
ABSTRACT
The centrosome acts as a protein platform from which proteins are deployed to function throughout the cell cycle. Previously, we have shown that the prolyl isomerase Cyclophilin A (CypA) localizes to the centrosome in interphase and re-localizes to the midbody during mitosis where it functions in cytokinesis. In this study, investigation of CypA by SDS-PAGE during the cell cycle reveals that it undergoes a mobility shift during mitosis, indicative of a post-translational modification, which may correlate with its subcellular re-localization. Due to the lack of a phospho-specific antibody, we used site-directed mutagenesis to demonstrate that the previously identified serine 77 phosphorylation site within CypA is important for control of CypA centrosome localization. Furthermore, CypA is shown to interact with the mitotic NIMA-related kinase 2 (Nek2) during interphase and mitosis, while also interacting with the Nek2-antagonist PP1 during interphase but not during mitosis, suggesting a potential role for the Nek2-PP1 complex in CypA phospho-regulation. In support of this, Nek2 is capable of phosphorylating CypA in vitro. Overall, this work reveals that phosphorylation of CypA at serine 77 is important for its release from the centrosome during mitosis and may be regulated by the activity of Nek2 and PP1 during the cell cycle.
Introduction
The centrosome is well established as the major microtubule-organizing center (MTOC) in mammalian cells that regulates cell shape, polarity and spindle pole structure [Citation1]. Each centrosome is comprised of a pair of cylindrical microtubule-based structures known as centrioles that are surrounded by a protein rich pericentriolar matrix (PCM). The centrosome undergoes a cycle of duplication, maturation and disjunction, which occurs in tandem with the cell cycle. Thus, at the onset of S phase a cell contains one centrosome that is duplicated during S phase, providing the cell with two centrosomes upon completion. At mitotic entry, the centrosome pair separate and form the mitotic spindle poles from which microtubules radiate and capture chromosomes on the mitotic spindle [Citation2–5]. In recent years, it has been proposed that centrosomes act as signaling centers due to the abundance of kinases and phosphatases that locate there [Citation6] including the NIMA-related kinases (Neks) [Citation7], cyclin-dependent kinases (Cdks) [Citation8], polo-like kinases (Plks) [Citation9] aurora kinases [Citation10], protein phosphatase 1 and 2 (PP1/2) [Citation11,Citation12] and cdk-activating phosphatases [Citation13].
Cyclophilin A (CypA) is an 18 kDa peptidyl-prolyl isomerase with functions in protein folding and trafficking, T-cell function and viral infection [Citation14–16]. CypA is over-expressed in a variety of human cancer cells including hematopoietic cancer [Citation17], prostate [Citation18], lung [Citation19] and oral squamous cell carcinoma [Citation20], and has been shown to promote proliferation of a variety of cancers [Citation21–24]. It has been previously demonstrated that CypA localizes to the centrosome in human cancer cells during interphase and mitotic entry, and is subsequently displaced from the centrosome, re-localizing to the midzone during mitosis and the midbody during cytokinesis where it plays a role in abscission of the intercellular bridge joining two daughter cells [Citation17]. However, the spatial and temporal regulation of CypA during the cell cycle remains to be elucidated.
Nek2 is a member of the NIMA-related serine/threonine kinase family that is conserved across eukaryotic species. Nek2 is located to the centrosome and plays a key role in regulation of centrosome structure [Citation25]. The Mst2 kinase phosphorylates Nek2 which controls its centrosome localization [Citation26]. Nek2 is unphosphorylated during interphase, however, during entry into mitosis the Mst2 kinase phosphorylates Nek2, mediating its centrosome localization. At the centrosome, Nek2 phosphorylates C-Nap1 and Rootletin within a proteinaceous linker that tethers two mature centrosomes resulting in C-Nap1 and Rootletin displacement from the centrosome and dissolution of the linker leading to centrosome separation, mitotic onset and mitotic spindle pole formation [Citation27,Citation28]. Protein phosphatase 1 (PP1) counteracts Nek2 through its dephosphorylation and inactivation [Citation29,Citation30], thereby promoting centrosome cohesion [Citation31].
In recent years additional Nek2 substrates have been identified. Nek2 phosphorylates Cep68 [Citation32], Centlein [Citation33], Disheveled and β-catenin to control centrosome separation [Citation34,Citation35], whereas phosphorylation of Nip2/Centrobin controls microtubule stabilization [Citation36], and phosphorylation of Hec1 and Mad2 is essential for spindle assembly checkpoint (SAC) function [Citation37,Citation38]. Collectively, these studies highlight centrosomal Nek2 as a master regulator of a variety of cell cycle functions. The identity of additional Nek2 substrates at the centrosome, together with an understanding of their functional significance and regulation, will expand our knowledge of core centrosome signaling networks.
Building on the work of Bannon et al., 2012, in this study the importance of a previously identified phosphorylation site, serine 77 [Citation39], in the displacement of CypA from the centrosome is presented for the first time. We also demonstrate that CypA co-immunoprecipitates with Nek2, and its antagonistic partner PP1, providing support for the existence of a Nek2-PP1-CypA complex. Furthermore, CypA is phosphorylated by Nek-2 in vitro. Collectively this data supports a model whereby Nek2-mediated phosphorylation of CypA controls its subcellular localization during mitosis.
Materials and methods
Cell culture, transfection and synchronization
All cells were obtained from the European Collection of Cell Cultures (Porton Down, Wiltshire, UK) and grown in RPMI-1640 GlutaMAX medium (Gibco) containing 100 µg/mL penicillin/streptomycin (Sigma) and 10% (v/v) fetal bovine serum (FBS) (Gibco). JurkatCypA-/- cells were obtained through the AIDS Research and Reference Reagent Program and were created as described previously [Citation17]. Midbody enrichment was achieved by treatment of cells with 160 nM nocodazole (Sigma) for 16 hr followed by washing in PBS before release into complete media. Synchronization using double-thymidine block (2 µM) followed by treatment with the Cdk1 inhibitor RO-3306 (9 µM) was achieved as outlined in . Plasmid transfections in Jurkat and K562 cells were performed using the Amaxa biosystems Cell Line Nucelofector® Kit V as per the manufacturer’s guidelines. Used plasmids are summarized in the Supplementary Information (SI), Table S1.
Site-directed mutagenesis
Site-directed mutagenesis (SDM) was performed using Phusion® High-Fidelity DNA Polymerase from New England BioLabs® as per the manufacturer’s recommendations. Thermocycling conditions were as follows: initial denaturation at 98°C for 30 s, 16–20 cycles of 98°C 5–10 s; 45-72°C* 10–30 s; 72°C for 30 s/kb plasmid, followed by a final extension step at 72°C for 10 min before hold at 4°C. The annealing temperature at step * was determined on the basis of each primer set; Tm was calculated by either using the New England BioLabs® Tm calculator (found at: https://tmcalculator.neb.com/#!/main) or chosen using the guidelines listed in the troubleshooting section of the Phusion® protocol found online at https://international.neb.com/Protocols/0001/01/01/pcr-protocol-m0530. The resulting PCR products were digested with DpnI (New England BioLabs®) at 37°C for 1 hr, followed by analysis through gel electrophoresis. Successful SDM reactions were ligated using T4 DNA Ligase (Promega) for 5 min at room temperature before chemical transformation into XL1 Escherichia coli for subsequent miniprep preparation of the plasmid (QIAprep Spin Miniprep Kit, QIAGEN) and confirmation of mutagenesis by sequencing. All sequencing was performed by Eurofins Genomics.
Immunoblotting and immunofluorescence
Immunoblotting was performed as described previously [Citation17]. All antibodies used are listed in SI, Table S1. For co-immunoprecipitations, proteins were visualized by enhanced chemiluminescence solution (Pierce) with Fuji Medical Super HR-T 30 autoradiography film. For all other blots, proteins were visualized by probing with DyLightTM Fluor 688 (mouse) or 800 (rabbit) conjugate secondary antibodies (ThermoFisher) and imaged using the LI-COR® Biosciences Odyssey® Infrared Imaging System. All original uncropped blots presented within this manuscript can be found within the SI.
For immunofluorescence, fixation and immunostaining was performed as described previously in reference 17. Cells were incubated with the relevant primary antibodies which were detected with either anti-rabbit or anti-mouse Alexa Fluor 594 or 488 secondary antibodies (Invitrogen). Nuclei were stained with DAPI (Sigma). Coverslips were mounted using Dako Cytomation fluorescent mounting medium (Dako Inc.). Stained cells were visualized using an Olympus Fluoview FV1000 confocal microscope with a 60× oil objective lens (numerical aperture (NA) 1.4) and images were acquired by FV10-ASW 3.0 software, with further analysis performed using Fiji. Significant differences observed in protein localization for each mutant to the centrosome and midbody were calculated relevant to EGFP-CypAWT localization at each by a two-tailed, two-sample t-test assuming unequal variance. All error bars presented represent standard error from the mean (SEM), unless stated otherwise.
Flow cytometry
Samples were prepared for flow cytometry as described previously in reference 17. The cell cycle profile was analyzed using the BD AccuriTM C6 Flow Cytometer. De NovoTM FCS Express (Ver 5 & 6) software was used for DNA multicycle analysis to calculate the percentage of cells present in the three main phases of the cell cycle (%G1, %S and %G2/M). For calculation of these percentages, the appropriate model was chosen for each sample based on the resulting Chi square values as recorded by the software.
Purification of recombinant protein
Escherichia coli BL21 Rosetta cells (Merck Millipore) were transformed with pD444-CypAWT as well as the equivalent phospho-defective plasmids generated through SDM (see above). A 10 mL overnight (2×TY broth (Fisher Scientific), 2% (w/v) glucose (Sigma), 50 µg/µL ampicillin (Formedium), 34 µg/µL chloramphenicol (Sigma)) was generated from a single colony and transferred to a 400 mL culture (2×TY broth, 0.1% (v/v) glucose, ampicillin, chloramphenicol) and shaken at 37°C until an OD600 nm of 0.6 minimum was reached. IPTG (Formedium) was added to a final concentration of 0.1 mM and the culture was grown for a further 5 hr at 30°C shaking. Cells were collected by centrifugation (6,000 RPM for 20 min) and pellets were frozen at −20°C for a minimum of 1 hr. Pellets were thawed and resuspended in IMAC lysis buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, pH 8) before sonication (5 × 5 10 s pulses with 10 s interval between each pulse). The supernatant was cleared by centrifugation at 10,000 × g at 4°C for 30 min. The clarified supernatant was passed through 1 mL (50% slurry) of HisPur Ni-NTA (ThermoFisher) resin equilibrated in IMAC lysis buffer in a gravity flow column. The bound protein was washed twice (50 mM sodium phosphate, 300 mM NaCl, 50 mM imidazole, pH 8) before elution in 4 × 500 µL fractions in elution buffer (50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazole, pH 8). Eluates were immediately subjected to size exclusion chromatography (SEC) using a 10/300 Tricorn column (GE Healthcare) packed with Superdex 75 prep grade resin (GE Healthcare) (HTEP: 0.0165, N: 1.788, h: 4.85) at a flow rate of 0.5 mL/min. The column was washed with 2× column volumes (CV) of filtered and degassed H2O, and 2× CV of running buffer (PBS, pH 7.4) before samples were applied manually using an 800 µL sample loop. Recombinant CypA eluted within 1.5× CV and was supplemented with 10% glycerol before long-term storage at −80°C.
Co-immunoprecipitation
Immunoprecipitation of exogenously expressed EGFP-tagged proteins was performed using GFP-Trap© (ChromoTek GmbH) beads according to the manufacturer’s protocol. Mammalian cells were transiently transfected with the relevant EGFP-tagged construct as described and incubated at 37°C in 5% CO2 for a period of 24–72 hr. Cells were collected by centrifugation at 430 RCF for 3 min, washed once in 1 mL of PBS and lysed in 200 µL co-IP lysis buffer (10 mM Tris/HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, supplemented with 1 mM PMSF, 1 µg/mL aprotinin, 1 µg/mL leupeptin, 1 µg/mL pepstatin A) and lysed by centrifugation at max speed for 30 min at 4°C in a bench-top centrifuge. Protein concentration was determined by Bradford assay (Sigma) and 1.5 mg of protein was subsequently diluted with Dilution/Wash buffer (10 mM Tris/HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA) to a final volume of 500 µL. GFP-Trap© beads were equilibrated, with a 25 µL bead slurry being used for each pull-down. Following equilibration, beads were then added to the diluted protein lysates and tumbled end-over-end for a minimum of 1 hr at 4°C. The resulting post-bind (PB) was kept while bound proteins were eluted from the beads by the addition of 50 µL 4× Laemmli sample buffer and heating at at 95°C for 10 min. SDS-PAGE and Western Blot were used to determine both pull-down efficiency and co-immunoprecipitation of proteins of interest.
In vitro kinase assay
Kinase assays were set up with commercial or purified recombinant proteins. For each kinase assay, either active or kinase-inactive (“kinase-dead”, KD) 0.4 µg Nek2 (Abcam, ab60342) was incubated with 2 µg Casein (Sigma) as a positive control or 2 µg of the protein of interest and made up to 30 µL using kinase reaction buffer (50 mM HEPES pH 7.5, 1 mM MgCl2, 2.5 mM EGTA, 0.1 mM dithiothreitol (DTT), 0.1 mM Na3VO4) containing 100 µM ATP and 1 µCi of [γ- Citation3232P] ATP (Perkin-Elmer). KD-Nek2 was created by incubating 0.4 µg active Nek2 with 5 mM DTT and incubating at 95°C for 10 min prior to assay set-up. The reaction was incubated at 30°C for 1 hr and stopped by the addition of 4× Laemmli sample buffer followed by heating at 95°C for 5 min. The samples were resolved by SDS-PAGE and examined by Western blot once the radioactivity of the isotope had sufficiently decayed. Initial analysis of [γ-Citation3232P] ATP incorporation was examined by autoradiography (Fuji Medical Super HR-T 30 autoradiography film). Significant differences observed in phosphorylation levels for each mutant were calculated by densitometry of their respective signals to that of His-CypAWT using a two-tailed, two sample t-test assuming unequal variance.
Isomerase assay
Peptidyl-prolyl cis/trans isomerase activity was determined using a protease-coupled assay in a multi-mode CLARIOStar microplate reader (BMG Labtech). The substrate peptide, N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Sigma), was dissolved in tetrafluoroethylene/lithium acetate (500 mM) and used at a final concentration of 80 µM. Each reaction well was made up with 50 µL isomerase buffer (100 mM NaCl, 40 mM HEPES, pH 7.9), 2 µL chymotrypsin (1 mM in 2 mM calcium chloride and 1 mM HCl, final concentration of 10 µM), 100 nM of the recombinant protein being assessed and H2O to a total reaction volume of 200 µL following substrate addition. Each reaction well was mixed gently by pipetting prior to substrate injection. Hydrolysis of substrate was detected at 390 nm immediately after substrate addition, with readings taken every 1 s for 120 s.
Results
CypA undergoes phosphorylation during mitosis that correlates with centrosome displacement and midbody recruitment during mitosis
In agreement with previous research [Citation17], we demonstrate that EGFP-CypAWT localizes to the centrosome and midbody in malignant hematopoietic cells (Supplementary Information (SI), Fig. S1), however detail of CypA localization to these structures remains unknown. The dissociation of CypA from the centrosome and re-localization to the midbody during mitosis is essential to allow for its function in timely abscission of the intercellular bridge during cytokinesis [Citation17]. Protein localization to distinct subcellular sites is often controlled by phosphorylation [Citation40]. In this study, investigation of a number of malignant hematopoietic cell lines including KYO.1, Lama84 and K562 cells revealed that CypA indeed undergoes a mobility shift when resolved by SDS-PAGE, the most striking of which is visible in K562 cells which appears to exist almost completely as a slower mobility form (). The decreased mobility detected is suggestive of post-translational modification(s) and is in agreement with other reports of CypA acetylation in response to reactive oxygen species in vascular smooth muscle cells (VSMCs), and phosphorylation in other non-malignant HEK293 cells, cancerous HeLa cells and K562 leukemic cells [Citation39,Citation41,Citation42]. In this study, in light of the cell-cycle dependent localization of CypA to either the centrosome or midbody, the mobility of CypA was investigated during mitotic progression upon release from the CDK1 inhibitor RO-3306 (). Results reveal that in asynchronous K562 cells, CypA exists as a doublet with the majority present in a slower mobility form. The mobility change in CypA is too small to represent the truncated 11 kDa CypA isoform. However, examination of mitotic cell extracts reveal that CypA undergoes additional modification evident through a further mobility shift up to 120 mins following release from RO-3306. It is noted that almost the total amount of CypA detected is subjected to enhanced slower mobility during mitosis. Densitometry analysis of 3 independent experiments demonstrates that the mobility shift in CypA correlates with cyclin B1 and Nek2 expression from 0–120 min following release from RO-3306. Furthermore, the slower mobility form of CypA is lost at 180 min following release from RO-3306, which correlates with decreasing cyclin B1 levels (Fig S1B, S1C and SI S3). Collectively this data suggests that within these cells, CypA exists in multiple forms with varied mobility detected by SDS-PAGE suggestive of differentially post-translational modified forms, including post translational modification specifically during mitosis.
Figure 1. Phosphorylation of CypA at Ser77 regulates subcellular localization.
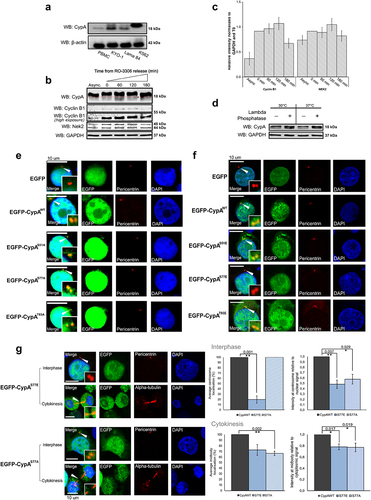
The modification of CypA during mitosis correlates with expression of the mitotic kinase Nek2, which also decreases 180 min following release from RO-3306. (). To investigate phosphorylation as a candidate post-translational modification during mitosis, K562 mitotic extracts prepared following release from RO-3306 for 60 min were treated with Lambda phosphatase and resolved on high percentage SDS-PAGE gels (). CypA within the untreated cell extracts presents as a doublet representing differentially modified forms, as observed previously. Importantly, phosphatase treatment was found to alter the ratio of doublet bands and decrease the fraction of slower mobility CypA demonstrating that phosphorylation contributes to the reduced mobility of CypA in these cells. The finding that the phosphatase treatment did not completely remove the slower mobility form may be due to insufficient active enzyme present in the assay, however, it may also indicate that CypA undergoes alternative modification also, such as acetylation [Citation43]. Overall, these results reveal that phosphorylation contributes to CypA post-translational modifications during mitosis.
The altered mobility of CypA detected during mitosis in this study coincides with previously reported subcellular localization of endogenous CypA [Citation17] and exogenous (pEGFP-CypAWT, SI, Fig. S1) CypA from the centrosome to midbody, and suggests that post-translational modification may control CypA localization during mitosis. Literature reports have already revealed that CypA is phosphorylated at Ser77 and Thr93 in K562 and HeLa cells [Citation39,Citation42], however the functional significance of phosphorylation at these sites, or the kinase involved, has not been investigated. To address this, examination of amino acids surrounding Ser77 and Thr93 using the Eukaryotic Linear Motif resource (ELM, available at http://elm.eu.org/ [Citation44] revealed that they lie within recognition sites for the mitotic kinase Nek2 (S77 (GGKSIY), T93 (LKHTGP)), together with Ser51 (YKGSCF) that has not previously been reported as a putative CypA phosphorylation site [Citation39,Citation42]. Nek2 is reported to induce phosphorylation-mediated dissociation of a number of centrosome proteins [Citation27,Citation45–47]. Furthermore, we found that the expression levels of Nek2 correlate with altered CypA mobility (), supporting the possibility that these CypA sites may be phosphorylated by Nek2 during mitosis to control localization.
Phosphorylation at Ser77 is important for centrosome displacement and midbody localization of CypA
To investigate the functional significance of phosphorylation at Ser51, Ser77 or Thr93 in mediating centrosome localization of CypA, K562 cells were transfected with pEGFP-CypAWT or a series of single-site mutants where Ser51, Ser77 and Thr93 were mutated to phospho-defective or phospho-mimetic forms by the introduction of alanine or glutamine respectively. The subcellular localization of the wild-type and mutant proteins was investigated by immunofluorescence and confocal microscopy. Results reveal that EGFP-CypAWT and all three phospho-defective mutants are capable of centrosome localization (). However, it was noted that while EGFP-CypAS77A was capable of binding to the centrosome it displayed a decrease in fluorescence intensity when compared to EGFP-CypAWT (p-value <0.05) (). We next investigated centrosome localization of phospho-mimetic CypA. It was found that EGFP-CypAS51E and EGFP-CypAT93E localized to the centrosome similar to wild-type CypA (), however, in contrast, the EGFP-CypAS77E phospho-mimetic could not be detected at the centrosome in 80% of the cell population. Furthermore, in the proportion of cells where EGFP-CypAS77E was bound, they displayed a significant decrease in fluorescence intensity at the centrosome (p-value <0.01) () suggesting weak binding. Overall, this data suggests that phosphorylation of CypA on Ser53 and Thr93 is not important for centrosome localization, however, the phosphorylation status of Ser77 is important for localization to the centrosome.
To investigate the role for Ser77 on midbody recruitment of CypA, the localization of the EGFP-CypA Ser77 phospho-mimetic and phospho-defective mutants was examined. Similar to findings at the centrosome, EGFP-CypAS51E and EGFP-CypAT93E bound to the midbody similar to wild type (Fig. S2). Qualitative results reveal aberrant localization of EGFP-CypAS77A to the midbody (p-value <0.01), whereas EGFP-CypAS77E localized to the midbody similar to wild-type protein (). When normalized to nuclear EGFP signal however, a decrease in signal intensity at the midbody was observed for both EGFP-CypAS77A and EGFP-CypAS77E (). This may be due to mutation of the native serine amino acid to alanine or glutamine, which does not fully recapitulate protein-protein interactions mediated by native amino acid, in unphosphorylated and phosphorylated form, thereby leading to weaker binding. Alternatively, we cannot rule out the possibility that additional modifications or interactions are required for stable localization of CypA to the midbody.
So far, these results demonstrate that mutation of S51 or T93 to alanine or glutamine does not alter the ability of CypA to localize to the centrosome or midbody, whereas mutation of S77 alone impacts localization to these sites. This data reveals that S77 is important in regulating CypA subcellular localization and suggests a model whereby CypA that is unphosphorylated at Ser77 localizes to the centrosome, and phosphorylation of Ser77 during mitosis mediates centrosome displacement.
Nek2 and PP1 as potential regulators of CypA localization during the cell cycle
Results so far reveal a novel function for Ser77 in control of CypA localization during mitosis (). We also reveal that S77 lies within a Nek2 binding motif and that CypA phosphorylation during mitosis correlates with increased Nek2 expression (). We next investigated the ability of CypA and Nek2 to interact during mitosis by co-immunoprecipitation studies from mitotic cells following release from RO-3306. Cells were unsynchronized or synchronized by a double thymidine block followed by release into RO-3306 (). Flow cytometry was used to confirm enrichment of cells in G2/M () and Western blotting were used to confirm Nek2 expression during interphase (, lane 1), which is elevated during mitosis and correlates with cyclin B1 expression (). Results reveal that Nek2 co-immunoprecipitates with EGFP-CypAWT during mitosis (). An interaction between CypA and Nek2 was investigated during interphase and interestingly, it was found that Nek2 also co-immunoprecipitates with CypA during interphase when both are associated with the centrosome () [Citation7,Citation17]. Taken together, these results reveal that CypA interacts with Nek2 throughout the cell cycle.
Figure 2. CypA interacts with Nek2 and PP1 during the cell cycle.
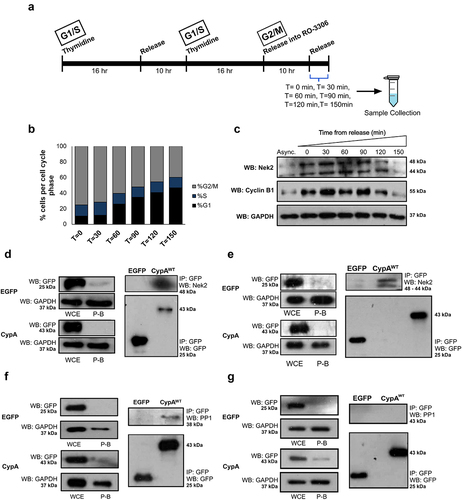
Nek2 exists in a complex with PP1 at the centrosome, and Nek2 kinase activity is regulated by phosphorylation and dephosphorylation by the opposing activity of PP1 and the Hippo pathway kinase Mst2 [Citation26]. CypA contains a putative PP1 docking motif (RVXF) at residues 123–130 (DGKHVVFG) that is important for PP1 binding [Citation44]. To investigate the possibility that CypA interacts with PP1, EGFP-CypAWT was immunoprecipitated from K562 cells during interphase and mitosis as before. Results reveal that PP1 co-immunoprecipitates with EGFP-CypAWT during interphase () suggesting that it may form part of a Nek2-PP1 complex. However, PP1 does not co-immunoprecipitate with CypA during mitosis (), in contrast to Nek2 which interacts with CypA during interphase and mitosis, as outlined (). This data suggests that during interphase CypA may exist in a complex with Nek2 and PP1 at the centrosome and that during mitosis, PP1 is displaced from the complex. Based on the data presented, it is possible that displacement of PP1 during mitosis, which is also coupled with elevated Nek2 expression, leads to increased Nek2 activity toward CypA.
Nek2 phosphorylates recombinant CypA in vitro
Having determined the importance of the putative Nek2 phosphorylation site Ser77 in the subcellular localization of CypA and highlighted the existence of a potential regulatory Nek2-PP1-CypA complex, we examined Nek2 as the candidate kinase responsible for CypA phosphorylation during mitosis.
An in vitro kinase assay was performed whereby wild-type Nek2, or kinase inactivated Nek2 (referred to as kinase-dead Nek2 (KD-Nek2)) was incubated with recombinant CypA and [ƴ-Citation3232P]-ATP (). Casein was included as a positive control [Citation45,Citation46]. Nek2 autophosphorylation was evident consistent with published data [Citation47], and absent from the KD-Nek2 samples. Results reveal CypA phosphorylation in the presence of active Nek2, and not KD-Nek2, demonstrating that it is a novel Nek2 substrate in vitro. Furthermore, multiple phosphorylation bands are detected on CypA suggesting that it is phosphorylated on multiple sites in vitro, similar to Casein as previously reported [Citation45,Citation46].
Figure 3. CypA is phosphorylated by Nek2 in vitro.
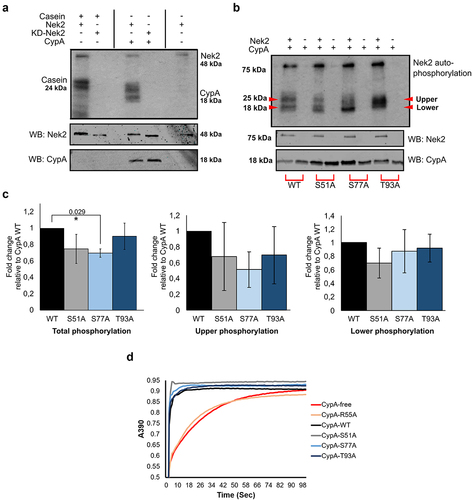
Next, Ser51, Ser77 and Thr93 were investigated as candidate Nek2 phosphorylation sites. Recombinant wild-type CypA (His-CypAWT) and single-site phospho-defective mutants were incubated in an in vitro kinase assay with Nek2 as before (). Results show that the single mutants undergo phosphorylation, albeit at levels less than CypAWT, and interestingly exhibit multiple banding patterns of phosphorylation depending on the mutant (). Densitometry analysis of total banding profile revealed that CypAS77A was significantly reduced compared to wild-type (). Further analysis of the intensity of upper and lower CypA forms reveals that the upper phosphorylation banding pattern was reduced on CypAS77A whereas the lower banding pattern was unchanged. Overall, this data demonstrates that Nek2 can phosphorylate CypA in vitro, and mutation of Ser77 in the CypAS77A protein results in a reduction in total phosphorylation levels. Furthermore, the presence of multiple banding patterns for each phospho-mutant, together with the inability to completely ablate phosphorylation by the alteration of a single site is consistent with the idea that CypA may be phosphorylated at multiple sites by Nek2.
We have previously demonstrated that CypA isomerase activity is not required for centrosome or midbody localization, however, it is important for timely abscission during cytokinesis [Citation17]. To examine the importance of Ser51, Ser77 and Thr93 for CypA enzymatic activity, the isomerase action of recombinant His-CypAWT, the isomerase defective His-CypAR55A mutant and Ser/Thr mutants, His-CypAS51A, His-CypAS77A and His-CypAT93A were tested in an α-chymotrypsin-based assay using the synthetic substrate N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide as described [Citation48,Citation49]. His-CypAWT accelerated the substrate cleavage whereas the His-CypAR55A isomerase inactive mutant did not (). Furthermore, it was found that the site-specific Ser/Thr mutants catalyzed isomerization similar to wild-type CypA, demonstrating that these residues are not important for CypA enzyme activity, and is consistent with the hypothesis that phosphorylation regulates subcellular localization independent of isomerase activity [Citation17].
CypA does not affect the ability of Nek2 or PP1 to localize to the centrosome or midbody
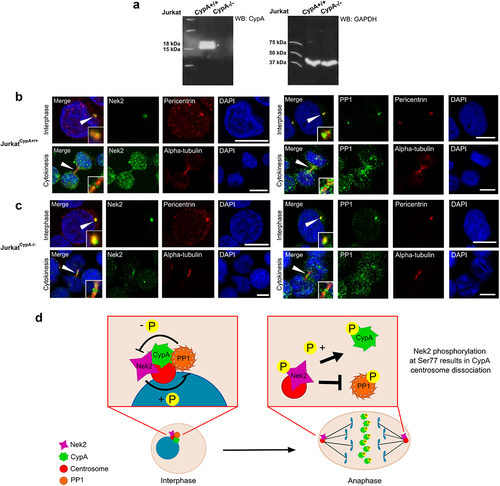
We and others have reported that CypA and Nek2 are centrosomal proteins that re-localize to the midbody during the cell cycle [Citation7,Citation17]. Similarly, PP1 has also been shown to undergo centrosome to midbody localization [Citation11]. This study has revealed novel interactions between CypA and Nek2 (), and CypA and PP1 (), and that CypA is a novel Nek2 substrate (). CypA is known to play a role in protein trafficking [Citation14,Citation50], therefore the possibility that CypA is required for Nek2 or PP1 trafficking to the midbody was finally examined. Subcellular localization of endogenous Nek2 and PP1 was examined in JurkatCypA+/+ and JurkatCypA-/- cells () at both interphase and cytokinesis using immunofluorescence and confocal microscopy (). In the presence or absence of CypA, it was found that both Nek2 and PP1 are capable of centrosome and midbody localization, confirming that while the interplay between Nek2 and PP1 may regulate CypA release from the centrosome, this release of CypA does not impact typical localization patterns of either protein.
Figure 4. CypA does not affect Nek2 or PP1 localization to the midbody.
Discussion
In recent years it has been proposed that the centrosome acts as a protein platform and signaling hub that controls important cellular events including cell cycle progression, DNA damage response, as well as cell shape, polarity and motility [Citation6,Citation51]. This is supported by evidence that cell cycle regulatory proteins such as polo-like kinase 1 (Plk1), cyclic AMP-dependent kinase, Aurora A, breast cancer type 2 susceptibility protein (BRCA2) and Cdk1 are localized at the centrosome [Citation52–55]. Furthermore, acentrosomal cells exhibit defects during cytokinesis suggesting that these signaling centers are not only essential during the early stages of mitosis but could provide necessary molecular cues for successful cytokinetic abscission [Citation56,Citation57].
A number of centrosome proteins are displaced during the cell cycle and subsequently recruited to the midbody where they function in abscission of the intercellular bridge, suggesting the co-ordinated deployment of proteins between these structures. For example, Plk1 which is responsible for mitotic entry, centrosome separation and maturation as well as microtubule-kinetochore attachment [Citation58] undergoes centrosome-to-midbody localization to enable the execution of multiple Plk1 functions during the cell cycle. Furthermore, Nek2 and PP1 also undergo centrosome-to-midzone and midbody localization during the cell cycle [Citation7,Citation11]. Centrosomal proteins such as Centriolin and centrosome protein 55 (Cep55) re-localize to the midbody where they mediate abscission of the intercellular bridge via recruitment of vesicle trafficking and fusion machinery [Citation59,Citation60]. Centriolin, a component of the maternal centriole, has been shown to localize to the midbody as a result of movement along the nuclear envelope or microtubules containing α-tubulin [Citation61], whereas in the case of Cep55 this re-localization is as a result of phosphorylation by Plk1 [Citation62]. The depletion of centrosome proteins such as Plk1, Cep55, BRCA2 and Centriolin leads to cytokinetic defects [Citation58,Citation60,Citation63,Citation64], highlighting their importance for the completion of cell division.
Transition through mitosis requires a number of important events including the timely onset of anaphase, central spindle formation, cleavage furrow ingression and cell abscission. We have previously shown that CypA is a centrosome protein during interphase and forms part of the bipolar spindle poles during early mitosis. During anaphase onset, CypA is displaced from the spindle poles and transitions to the midzone during telophase and eventually to the midbody during cytokinesis where it contributes to the timely completion of division, supporting the centrosome as a signaling center involved in the regulation of cell division [Citation17]. However, a better understanding of the regulatory factors that control the spatial and temporal mobility of centrosomes and their components during the cell cycle is required.
CypA is over-expressed in cancer cells and loss of CypA expression significantly reduced colony formation and suppressed proliferation [Citation24] thereby suggesting that CypA provides cancer cells with a mechanism for rapid proliferation. Consistent with that, a recent publication revealed that a novel cyclophilin inhibitor decreased cell proliferation and tumor growth in models of hepatocellular carcinoma [Citation65]. Previously, we revealed that the isomerase activity of CypA is essential for abscission of the intercellular bridge, however it is not required for centrosome localization or subsequent midbody re-localization [Citation17]. A better understanding of the mechanisms that regulate CypA localization to the centrosome, together with signals that mediate centrosome dissociation and midbody recruitment will provide insight into signal cascades exploited by cancer cells for accelerated growth.
In this study, it is shown that CypA undergoes a mobility shift during mitosis that is indicative of a post-translational modification, and which may correlate with its subcellular re-localization. Building on this, using site directed mutagenesis we reveal that the previously identified phosphorylation site, Ser77, is important in regulating subcellular localization of CypA. Results suggest that CypA unphosphorylated at Ser77 localizes to the centrosome and that phosphorylation during mitosis causes centrosome displacement, allowing for subsequent midbody recruitment. Furthermore, mutation of Ser77 to an unphosphorylated form does not impact CypA isomerase activity, which supports findings by Bannon et al. that CypA localization occurs independent of its isomerase activity [Citation17]. In addition, it was found that mutation of Ser51 or Thr93 to phospho-mimetic or -defective residues does not influence CypA centrosome or midbody association. Thus, we propose that a single phosphorylation event at Ser77 controls localization of CypA at the centrosome specifically. However, this should be confirmed using a phosphorylation specific antibody directed to Ser77, which is currently not commercially available and is a limitation of the study. Furthermore, although interacting partner(s) of CypA that are responsible for its centrosome docking remain to be identified, it is possible that phosphorylation at Ser77 causes a conformational change within CypA that favors its centrosome displacement. From our evidence that CypA S77E is capable of midbody localization, although with reduced intensity, suggests that the serine to glutamine mutation does not recapitulate natively phosphorylated residue, or that additional unidentified factors also contribute to CypA midbody binding.
Previous mass spectrometry studies have shown that CypA is indeed phosphorylated at Ser77 and Thr93 [Citation39,Citation42] (MS data summarized in SI Table 3 and 4). Our computational modeling revealed that these residues, together with Ser51, represent putative Nek2 phosphorylation sites [Citation44,Citation66,Citation67]. Furthermore, we demonstrate that increased levels of CypA phosphorylation are observed during mitotic progression, coinciding with increase Nek2 expression levels. Moreover, we provide evidence of an interaction between CypA and Nek2 during interphase and mitosis, and demonstrate that Nek2 can phosphorylate CypA at multiple residues in vitro, including Ser77.
The Nek2 kinase regulates phosphorylation-induced displacement of numerous centrosome proteins [Citation27,Citation31,Citation45,Citation68–70]. Nek2 activity is tightly controlled by the upstream kinase Mst2, and the Nek2 binding partner protein phosphatase 1 (PP1), the latter of which de-phosphorylates and inactivates Nek2 and its substrates [Citation29,Citation30]. Data from this study suggests that CypA phosphorylation is also regulated by the interplay between Nek2 and PP1. It was found that CypA and Nek2 interact during interphase and mitosis suggesting that proximity of the two proteins alone is insufficient for phosphorylation and displacement of CypA. CypA was also found to interact with PP1 during interphase suggesting that CypA exists in a complex with Nek2-PP1 at the centrosome during interphase consistent with other Nek2 substrates such as C-Nap1 [Citation45]. In contrast to its interaction with Nek2, CypA does not interact with PP1 during mitosis. Loss of PP1 from the complex during mitosis may represent the removal of an inhibitory block on Nek2, and coupled with increased Nek2 expression levels, could enhance Nek2 activity toward CypA Ser77 to promote centrosome displacement and midbody recruitment (), which is important for timely abscission of the intercellular bridge during cytokinesis and completion of cell division [Citation17]. Overall, this work describes a novel signal transduction cascade linking the centrosome and midbody that involves the prolyl isomerase CypA and is important for control of cell division.
Author contributions
RL Gorry, PT Lavin and MM Mc Gee designed the project, with RL Gorry, PT Lavin, K Brennan and R Sheridan performing the experiments. All authors discussed the results, with analysis performed by RL Gorry and PT Lavin. K Brennan helped with the molecular modeling of CypA as well as the design and optimization of the isomerase assay. PT Lavin created the phospho-defective S/T to A mutants in EGFP-CypA. RL Gorry and PT Lavin performed all confocal microscopy. R Sheridan and K Brennan performed the mobility shift western blots. RL Gorry generated the S/T to E mutants in EGFP-CypA as well as the S/T to A mutants in D444-SR-CypA and purified all recombinant forms of the protein used in the isomerase and kinase assays. RL Gorry and MM Mc Gee wrote the manuscript.
Supplemental Material
Download Zip (22.6 MB)Acknowledgements
We thank Dr Alfonso Blanco and Dr Fergal O’Meara in UCD Conway Institute, as well as Prof Jeremy C Simpson of the School of Biology and Environmental Science (SBES) for technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and/or its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2023.2167430.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Bornens M. The centrosome in cells and organisms. Science. 2012;335:422–426.
- Meraldi P, Nigg E. The centrosome cycle. FEBS Lett. 2002;521:9–13.
- Schatten H. The mammalian centrosome and its functional significance. Histochem Cell Biol. 2008;129:667–686.
- Azimzadeh J, Bornens M. Structure and duplication of the centrosome. J Cell Sci. 2007;120:2139–2142.
- Schatten H, Sun Q-Y. Functions and dysfunctions of the mammalian centrosome in health, disorders, disease, and aging. Histochem Cell Biol. 2018;150:303–325.
- Arquint C, Gabryjonczyk A-M, Nigg EA. Centrosomes as signalling centres. Philos Trans R Soc B Biol Sci. 2014;369:20130464.
- Ha Kim Y, Yeol Choi J, Jeong Y, et al. Nek2 localizes to multiple sites in mitotic cells, suggesting its involvement in multiple cellular functions during the cell cycle. Biochem Biophys Res Commun. 2002;290:730–736.
- Ferguson RL, Maller JL. Centrosomal localization of cyclin E-Cdk2 is required for initiation of DNA synthesis. Curr Biol. 2010;20:856–860.
- Jiang N, Wang X, Jhanwar-Uniyal M, et al. Polo box domain of Plk3 functions as a centrosome localization signal, overexpression of which causes mitotic arrest, cytokinesis defects, and apoptosis. J Biol Chem. 2006;281:10577–10582.
- Luca MD, Lavia P, Guarguaglini G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle. 2006;5:296–303.
- Trinkle-Mulcahy L, Andrews, P. D., Wickramasinghe, S., Sleeman, J., Prescott, A., Lam, Y. W, and Lamond, A. I. Time-lapse imaging reveals dynamic relocalization of PP1γ throughout the mammalian cell cycle. Mol Biol Cell. 2003;14:107–117.
- Mohr L, Buheitel, J., Schöckel, L., Karalus, D., Mayer, B., and Stemmann, O. An alternatively spliced bifunctional localization signal reprograms human shugoshin 1 to protect centrosomal instead of centromeric cohesin. Cell Rep. 2015;12:2156–2168.
- Boutros R, Ducommun B. Asymmetric localization of the CDC25B phosphatase to the mother centrosome during interphase. Cell Cycle. 2008;7:401–406.
- Kim H, Oh, Y., Kim, K., Jeong, S., Chon, S., Kim, D.,and Choe, W. Cyclophilin a regulates JNK/p38-MAPK signaling through its physical interaction with ASK1. Biochem Biophys Res Commun. 2015;464:112–117.
- Braaten D, Luban J. Cyclophilin a regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 2001;20:1300–1309.
- Brazin KN, Mallis RJ, Fulton DB, et al. Regulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclophilin a. Proc Natl Acad Sci. 2002;99:1899–1904.
- Bannon JH, O’Donovan DS, Kennelly SME, et al. The peptidyl prolyl isomerase cyclophilin a localizes at the centrosome and the midbody and is required for cytokinesis. Cell Cycle. 2012;11:1340–1353.
- Li M, Zhai, Q., Bharadwaj, U., Wang, H., Li, F., Fisher, W. E., and Yao, Q. Cyclophilin a is overexpressed in human pancreatic cancer cells and stimulates cell proliferation through CD147. Cancer. 2006;106:2284–2294.
- Yang H, Chen, J., Yang, J., Qiao, S., Zhao, S, and Yu, L Cyclophilin a is upregulated in small cell lung cancer and activates ERK1/2 signal. Biochem Biophys Res Commun. 2007;361:763–767.
- Fillies T, Werkmeister, R., van Diest, P. J., Brandt, B., Joos, U., and Buerger, H. HIF1-alpha overexpression indicates a good prognosis in early stage squamous cell carcinomas of the oral floor. BMC Cancer. 2005;5:84.
- Howard BA, Furumai, R., Campa, M. J., Rabbani, Z. N., Vujaskovic, Z., Wang, X. F., and Patz, E. F. Jr Stable RNA Interference–Mediated suppression of cyclophilin a diminishes non–small-cell lung tumor growth in vivo. Cancer Res. 2005;65:8853–8860.
- Feng W, Xin Y, Xiao Y, et al. Cyclophilin a enhances cell proliferation and xenografted tumor growth of early gastric cancer. Dig Dis Sci. 2015;60:2700–2711.
- Zheng J, Koblinski JE, Dutson LV, et al. Prolyl isomerase cyclophilin a regulation of janus-activated kinase 2 and the progression of human breast cancer. Cancer Res. 2008;68:7769–7778.
- Qian Z, Zhao X, Jiang M, et al. Downregulation of cyclophilin a by siRNA diminishes non-small cell lung cancer cell growth and metastasis via the regulation of matrix metallopeptidase 9. BMC Cancer. 2012;12:442.
- Fry AM. The Nek2 protein kinase: a novel regulator of centrosome structure. Oncogene. 2002;21:6184–6194.
- Ardestani A, Lupse B, Maedler K. Hippo signaling: key emerging pathway in cellular and whole-body metabolism. Trends Endocrinol Metab. 2018;29:492–509.
- Agircan FG, Schiebel E, Mardin BR. Separate to operate: control of centrosome positioning and separation. Philos Trans R Soc B Biol Sci. 2014;369:20130461.
- Mardin BR, Lange C, Baxter JE, et al. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat Cell Biol. 2010;12:1166–1176.
- Eto M, Elliott E, Prickett TD, et al. Inhibitor-2 regulates protein phosphatase-1 complexed with NimA-related kinase to induce centrosome separation. J Biol Chem. 2002;277:44013–44020.
- Mi J, Guo C, Brautigan DL, et al. Protein phosphatase-1α regulates centrosome splitting through Nek2. Cancer Res. 2007;67:1082–1089.
- Fry AM, Bayliss R, Roig J. Mitotic Regulation by NEK Kinase Networks. Front Cell Dev Biol. 2017;5:102.
- Man X, Megraw TL, Lim YP. Cep68 can be regulated by Nek2 and SCF complex. Eur J Cell Biol. 2015;94:162–172.
- Fang G, Zhang D, Yin H, et al. Centlein mediates an interaction between C-Nap1 and Cep68 to maintain centrosome cohesion. J Cell Sci. 2014;127:1631–1639.
- Bahmanyar S, Kaplan DD, Deluca JG, et al. β-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008;22:91–105.
- Mbom BC, Siemers KA, Ostrowski MA, et al. Nek2 phosphorylates and stabilizes β-catenin at mitotic centrosomes downstream of Plk1. Mol Biol Cell. 2014;25:977–991.
- Jeong Y, Lee J, Kim K, et al. Characterization of NIP2/centrobin, a novel substrate of Nek2, and its potential role in microtubule stabilization. J Cell Sci. 2007;120:2106–2116.
- Wei R, Ngo B, Wu G, et al. Phosphorylation of the Ndc80 complex protein, HEC1, by Nek2 kinase modulates chromosome alignment and signaling of the spindle assembly checkpoint. Mol Biol Cell. 2011;22:3584–3594.
- Liu Q, Hirohashi Y, Du X, et al. Nek2 targets the mitotic checkpoint proteins Mad2 and Cdc20: a mechanism for aneuploidy in cancer. Exp Mol Pathol. 2010;88:225–233.
- Zhou H, Di Palma S, Preisinger C, et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. 2013;12:260–271.
- Bauer NC, Doetsch PW, Corbett AH. Mechanisms regulating protein localization. Traffic. 2015;16:1039–1061.
- Xue C, Sowden MP, Berk BC. Extracellular and intracellular cyclophilin A, native and post-translationally modified, show diverse and specific pathological roles in diseases. Arterioscler Thromb Vasc Biol. 2018;38:986–993.
- Olsen JV, Vermeulen M, Santamaria A, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3–ra3.
- Soe NN, Sowden M, Baskaran P, et al. Acetylation of cyclophilin a is required for its secretion and vascular cell activation. Cardiovasc Res. 2014;101:444–453.
- Gouw M, Michael S, Sámano-Sánchez H, et al. The eukaryotic linear motif resource – 2018 update. Nucleic Acids Res. 2018;46:D428–434.
- Helps NR, Luo X, Barker HM, et al. NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem J. 2000;349:509–518.
- Kofron JL, Kuzmic P, Kishore V, et al. Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry. 1991;30:6127–6134.
- Song F, Zhang X, Ren XB, et al. Cyclophilin a (CyPA) induces chemotaxis independent of its peptidylprolyl Cis-trans isomerase activity. J Biol Chem. 2011;286:8197–8203.
- Hames RS. APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 2001;20:7117–7127.
- Nigg EA, Holland AJ. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol. 2018;19:297–312.
- Nigro P, Pompilio G, Capogrossi MC. Cyclophilin A: a key player for human disease. Cell Death Dis. 2013;4:e888.
- Loffler H, Fechter A, Matuszewska M, et al. Cep63 recruits Cdk1 to the centrosome: implications for regulation of mitotic entry, centrosome amplification, and genome maintenance. Cancer Res. 2011;71:2129–2139.
- Piel M. Centrosome-dependent exit of cytokinesis in animal cells. Science. 2001;291:1550–1553.
- Khodjakov A, Rieder CL. Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J cell Biol. 2001;153:237–242.
- Schmucker S, Sumara I. Molecular dynamics of PLK1 during mitosis. Mol Cell Oncol. 2014;1:e954507.
- Gromley A, Yeaman, C, Rosa, J., Redick, S., Chen, C. T., Mirabelle, S., and Doxsey, S. J. Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell. 2005;123:75–87.
- Jonsdottir AB, Dirks, R. W., Vrolijk, J., Ögmundsdottir, H. M., Tanke, H. J., Eyfjörd, J. E., and Szuhai, K Centriole movements in mammalian epithelial cells during cytokinesis. BMC Cell Biol. 2010;11(34): DOI:10.1186/1471-2121-11-34
- Zhao W, Seki A, Fang G. Cep55, a microtubule-bundling protein, associates with centralspindlin to control the midbody integrity and cell abscission during cytokinesis. Mol Biol Cell. 2006;17:3881–3896.
- Takaoka M, Saito H, Takenaka K, et al. BRCA2 phosphorylated by PLK1 moves to the midbody to regulate cytokinesis mediated by nonmuscle myosin IIC. Cancer Res. 2014;74:1518–1528.
- Martinez-Garay I, Rustom A, Gerdes H-H, et al. The novel centrosomal associated protein CEP55 is present in the spindle midzone and the midbody. Genomics. 2006;87:243–253.
- Lavin PTM, Mc Gee MM. Cyclophilin function in cancer; lessons from virus replication. Curr Mol Pharmacol. 2015;9:148–164.
- Di Agostino S, Rossi P, Geremia R, et al. The MAPK pathway triggers activation of Nek2 during chromosome condensation in mouse spermatocytes. Development. 2002;129:1715–1727.
- Fabbro M, Zhou, B. B., Takahashi, M., Sarcevic, B., Lal, P., Graham, M. E., and Khanna, K. K. Cdk1/erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev Cell. 2005;9:477–488.
- Di Agostino S, Fedele, M., Chieffi, P., Fusco, A., Rossi, P., Geremia, R., and Sette, C. Phosphorylation of high-mobility group protein A2 by Nek2 kinase during the first meiotic division in mouse spermatocytes. Mol Biol Cell. 2004;15:1224–1232.
- Du J, Cai, X., Yao, J., Ding, X., Wu, Q., Pei, S., and Yao, X. The mitotic checkpoint kinase NEK2A regulates kinetochore microtubule attachment stability. Oncogene. 2008;27:4107–4114.
- Simón Serrano S, Tavecchio, M., Grönberg, A., Sime, W., Jemaà, M., Moss, S., and Massoumi, R. Novel cyclophilin inhibitor decreases cell proliferation and tumor growth in models of hepatocellular carcinoma. Cancers (Basel). 2021;13:3041.
- Zhang X-C, Wang W-D, Wang J-S, et al. Ppiase independent chaperone-like function of recombinant human Cyclophilin a during arginine kinase refolding. FEBS Lett. 2013;587:666–672.
- Alexander J, Lim, D., Joughin, B. A., Hegemann, B., Hutchins, J. R., Ehrenberger, T., and Yaffe, M. B. Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci Signal. 2011;4:ra42–ra42.
- Mayor T, Stierhof Y-D, Tanaka K, et al. The centrosomal protein C-Nap1 is required for cell cycle–regulated centrosome cohesion. J cell Biol. 2000;151:837–846.
- Bahe S, Stierhof Y-D, Wilkinson CJ, et al. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J cell Biol. 2005;171:27–33.
- Graser S, Stierhof Y-D, Nigg EA. Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J Cell Sci. 2007;120:4321–4331.