downregulation of NAV2-AS5, upregulation of miR-8082, and downregulation of TNFAIP3 interacting protein 2 (TNIP2). Moreover, we found that both overexpression of NAV2-AS5 and miR-8082 inhibitor promoted cell proliferation, inhibited apoptosis, and released inflammatory cytokines in LPS-treated chondrocytes. MiR-8082 was predicted to be a target of both NAV2-AS5 and TNIP2. In addition, rescue experiments showed that silencing of TNIP2 reversed the effects of the miR-8082 inhibitor on proliferation, cell cycle, apoptosis, and inflammatory factors in sh-NAV2-AS5-treated chondrocytes. In conclusion, these findings indicate that NAV2-AS5 relieves chondrocyte inflammation by targeting miR-8082/TNIP2 in OA, which provides a new theoretical basis for OA therapy.
Introduction
Osteoarthritis (OA) is a degenerative joint disease characterized by gradual loss of articular cartilage and subchondral bone, together with varying degrees of synovial inflammation [Citation1]. Due to the aging population and increasing obesity rates, the incidence of OA has gradually risen [Citation2]. The World Health Organization has reported that 9.6% of men and 18.0% of women over 60 years of age have OA [Citation3]. OA, which mainly manifests as joint pain, swelling, and stiffness, can affect sleep and lead to mental discomfort. Meanwhile, as one of the main causes of disability in adults, OA seriously disturbs the lives of patients and reduces their quality of life [Citation4]. At present, relieving pain, protecting against and delaying disease progression, and maintaining joint function have become the mainstay treatment modalities for patients with OA. However, these treatment methods are limited in clinical settings. Therefore, understanding OA pathogenesis is essential. However, the pathogenesis of OA is complex, and its specific pathological mechanisms remain unclear. Therefore, exploring effective ways to prevent and treat OA based on its pathogenesis represents a direction for future research.
Previous studies have shown that OA is a non-inflammatory joint disease. However, various reports have shown that multiple immune cells, pro-inflammatory cytokines, complement proteins, and other parts of the immune system play crucial roles in the occurrences of OA [Citation5]. During the development of OA, inflammation involving joint tissues includes the production of cytokines, low-grade cell infiltration, and activation of joint cells such as synovial cells and articular cartilage cells [Citation6]. Studies have shown that interleukin (IL)-1 binding to its cognate receptor induces matrix metalloproteinases to degrade articular cartilage, reduce proteoglycan and collagen synthesis, and induce cartilage and synovial cells to produce inflammatory mediators, including IL-6 and IL-8, further leading to chondrocyte destruction [Citation7]. In addition, IL-1, IL-6, and tumor necrosis factor (TNF)-α are all highly expressed in patients with OA. The levels of IL-1, IL-6, and TNF-α, which are positively correlated with the severity of inflammation, may be used as reference indicators to determine disease progression in patients with OA [Citation8,Citation9].
Many studies have shown that the occurrence and development of OA are inseparable from long noncoding RNAs (lncRNAs), which play vital roles in physiological and pathological processes [Citation10]. For instance, lncRNA MALAT-1 reduces IL-1β-induced cell apoptosis and matrix metabolism disorder, and inhibits inflammation in chondrocytes by blocking activation of the JNK signaling pathway [Citation11]. Chen et al. integrated the analysis of critical mRNAs and lncRNAs in OA and found NAV2-AS5 significantly differentially expressed mRNAs between patients with OA and healthy controls [Citation12]. NAV2-AS5 is a long chain, non-coding RNA composed of 3,336 nucleotides, but novel data on the molecular mechanisms and functional roles of lncRNA NAV2-AS5 in OA have not yet been reported. This study explored the mechanism of action of lncRNA NAV2-AS5 in OA. We found that lncRNA NAV2-AS5 regulates inflammatory responses in OA through the miR-8082/TNIP2 signaling pathway.
Materials and methods
Clinical samples and ethics statement
Thirty male osteoarthritis patients with an average age of 53.45 ± 5.18 years old and thirty healthy males with an average age of 54.73 ± 6.15 years old were recruited from the Second Affiliated Hospital of the Air Force Medical University during total knee replacement surgeries. This study was approved by the Ethics Committee of the Second Affiliated Hospital of the Air Force Medical University and carried out in accordance with the Declaration of Helsinki. All participants provided signed informed consent prior to the study. Cartilage tissue samples were collected and stored in −80°C freezer until usage.
Cell culture and transfection
Human chondrocyte cell line was purchased from ATCC (Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum (Bovogen, Melbourne, Australia) and 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator.
Full-length NAV2-AS5 genomic DNAs were inserted into pcDNA3.1 vector to establish the NAV2-AS5 expression vector (GeneBay, Nanjing, China). The miR-8082 inhibitor, negative control oligonucleotide (NC inhibitor), short hairpin RNA of NAV2-AS5 or TNIP2 (sh-NAV2-AS5, sh-TNIP2), and sh-NC were obtained from GeneBay. Chondrocytes (1 × 105/mL) were transfected with 10 nM pc-NAV2-AS5, 10 nM pc-NC, 30 nM miR-8082 inhibitor, 30 nM NC inhibitor, 10 nM sh-NAV2-AS5, 10 nM sh-TNIP2, and 10 nM sh-NC, separately or jointly, using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) for 48 h.
LPS treatment of chondrocytes
LPS from Escherichia coli O111: B4 was obtained from Sigma-Aldrich. LPS concentrations ranging from 0 to 20 g/mL were used to treat the chondrocytes for 12 h to induce inflammatory injury.
Quantitative reverse transcription polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the samples using TRIzol reagent (Solarbio, Beijing, China). The PrimeScript RT reagent kit (TaKaRa, Shiga, Japan) was used to reverse-transcribe 1 μg of total RNA. Quantitative real-time PCR (RT-qPCR) was performed using the SYBR Green PCR Kit (Vazyme, Nanjing, China) on an ABI 7500 Rapid real-time PCR system (Applied Biosystems, CA, USA), according to the manufacturer’s protocol. Quantitative measurements were obtained using the 2−ΔΔCT method. The primers for RT-qPCR were as follows: hsa-NAV2-AS5: 5’-GGCACATTTAGGCAGAGAGG-3’ (forward) and 5’-CAGAGGTCACACACCCACAA-3’ (reverse); hsa-TNIP2: 5’-CTAAAGAGGCGGCAGGTCCCTC-3’ (forward) and 5’-CAAGATGACCTTCCAGTGAC-3’ (reverse); hsa-β-actin: 5’-CTATCGGCAATGAGCGGTTC-3’ (forward) and 5’-AGGAGCCAGGGCAGTAATCT-3’ (reverse); hsa-U6: 5’-GTGATCACTCCCTGCCTGAG-3’ (forward) and 5’-GGACTTCACTGGACCAGACG-3’ (reverse). β-actin (ACTB) and U6 were used as controls.
Cell counting kit-8 assay and 5-ethynyl-2’-deoxyuridine (EdU) staining
According to previous studies, Cell Counting Kit-8 (CCK-8, Yeasen, Shanghai, China) and EdU assays (Solarbio) were used to examine chondrocyte proliferation [Citation13,Citation14]. For CCK-8 assays, the cells were incubated in 96-well plates for 24, 48, or 72 h. Then, the cells were treated with the CCK-8 solution at 37°C for 3 h. Subsequently, the optical density was detected using an ultraviolet spectrophotometer (Thermo Fisher Scientific, Inc.). For EdU staining, chondrocytes were seeded in 24-well plates and incubated for 24 h. Cells were then stimulated with EdU solution for 2 h, followed by fixation. Subsequently, the cells were stained with 1 × Apollo stain for 30 min in the dark. Finally, cell nuclei were stained with 4’,6-diamidino-2-phenylindole for 5 min and photographed under a fluorescence microscope (Leica, Germany).
Western blot analysis
Proteins were extracted from samples using RIPA lysis buffer plus PMSF, and the concentrations were assessed using the BCA method. Protein samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto PVDF membranes. After blocking with skim milk at room temperature for 1 h, the membranes were incubated with primary antibodies overnight and then with secondary antibody at room temperature for 1 h. Finally, immunoreactive bands were visualized by enhanced chemiluminescence (Pierce, Rockford, IL, USA). Anti-PCNA (ab29, 1:1,000), anti-Ki-67 (ab243878, 1:1,000), anti-cyclin A1 (ab53699, 1:500), anti-cyclin B1 (ab181593, 1:2,000), anti-cyclin D2 (ab230883, 1:800), anti-p27 (ab113075, 1:2,000), anti-Bax (ab32503, 1:2,000), anti-Bcl-2 (ab182858, 1:2,000), anti-cleaved caspase 3 (ab32042, 1:500), anti-cleaved-caspase 9 (ab2324. 1:500), and anti-β-actin (ab8226, 1:2,000) antibodies were obtained from Abcam (Cambridge, UK).
Flow cytometry
Cell cycle and apoptotic status were detected using flow cytometry. In brief, cells (4 × 106/mL) were inoculated and incubated at 37°C under 5% CO2 and cultured on the wall. After 48 h of incubation, half of the collected cells were blown into suspension with 1 mL phosphate-buffered saline (PBS) to detect apoptosis, followed by incubation with 5 μL Annexin V-Alexa Fluor 647 for 15 min and treatment with 5 μL PI. Finally, the apoptosis rate was measured using a flow cytometer to detect apoptotic cells (Beckman Coulter, CA, USA). The remaining cells were resuspended in 250 μL PBS and 95% ethanol pre-cooled to −20°C was slowly added and then, incubated for 30 min to detect cell cycle. Next, 0.2 mL RNase A (1 mg/mL) was added for 30 min at 37°C. Then, 0.3 mL of PI (100 g/mL) was added, and staining was performed for 20 min in the dark. Fluorescence intensity was measured using FACSCalibur flow cytometry (488 nm). The cell cycle was analyzed using Cell Quest and Modfit software to determine the cell cycle distribution.
Enzyme-linked immunosorbent assay (ELISA)
Levels of TNF-α, IL-1β, and IL-6 in the cellular supernatant were examined using ELISA (Mlbio, Shanghai, China) following the manufacturer’s instructions. In brief, the prepared sample homogenates and standard solution were delivered to 96-well plates and incubated at 37°C for 90 min. After the colorimetric reaction, the optical absorbance was measured. The Bradford method was used to determine protein concentrations, and a series of standard dilutions was used to construct a calibration curve.
Luciferase reporter assay
NAV2-AS5-WT or NAV2-AS5-MUT were co-transfected with miR-8082 mimic or NC mimic into chondrocytes using Lipofectamine 3000 (Glpbio, Montclair, CA, USA). TNIP2-WT or TNIP2-MUT were transfected with miR-8082 mimic or NC mimic into chondrocytes. After 48 h of transfection, luciferase activity was detected using a dual-luciferase reporter assay system (Promega).
RNA binding protein immunoprecipitation (RIP)
According to previous studies [Citation13,Citation15], the relationship between NAV2-AS5 and miR-8082 was identified using an RNA Immunoprecipitation Kit (GeneCreate, Wuhan, China) following the manufacturer’s protocol. Briefly, the cells were stimulated with miR-8082 mimic and its corresponding control. The cells were then treated with the RIP lysis buffer and protease and RNase inhibitors. The cell lysate was collected and incubated with magnetic beads (Millipore) coated with anti-Ago2 antibody at 4°C for 4 h. Finally, RT-qPCR was used to assess the expression of NAV2-AS5, with IgG acting as an internal control.
Statistical analysis
All results are presented as the mean value ± standard deviation. Statistical significance between two groups was assessed using unpaired Student’s t-test, and multiple groups were evaluated using one-way analysis of variance (ANOVA) with Bonferroni post-hoc test correction. P < 0.05 was considered significant.
Results
NAV2-AS5 is downregulated in OA tissue and LPS-treated chondrocytes
To assess the potential regulatory roles of NAV2-AS5 in OA, RT-qPCR was used to measure the levels of NAV2-AS5 in OA tissues and LPS-treated chondrocytes. As shown in and 1(b), the expression levels of NAV2-AS5 were dramatically downregulated in OA tissue and LPS-treated chondrocytes compared with those in the corresponding control group, suggesting that NAV2-AS5 was associated with OA.
Figure 1. NAV2-AS5 is downregulated in OA tissue and LPS-stimulated chondrocyte. (a) Relative expression of NAV2-AS5 in OA tissue and normal tissue,*p < 0.05, OA tissue compared with the normal tissue. (b) Relative expression of NAV2-AS5 in LPS-treated chondrocyte and normal group. **P < 0.01 LPS-treated chondrocyte compared with the normal group.
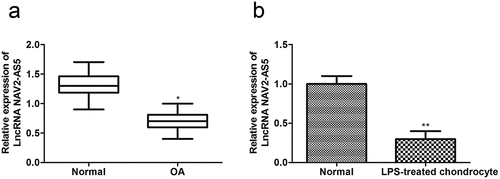
NAV2-AS5 overexpression facilitates cell proliferation and hampers apoptosis in LPS-stimulated chondrocytes
To test the effects of NAV2-AS5 on cell proliferation and apoptosis of LPS-treated chondrocytes, pc-NAV2-AS5 was used to transfect LPS-treated chondrocytes. RT-qPCR indicated that NAV2-AS5 was overexpressed in chondrocytes after transfection with pc-NAV2-AS5 (). CCK-8 assay results indicated that cell viability was inhibited by LPS, while overexpression of NAV2-AS5 partly covered the functions of LPS (). In addition, EdU staining demonstrated that LPS markedly reduced the number of EdU-positive cells, whereas NAV2-AS5 overexpression reduced the effects of LPS on chondrocytes (). Furthermore, western blot analysis showed that upregulation of NAV2-AS5 rescued downregulation of the proliferation-related proteins PCNA and Ki-67 induced by LPS (). Moreover, flow cytometric analysis was used to detect whether NAV2-AS5 was involved in chondrocyte cell cycle progression. These results illustrated that NAV2-AS5 upregulation decreased the percentage of LPS-induced G0/G1 phase cells and reduced the percentage of S phase cells (). In addition, LPS decreased the expression levels of cell cycle – related proteins cyclin A1, cyclin B1, and cyclin D2, and increased p27 expression in chondrocytes, while overexpression of NAV2-AS5 reduced the function of LPS in the cell cycle (). Furthermore, flow cytometric analysis and western blot analysis revealed that LPS positively affected apoptosis, including enhancement of the number of apoptotic cells, upregulation of Bax, Cleaved-caspase-9, and Cleaved-caspase-3 expression, and downregulation of Bcl-2 expression, which was reduced by NAV2-AS5 overexpression (). These results revealed that overexpression of NAV2-AS5 promoted cell proliferation and inhibited apoptosis in LPS-treated chondrocytes.
Figure 2. Overexpression of NAV2-AS5 promotes proliferation and inhibits apoptosis in LPS-treated chondrocytes. (a) Relative expression of NAV2-AS5. (b) Cell viability detected by CCK-8 assay. (c) EdU-positive cells assessed by EdU staining. (d) The expression of PCNA and Ki-67. (e) The cell cycle was examined by flow cytometry analysis. (f) The expression of CyclinA1, CyclinB1, CyclinD1, and p27. (g) Cell apoptosis was assessed by flow cytometry. (h) The protein expression of Bax, Bcl-2, Cleaved-caspase-3, and Cleaved-caspase-9. **P < 0.01, pc-NC+LPS group compared with the control LPS group, #P < 0.05, ##P < 0.01 pc-NAV2-AS5+LPS compared with the control LPS group.
NAV2-AS5 overexpression inhibits the release of inflammatory factors in LPS-treated chondrocytes
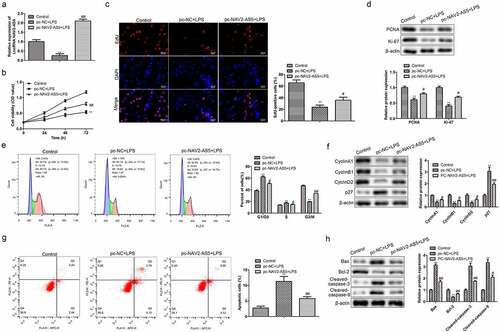
Early studies considered OA to be a non-inflammatory disease, but reports have found that inflammatory factors play major roles in OA occurrence. We detected the levels of inflammatory cytokines, including TNF-α, IL-1β, and IL-6, and found that overexpression of NAV2-AS5 significantly inhibited LPS-induced release of TNF-α, IL-1β, and IL-6 by chondrocytes ()). These results indicated that overexpression of NAV2-AS5 had anti-inflammatory effects in OA.
Figure 3. Overexpression of NAV2-AS5 inhibits the release of inflammatory factors in LPS-treated chondrocytes. (a–c). The content of TNF-α, IL-1β, and IL-6 was detected by ELISA. **P <0.01, pc-NC+LPS group compared with the control LPS group, #P <0.05, ##P <0.01 pc-NAV2-AS5+LPS compared with the control LPS group.
MiR-8082 targets 3’-UTR of NAV2-AS5
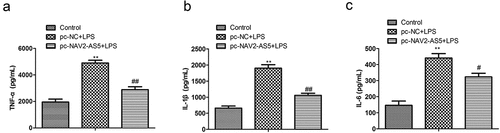
To examine the molecular mechanism by which NAV2-AS5 regulates the occurrence of OA, miRNAs targeting NAV2-AS5 were predicted using bioinformatics algorithms (DIANA). We found that miR-8082 significantly interacted with NAV2-AS5. Potential binding sites involving NAV2-AS5 and miR-8082 are displayed in . To confirm miR-8082 binding to NAV2-AS5, we transfected NAV2-AS5–WT and NAV2-AS5-MUT vectors, both of which were co-transfected with miR-8082 mimic and NC mimic. Luciferase activity was significantly decreased in co-transfected miR-8082 mimics and NAV2-AS5-WT cells compared with that in co-transfected NC mimics and NAV2-AS5-WT cells, while luciferase activity was not significantly different in transfected NAV2-AS5-MUT cells (). In addition, the expression of miR-8082 was determined using RT-qPCR in chondrocytes transfected with sh-NAV2-AS5 or sh-NC. The results showed that knockdown of NAV2-AS5 significantly promoted the expression of miR-8082 (). RIP detection results demonstrated that compared with those in the IgG group, the enrichment levels of NAV2-AS5 and miR-8082 in the Ago2 RIP group were increased (). Subsequently, we examined the expression of miR-8082 in OA tissues and found that, compared with normal tissues, miR-8082 was upregulated (). The expression of NAV2-AS5 was negatively correlated with the expression of miR-8082 (P < 0.001, R2 = 0.4188) (). These data suggested that NAV2-AS5 is physically associated with miR-8082.
Figure 4. MiR-8082 targets 3’-UTR of NAV2-AS5. (a) The binding sites between miR-8082 and NAV2-AS5 were predicted by DIANA. (b) Luciferase reporter assay, **P <0.01, miR-8082 mimic group compared with the NC mimic group. (c) The expression of miR-8082 detected by qRT-PCR, **P <0.01, the sh-NAV2-AS5 group compared with the sh-NC group. (d) The enrichment of NAV2-AS5 and miR-8082, **P <0.01, Ago group compared with the IgG group. (e) The expression of miR-8082 was detected by qRT-PCR. (f) The correlation analysis between the expression of NAV2-AS5 and miR-8082. *P <0.05 OA group compared with the normal group.
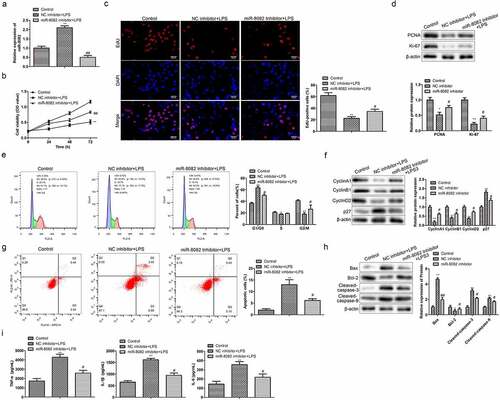
Knockdown of miR-8082 promotes cell proliferation and inhibits cell apoptosis and release of inflammatory factors in LPS-treated chondrocytes
To verify the effects of miR-8082 on chondrocyte proliferation, apoptosis, and inflammatory factors, miR-8082 inhibitor was used to treat LPS-induced cells. As shown in , miR-8082 inhibitor significantly inhibited the upregulation of miR-8082 in LPS-induced chondrocytes. CCK-8 and EdU assays revealed that miR-8082 inhibitor reversed the decline in cell viability and the reduction of EdU-positive cell numbers induced by LPS, suggesting that miR-8082 affects chondrocyte proliferation and regulates OA progression (). Consistently, the downregulation of PCNA and Ki-67 expression in LPS-induced chondrocytes was reversed by miR-8082 inhibitor (). In contrast, flow cytometry analysis revealed that knockdown of miR-8082 reversed the upregulation of LPS-induced G0/G1 phase cells and downregulated the percentage of S phase cells (). Similar to the effect of NAV2-AS5 overexpression, miR-8082 inhibitor also reversed the downregulation of LPS-induced cyclin A1, cyclin B1, and cyclin D2, and upregulation of p27 (). In addition, the reduction in apoptotic cell incidence, increased Bax, Cleaved-caspase-9, and Cleaved-caspase-3 expression, and decreased Bcl-2 expression in the LPS group were rescued by miR-8082 inhibitor (). Moreover, miR-8082 inhibitor inhibited the release of TNF-α, IL-1β, and IL-6 from LPS-induced chondrocytes ().
Figure 5. Knockdown of miR-8082 promotes cell proliferation, inhibits cell apoptosis, and releases inflammatory factors in LPS-treated chondrocytes. (a) Relative expression of miR-808. (b) Cell viability. (c) EdU-positive cells assessed by EdU staining. (d) The protein expression of PCNA and Ki-67.(e) The cell cycle. (f) The protein expression of CyclinA1, CyclinB1, CyclinD1, and p27.(g) Cell apoptosis. (h) The protein expression of Bax, Bcl-2, Cleaved-caspase-3, and Cleaved-caspase-9. I. The content of TNF-α, IL-1β, and IL-6 was detected by ELISA. **P <0.01, *P <0.05, NC inhibitor+lps compared with the control group; #P <0.05, ##P <0.01, miR-8082 inhibitor+lps compared with the control group.
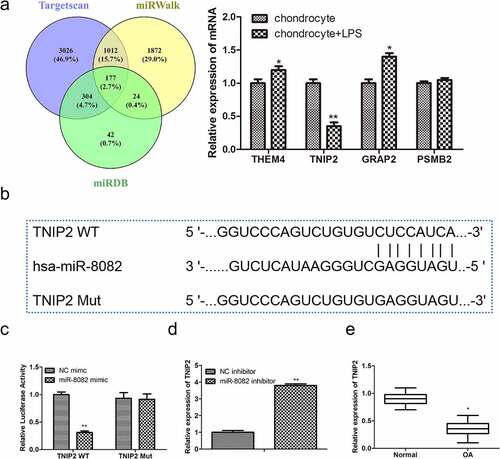
TNIP2 is a target gene of miR-8082
To explore the mechanisms by which miR-8082 functions in OA progression, target genes of miR-8082 were predicted using TargetScan, miRDB, and miRWalk, and we found that 177 genes had shared intersections among them. We selected the top four genes (THEM4, TNIP2, GRAP2, and PSMB2) with the highest correlation with OA, and RT-qPCR analysis showed that only TNIP2 was downregulated in LPS-induced chondrocytes (). Furthermore, there were binding sites identified for miR-8082 and TNIP2 (). Next, luciferase reporter assays were performed to confirm the relationship between miR-8082 and TNIP2. The results showed that miR-8082 mimic dramatically decreased luciferase activity in TNIP2-WT-transfected cells, whereas luciferase activity was unaltered when putative TNIP2 sites were mutated (). In addition, miR-8082 inhibitor significantly promoted the expression of TNIP2 (), which was otherwise downregulated in OA tissues compared with that in normal tissues (). The expression of TNIP2 was negatively correlated with the expression of miR-8082 (P < 0.001, R2 = 0.5690) (). These data demonstrated that TNIP2 is a target gene of miR-8082, which negatively regulates TNIP2 expression in OA.
Figure 6. TNIP2 is a target gene of miR-8082. (a) The target genes of miR-8082 were detected by Bioinformatics. (b) The binding sites between miR-8082 and TNIP2. **P <0.01, *P <0.05 LPS-induced chondrocytes compared with the control chondrocytes group. (c) Luciferase reporter assay, **P <0.01, miR-8082 mimic group compared with the NC mimic group. (d) The mRNA expression of TNIP2, **P <0.01, miR-8082 inhibitor group compared with the NC inhibitor group. (e) The mRNA expression of TNIP2. *P <0.05, OA group compared with the normal group. (f) The correlation analysis between the expression of TNIP2 and miR-8082.
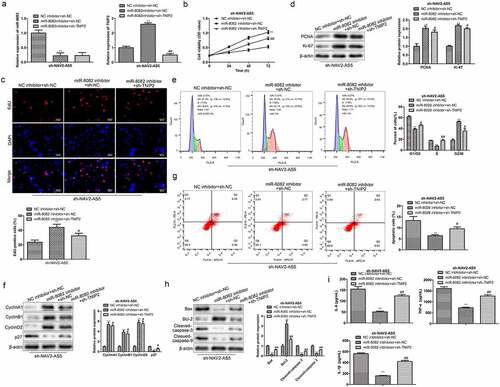
NAV2-AS5 modulates OA progression via the miR-8082/TNIP2 axis in vitro
To further verify whether NAV2-AS5 regulates OA progression through the miR-8082/TNIP2 axis, we transfected miR-8082 inhibitor and sh-TNIP2 into sh-NAV2-AS5-transfected chondrocytes, separately or simultaneously. Transfection efficiency was examined by RT-qPCR. The expression of miR-8082 was downregulated in the miR-8082 inhibitor group, and TNIP2 expression was reduced in the miR-8082 inhibitor+sh-TNIP2 group compared to that in the corresponding control group (). CCK-8 and EdU assays revealed that knockdown of TNIP2 inhibited cell proliferation induced by miR-8082 inhibitor in sh-NAV2-AS5-treated chondrocytes (). Correspondingly, the expression of cell proliferation-related proteins (PCNA and Ki-67) was reduced in the miR-8082 inhibitor+ sh-TNIP2 group compared with that in the miR-8082 inhibitor+sh-NC group (). Furthermore, TNIP2 silencing reversed the decrease in the percentage of G0/G1 phase cells induced by miR-8082 inhibitor and the increased percentage of S phase cells in sh-NAV2-AS5-transfected chondrocytes (). Furthermore, western blot analysis showed that miR-8082 inhibitor markedly upregulated the expression of cyclin A1, cyclin B1, and cyclin D2, and downregulated the expression of p27, while TNIP2 knockdown partly covered the functions of miR-8082 inhibitor in sh-NAV2-AS5-transfected chondrocytes (). Additionally, TNIP2 knockdown reversed the decrease in the number of apoptotic cells induced by miR-8082 inhibitor, downregulation of Bax, Cleaved-caspase-9, and Cleaved-caspase-3, and upregulation of Bcl-2 in sh-NAV2-AS5-transfected chondrocytes (). Moreover, inhibition of miR-8082 inhibitor on the release of inflammatory factors (TNF-α, IL-1β, and IL-6) in sh-NAV2-AS5-transfected chondrocytes was improved by sh-TNIP2 (). These data illustrate that NAV2-AS5 modulates OA progression via the miR-8082/TNIP2 axis.
Figure 7. NAV2-AS5 modulates chondrocyte proliferation, migration and inflammat -ion via miR-8082/TNIP2 axis. (a) Relative expression of miR-8082 and TNIP2. (b) Cell viability. (c) EdU-positive cells assessed by EdU staining. (d) The protein expression of PCNA and Ki-67. (e) The cell cycle. (f) The protein expression of CyclinA1, CyclinB1, CyclinD1 and p27. (g) Cell apoptosis. (h) The protein expression of Bax, Bcl-2, Cleaved-caspase-3 and Cleaved-caspase-9. (i) The content of TNF-α, IL-1β, and IL-6. **P < 0.01, *P < 0.05, miR-8082 inhibitor+sh-NC group compared with the control group (NC-inhibitor+sh-NC), #P < 0.05, ##P < 0.01, miR-8082 inhibitor+sh-TNIP2 group compared with the control group (NC-inhibitor+sh-NC).
Discussion
Several lncRNAs have been reported to participate in cancer regulation. However, there are few reports concerning NAV2-AS5, whose role in different types of cancer is not yet fully understood. Our results indicated that NAV2-AS5 is expressed at low levels in patients with OA. Degradation of the cartilage extracellular matrix and inflammation are two main characteristics of OA [Citation16]. In our study, LPS induced inflammation in human chondrocytes.
Interestingly, LPS stimulation not only decreased the expression of NAV2-AS5 and increased the expression of miR-8082 but also inhibited cell proliferation, promoted cell apoptosis, and induced the release of inflammatory factors by chondrocytes. However, NAV2-AS5 overexpression or miR-8082 knockdown reversed such effects of LPS on chondrocytes. Subsequently, NAV2-AS5 acts as a sponge for miR-8082, thereby modulating TNIP2 expression. Loss-of-function approaches demonstrated that NAV2-AS5 functioned in the progression of OA by targeting miR-8082/TNIP2. Previous reports have shown that the occurrence of OA was accompanied by abnormal expression of lncRNA [Citation17]. Numerous lncRNAs have been reported to be associated with the occurrence and development of OA. For instance, lncRNA-ROR is less expressed in OA chondrocytes than in normal chondrocytes, and knockdown of lncRNA-ROR inhibits apoptosis and promotes autophagy via the HIF1α/p53 axis [Citation18]. Thus, lncRNA-ROR may represent a new potential therapeutic target for OA. LncRNA LOXL-AS1 is upregulated in OA cartilage tissues, and knockdown of LOXL-AS1 hampers proliferation and accelerates apoptosis in chondrocytes via the miR-423-5p/KDM5C axis [Citation19]. NAV2-AS5, a long-chain non-coding RNA composed of 3,336 nucleotides, has not been reported in various diseases. In the present study, we found that NAV2-AS5 expression in OA was generally lower than that in normal tissues. Based on these results, we conclude that NAV2-AS5 might play an inhibitory role in the occurrence of OA, and might be correlated with the migration and apoptosis of OA chondrocytes. To confirm this, we further verified that NAV2-AS5 upregulation reversed the reduction of cell proliferation and elevation of cell apoptosis induced by LPS in chondrocytes using CCK-8, EdU assays and flow cytometry.
Inflammation, as an essential factor in OA, is closely related to the progression of cartilage loss and clinical symptoms of OA, including joint pain, swelling, stiffness, and indicators of inflammation [Citation20]. In the serum or synovial fluid of OA patients, IL-1β, TNF-α, and IL-6 are the primary mediators of catabolism of joint tissues [Citation21,Citation22]. IL-1β, a thoroughly studied cytokine, exerts many biological effects and plays an indispensable role in regulating inflammation [Citation23]. Kaneko et al. found that IL-1β and TNF-α induce the production of IL-6 in synovial cells and chondrocytes in OA, and IL-1α, TNF-α, and IL-6 are distributed in the same position. Meanwhile, IL-6 stimulates the production of IL-1β and TNF-α and aggravates inflammatory responses through the combined action of the two [Citation24]. TNF-α, which plays a crucial role in the degenerative process involving articular cartilage, interferes with the synthesis of cartilage collagen and the production of proteoglycan and affects cartilage destruction and synovitis in OA [Citation25]. In our study, overexpression of NAV2-AS5 inhibited the effects of LPS on the release of IL-1β, TNF-α, and IL-6 in chondrocytes, suggesting that NAV2-AS5 might improve OA by inhibiting inflammation. However, how does NAV2-AS5 regulate chondrocyte proliferation, apoptosis, and the release of inflammatory factors?
Previous studies have shown that lncRNAs and miRNAs affect the occurrence and development of many diseases, including OA, through transcriptional or post-transcriptional regulation. Many studies have revealed that lncRNA, a competing endogenous RNA (ceRNA), promotes cartilage degradation in human OA [Citation26]. For instance, Liu. et al. found that lncRNA-MSR regulates TMSB4 expression through competition with miRNA-152, thus affecting cartilage degradation [Citation27]. Lu. et al. found that lncRNA-CIR is involved in oxidative stress-related apoptosis of chondrocytes in OA by modulating the miR-130a/Bim axis [Citation28]. These results illustrate that lncRNAs play important roles in OA by targeting miRNAs. In this study, miR-8082 was verified to be a target of lncRNA NAV2-AS5. The level of miR-8082 was upregulated in OA tissues and LPS-treated chondrocytes, and miR-8082 was negatively regulated by lncRNA NAV2-AS5. In addition, we observed that miR-8082 knockdown significantly reversed the LPS-induced decrease in cell viability and increase in cell apoptosis and the release of inflammatory factors. These results indicated that lncRNA NAV2-AS5 modulates OA progression by sponging miR-8082.
To further study the regulatory mechanism involving miR-8082 in OA, we identified downstream target genes of miR-8082 and TNIP2. TNIP2 (also known as ABIN2) was first discovered in a yeast two-hybrid screen as a binding partner of the zinc finger protein A20 (A20), a negative regulator of NF-κB signaling [Citation29]. It has been reported that TNIP2, as an anti-inflammatory signaling molecule, negatively regulates allergic airway responses [Citation30]. In this research, TNIP2 mRNA levels were downregulated in OA tissues and LPS-induced chondrocytes. In addition, rescue experiments showed that knockdown of TNIP2 transposed the impact of miR-8082 inhibitor on cell proliferation, apoptosis, and inflammation in sh-NAV2-AS5-treated chondrocytes.
In conclusion, our study revealed that NAV2-AS5 regulates OA progression via the miR-8082–TNIP2 axis, indicating that NAV2-AS5 is a critical molecule involved in OA progression.
Ethics approval and consent to participate
This study was authorized by the Ethics committee of The Second Affiliated Hospital of The Air Force Medical University and carried out according to the Declaration of Helsinki.
Consent for publication
The authors consent for publication in the Journal.
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Supplemental Material
Download PDF (306.9 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2022.2154554
Additional information
Funding
References
- Pearson MJ, Philp AM, Heward JA, et al. Long intergenic noncoding RNAs mediate the human chondrocyte inflammatory response and are differentially expressed in osteoarthritis cartilage. Arthritis Rheumatol. 2016;68(4):845–856. DOI:10.1002/art.39520
- Bijlsma JW, Berenbaum F, Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. 2011;377(9783):2115–2126.
- Glyn-Jones S, Palmer AJR, Agricola R, et al. Osteoarthritis. Lancet. 2015;386(9991):376–387. DOI:10.1016/S0140-6736(14)60802-3
- Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380(9859):2163–2196. DOI:10.1016/S0140-6736(12)61729-2
- Kandahari AM, Yang X, Dighe AS, et al. Recognition of immune response for the early diagnosis and treatment of osteoarthritis. J Immunol Res. 2015;2015:192415.
- Wang X, Hunter D, Xu J, et al. Metabolic triggered inflammation in osteoarthritis. Osteoarthritis Cartilage. 2015;23(1):22–30.
- Vaamonde-Garcia C, Riveiro-Naveira RR, Valcárcel-Ares MN, et al. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012;64(9):2927–2936. DOI:10.1002/art.34508
- Wang T, He C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018;44:38–50.
- Liu YP, Li J, Xin SB, et al. Study the relevance between inflammatory factors and estradiol and their association with knee osteoarthritis in postmenopausal women. Eur Rev Med Pharmacol Sci. 2018;22(2):472–478.
- Song J, Ahn C, Chun CH, et al. A long non-coding RNA, GAS5, plays a critical role in the regulation of miR-21 during osteoarthritis. J Orthop Res. 2014;32(12):1628–1635.
- Gao GC, Cheng XG, Wei QQ, et al. Long noncoding RNA MALAT-1 inhibits apoptosis and matrix metabolism disorder in interleukin-1β-induced inflammation in articular chondrocytes via the JNK signaling pathway. J Cell Biochem. 2019;120(10):17167–17179.
- Chen L, Zhang Y, Rao Z, et al. Integrated analysis of key mRnas and lncRnas in osteoarthritis. Exp Ther Med. 2018;16:1841–1849.
- Hu J, Wang Z, Shan Y, et al. Long non-coding RNA HOTAIR promotes osteoarthritis progression via miR-17-5p/FUT2/β-catenin axis. Cell Death Dis. 2018;9(7):711. DOI:10.1038/s41419-018-0746-z
- He H, Liu X, Liu Y, et al. Human papillomavirus E6/E7 and long noncoding RNA TMPOP2 mutually upregulated gene expression in cervical cancer cells. J Virol. 2019;93(8). DOI:10.1128/JVI.01808-18.
- Chen H, Yang S, Shao R. Long non-coding XIST raises methylation of TIMP-3 promoter to regulate collagen degradation in osteoarthritic chondrocytes after tibial plateau fracture. Arthritis Res Ther. 2019;21(1):271.
- Yu C, Shi D, Li Z, et al. Retracted : long noncoding RNA CHRF exacerbates IL-6-induced inflammatory damages by downregulating microRNA-146a in ATDC5 cells. J Cell Physiol. 2019;234(12):21851–21859.
- Chen WK, Yu X-H, Yang W, et al. lncRnas: novel players in intervertebral disc degeneration and osteoarthritis. Cell Prolif. 2017;50(1):e12313. DOI:10.1111/cpr.12313
- Yang Z, Tang Y, Lu H, et al. Long non-coding RNA reprogramming (lncRNA-ROR) regulates cell apoptosis and autophagy in chondrocytes. J Cell Biochem. 2018;119(10):8432–8440. DOI:10.1002/jcb.27057
- Chen K, Fang H, Xu N. LncRNA LOXL1-AS1 is transcriptionally activated by JUND and contributes to osteoarthritis progression via targeting the miR-423-5p/kdm5c axis. Life Sci. 2020;258:118095.
- Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6(11):625–635.
- Wojdasiewicz P, Poniatowski LA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459.
- Imamura M, Ezquerro F, Marcon Alfieri F, et al. Serum levels of proinflammatory cytokines in painful knee osteoarthritis and sensitization. Int J Inflam. 2015;2015:329792.
- Tunjungputri RN, Li Y, de Groot P, et al. The inter-relationship of platelets with interleukin-1β-mediated inflammation in humans. Thromb Haemost. 2018;118(12):2112–2125. DOI:10.1055/s-0038-1675603
- Livshits G, Zhai G, Hart DJ, et al. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: the Chingford study. Arthritis Rheum. 2009;60(7):2037–2045. DOI:10.1002/art.24598
- Reboul P, Pelletier JP, Tardif G, et al. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J Clin Invest. 1996;97(9):2011–2019.
- Kang Y, Song J, Kim D, et al. PCGEM1 stimulates proliferation of osteoarthritic synoviocytes by acting as a sponge for miR-770. J Orthop Res. 2016;34(3):412–418. DOI:10.1002/jor.23046
- Liu Q, Hu X, Zhang X, et al. The TMSB4 pseudogene LncRNA functions as a competing endogenous RNA to promote cartilage degradation in human osteoarthritis. Mol Ther. 2016;24(10):1726–1733. DOI:10.1038/mt.2016.151
- Lu Z, Luo M, Huang Y. lncRNA-CIR regulates cell apoptosis of chondrocytes in osteoarthritis. J Cell Biochem. 2018;120(5):7229–7237.
- Van Huffel S, Delaei F, Heyninck K, et al. Identification of a novel A20-binding inhibitor of nuclear factor-κappa B activation termed ABIN-2. J Biol Chem. 2001;276(32):30216–30223.
- Ventura S, Cano F, Kannan Y, et al. A20-binding inhibitor of NF-κB (ABIN) 2 negatively regulates allergic airway inflammation. J Exp Med. 2018;215(11):2737–2747. DOI:10.1084/jem.20170852