
ABSTRACT
The retinoblastoma tumor suppressor (RB) prevents G1 to S cell cycle transition by inhibiting E2F activity. This function requires that RB remains un- or underphosphorylated (the so-called active forms of RB). Recently, we showed that active forms of RB cause widespread changes in nuclear architecture that are visible under a microscope. These phenotypes did not correlate with cell cycle arrest or repression of the E2F transcriptional program, but appeared later, and were associated with the appearance of autophagy or in IMR-90 cells with senescence markers. In this perspective, we describe the relative timing of these RB-induced events and discuss the mechanisms that may underlie RB-induced chromatin dispersion. We consider the relationship between RB-induced dispersion, autophagy, and senescence and the potential connection between dispersion and cell cycle exit.
Introduction
Chromatin is arranged carefully within the nucleus. Regions of chromatin are clustered together and positioned based on gene activity. These regions can be associated with nuclear landmarks such as the lamina or nucleoli (or they can be distanced from such structures), they can be either densely or loosely packed, and they are organized into three-dimensional domains called Topologically Associated Domains or TADs [Citation1,Citation2]. The way in which chromatin is organized influences gene expression and consequently affects cellular states. Changes in chromatin organization have been implicated in several diseases including cancer. Alterations include the repositioning of chromatin territories within the nucleus, the gain (or loss) of heterochromatin aggregates, and genome-wide changes in chromatin contacts [Citation3–5]. Some cancers show changes in 3D interactions, and there are examples of oncogenes and tumor suppressors that modify enhancer–promoter interactions [Citation4,Citation6]. Although it seems likely that tumorigenesis involves changes in nuclear architecture, to date, the events that connect the genes that are recurrently mutated in human cancer to the control of nuclear architecture are not fully understood.
RB is one of the best-known tumor suppressor genes, and it is a master regulator of cellular decisions that affect cell proliferation, cell cycle exit, and the onset of cell differentiation [Citation7–11]. Molecular studies show that RB limits cell proliferation by suppressing E2F-dependent transcription. In doing so, RB facilitates stable exit from the cell cycle and this role is particularly significant during cellular senescence [Citation12,Citation13]. In addition, in several cell types RB appears to actively promote differentiation. RB associates, in a cell-type specific manner, with regulators of differentiation, and these interactions modulate the transcription of their targets [Citation14,Citation15]. In certain contexts, RB has also been shown to help cells maintain lineage fidelity; the loss of this activity appears to be important for the malignant progression of tumor cells [Citation16–19].
How does RB promote these myriad effects? The conventional wisdom is that RB is recruited to specific promoters and that it acts by controlling the transcription of precisely targeted loci. However, the requirement that RB acts on hundreds of genes scattered throughout the genome raises the possibility that the coordinated regulation of RB targets might involve widespread changes in chromatin organization or higher-order changes in nuclear architecture. Such changes may help to facilitate the extensive rewiring of transcription that occurs when cells change cellular state.
Currently, there are just fragments of evidence supporting the idea that RB impacts the organization of chromatin. From epigenetic-profiling experiments, it was reported that RB loss from retinal cells leads to substantial changes in histone marks [Citation20]. RB has been reported to promote the deposition of heterochromatin histone marks [Citation21] and to promote the epigenetic activity of the polycomb repressor complexes [Citation22,Citation23]. More recent studies have shown that the treatment of breast cancer cell lines with CDK4/6 inhibitors also alters the accessibility of enhancer regions in an RB-dependent manner [Citation24]. Molecular studies have revealed several mechanisms that may connect RB to the organization of chromosomal domains. Physical and functional experiments linked RB to CapD3 and to the Condensin II complex [Citation25,Citation26]. Other studies linked RB to chromosome cohesion; loss-of-function experiments show that the removal of RB alters pericentromeric heterochromatin and reduces centromeric cohesion [Citation27–30]. In support of the idea that RB might have far-reaching effects, ChIP-seq experiments have identified many thousands of euchromatic RB-binding sites in the human genome. Very recently, it was found that RB does not just bind to promoters, but it also associates with AP-1-bound enhancers and with CTCF-bound insulators [Citation31]. Thus, rather than just targeting a few carefully selected promoters, RB is widely distributed in the nucleus, and there are good reasons to imagine that RB might have a broad impact on chromatin organization.
Comparing pairs of RB wild-type and RB knockout cells has been a widely used strategy to identify roles for RB, and much of RB research has focused on abnormalities seen in RB-deficient cells. However, if RB truly controls nuclear organization, then it ought to be possible to see RB-induced changes in nuclear organization in cells that express functional RB. The general absence of this type of data is perhaps one of the most significant gaps in the evidence for models proposing that RB controls aspects of nuclear organization. To date, the strongest evidence for RB-induced changes in nuclear organization has come from studies of senescence. Oncogene-induced senescence (OIS) requires RB and it involves major changes in nuclear organization [Citation12,Citation13]. In some cells, these changes involve the formation of Senescence-Associated Heterochromatin Foci (SAHFs), and FISH experiments indicate that SAHFs contain RB/E2F-regulated loci [Citation13]. However, there is no evidence that RB specifically accumulates at SAHFs, or even that it is directly involved in the recruitment of loci to SAHFs. Indeed, while functional RB appears to be generally required for senescence, SAHFs are not a universal feature of senescent cells. The link between RB and senescence might, alternatively, be explained by the idea that RB is needed for the repression of E2F-regulated genes and that stable cell cycle exit is necessary for senescence. Currently, it is uncertain whether RB is directly involved in any of the additional features of senescent cells, beyond cell cycle arrest.
With this background, the results recently reported by Krishnan et al. [Citation32] are particularly notable. This report provided two types of new information. First, the results show clear evidence of visible changes in multiple aspects of nuclear architecture following the activation of RB. Changes were quantified using probes against large regions of euchromatin or heterochromatin and against whole chromosomes. From these data, it is evident that RB-induced changes in chromatin organization do occur, and they are widespread. Interestingly, RB-induced changes were not limited to E2F target genes; changes were measured at multiple loci and occurred regardless of whether the regions detected contain or lack E2F/RB-binding sites. Indeed, the expression of the dominant-negative mutant DP1 that reduces E2F1 binding to chromatin does not affect the RB-induced chromatin changes. Second, this report revealed the cellular context in which RB-induced chromatin changes are visible. Interestingly, the changes in organization were not concurrent with RB-induced G1-arrest but appeared afterward. Effects were measured in cells that underwent senescence, but changes were also evident in cells that did not express senescence markers. Hence, the role of RB in chromatin organization is not simply a feature of cell cycle arrest, and it is not restricted to senescent cells. Instead, the effects of RB on chromatin organization are inducible and time dependent, and they appear in arrested cells. No significant differences were seen when populations of proliferating RB-expressing cells we compared with RB-mutant cells. The fact that RB-dependent changes were context dependent and accumulated in arrested cells may, in part, explain why they had not been noted before.
The timeline of chromatin dispersion induced by active RB
In much of the published work, Krishnan et al. used a constitutively active form of RB, a protein in which all 14 CDK in RB sites are mutated to alanine (ΔCDK RB).
This form of RB cannot be phosphorylated by CDKs and is thought to possess activities similar to unphosphorylated RB [Citation33]. ΔCDK RB is well known to target E2F and to inhibit the E2F transcription program, causing cells to accumulate in G1 [Citation34]. Heterochromatin and euchromatin regions were probed using oligopaints or FISH probes and were visualized by three-dimensional confocal imaging. ΔCDK RB caused the regions to disperse and scatter. The α-satellite heterochromatin regions of chromosomes 6 and 7 changed from a spherical compact focus to a non-compact amorphous and punctate foci; and two large regions on chromosome 19 q-arm changed to expanded, punctate foci. When whole chromosome probes were used, similar scattering of the FISH signals was observed, and chromosome 19 was observed to re-position toward the nucleolar periphery.
A well-controlled, doxycycline (DOX)-inducible expression system that allowed endogenous RB to be replaced by ΔCDK RB made it possible to explore the timeline of the changes seen following the expression of active RB (). Significantly, the RB-mediated chromatin dispersion appeared much later than cell cycle arrest. Dispersion/scattering was first seen 48 h after induction of active RB, almost 36 h after the repression of E2F targets and changes in cell cycle position became evident. Dispersion reached maximum value by 72 h and was not reversed by DOX washout after this point. These observations suggest that cells enter an altered state between 48 h and 72 h after ∆CDK RB expression, a state that involves a change in nuclear organization.
Figure 1. Timeline of Active RB expression and Dispersion. The illustration shows the major changes observed progressively 0, 24, 48, and 72 h after the DOX-induced replacement of the endogenous RB by exogenous active forms of RB. The active RB-mediated chromatin dispersion and induction of autophagy program occurs much later than active RB-induced G1 arrest and repression of E2F targets. Dispersion measures and autophagy response increase between 48 and 72 h.
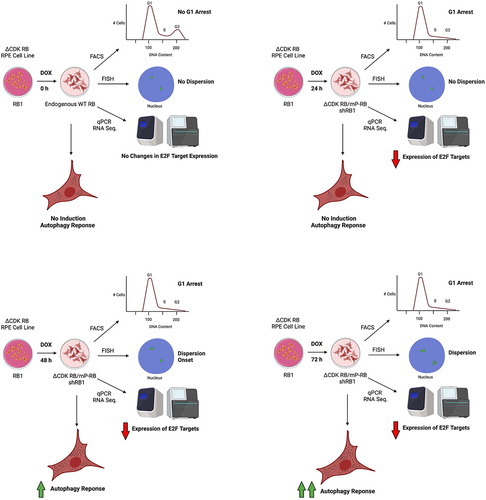
Much of the analysis was carried out in RPE1 cells and the results uncovered an unexpected advantage of this cell line for studies of ∆CDK RB-induced dispersion. ∆CDK RB does not readily induce markers of senescence in RPE1 cells. Indeed, after 72 h of ∆CDK RB expression, RPE1 cells failed to show any activation markers of senescence, such as positive β-galactosidase staining, or transcription activation of senescence-related genes. This illustrates that the ∆CDK RB-induced state is not equivalent to senescence. It also means that the RB-induced changes in chromatin organization seen in RPE1 cells cannot be an indirect consequence of other types of chromatin changes induced by senescence. However, when IMR-90 cells were treated with irradiation (IR) to induce senescence, the dispersion phenotype correlated with the appearance of β-galactosidase-positive cells and upregulated expression of specific markers of senescence.
Putting these two observations together, we see two potential interpretations. One possibility is that RB-induced dispersion represents an RB-dependent step that is a precursor to senescence. An alternative possibility is that the RB-induced changes in chromatin organization are just one component of the more extensive pan-nuclear events that re-organize chromatin in senescent cells, and RPE1 cells are unusual in allowing these RB-induced changes to be distinguished from other senescence-associated events. Further studies of RB-induced cell cycle exit will be needed to distinguish between these possibilities. In further experiments, it may also be possible to refine the timeline further and to pinpoint the precise time when cells switch to the dispersed state.
In the RB-replacement system in RPE1 cells, over time, clones of cells emerge that have lost the expression of ∆CDK RB. An experimental system that allows stable expression of active RB for 144 h and beyond may be necessary to carefully dissect the dynamics of RB-induced dispersion, the appearance of senescence signature, and other RB-induced phenotypes.
Specific forms of RB promote dispersion
The discovery of distinct mono-phosphorylated forms of RB has raised the question of whether the various forms of active RB that are expressed in G1 phase cells are functionally distinct. Fourteen different forms of active RB have been described that differ in the position of a CDK4/6-dependent, monophosphorylated residue [Citation33]. Mono-phosphorylation controls RB interaction with other proteins and changes the flavor of RB-dependent transcriptional regulation [Citation33,Citation34], but investigation of the functional differences between the various forms of active RB remains at an early stage [Citation34]. It is striking therefore that when individual monophosphorylated forms of RB (mP-RBs) were expressed in RPE1 cells, using the same inducible system used to express ΔCDK RB, they dispersed/scattered to very different degrees. RB isoforms were classified into high, medium, and low dispersers and scatterers based on how they changed heterochromatin and euchromatin regions, respectively. Interestingly, the dispersion seen with the mP-RBs did not correlate with the degree of cell cycle arrest or with the repression of the E2F transcription program.
Proteins that co-immunoprecipitate with each of the mP-RBs, as well as proteins bound by wild-type RB and ΔCDK RB, have previously been identified by mass spectrometry [Citation34]. Disappointingly, the RB-induced dispersion phenotype did not correlate with any of the known interactors across the panel of RB-associated proteins. It may be that the current lists of mP-RB-associated proteins are incomplete, it may be that the dispersion phenotype is too complex to be attributed to interactions with a single protein, and it may be necessary to profile mP-RB-associated proteins at a specific time point during the induction of dispersion to identify the critical targets. An alternative way to identify proteins involved in RB-induced dispersion is to test whether any of the known RB-associated complexes or activities are necessary for the phenotype, and this approach provided several intriguing leads. Known RB-interacting factors and activities were tested, and the results revealed that RB-induced dispersion can be suppressed by inhibition of Histone Deacetylases (HDACs) or by knockdown of TOPIIB. In contrast, the knockdown or inhibition of other factors (TOPIIA, CAPD3, and WAPL: two chromosome cohesion factors, EZH2 and RNA Polymerase II) did not influence dispersion positively or negatively, with the caveat that negative results in these types of experiments should be interpreted cautiously.
TOPIIB [Citation32,Citation35,Citation36] and HDACs [Citation34,Citation37,Citation38] both associate with active RBs, but it is unclear how they contribute to the conformational changes seen during dispersion. TOPIIB catalyzes topological changes in DNA [Citation39,Citation40]. We hypothesize that the visual changes in chromosome organization seen under the microscope likely involve either the creation, or resolution, of chromatin loops and tangles and TOPIIB may act on these. It is also uncertain how HDACs promotes dispersion. One possibility is that the HDACs inhibitors block RB-induced dispersion by blocking the activity of RB-associated complexes. Other studies have shown that HDACs have the ability to directly affect nuclear organization by inducing euchromatin association/relocation to the nuclear pore complex [Citation41] or by promoting compartment switching by promoting a repressive chromatin environment [Citation4]. Thus, while the simplest interpretation is that HDAC inhibitors act directly on RB-recruited complexes, they might also intersect very generally with RB at the level of nuclear organization. More information on the mechanism by which active RB regulates HDACs activity will be needed to distinguish between these models. The chromatin distribution of HDACs must be assessed after active RB-induced dispersion to define how changes in HDACs enzymatic activity relate to the repressive chromatin environment around RB-binding sites and the 3D landscape of chromatin contacts.
To identify additional proteins that help RB to promote dispersion, it is likely that comprehensive screens will be necessary. Approaches using CRISPR, RNAi technologies, or compound libraries all have the potential to identify components that affect chromatin structure or chromosome topology and proteins that link directly to RB. A key factor to consider for such a screen is the timing of intervention. In the published data, it was necessary for inhibition/knockdown to be done at, or before, the time that dispersion first became apparent (48 h after DOX induction) to obtain good suppression. Once dispersion had been established (72 h after induction), it was not possible to reverse or suppress the change by targeting any of the factors examined, including HDAC inhibition and TOPIIB knockdown. Indeed, the fact that DOX washouts could not completely reverse dispersion suggests that dispersion may be an irreversible or “locked-in” state. Nevertheless, by targeting proteins between 48 and 72 h after ΔCDK RB expression, it should be possible to generate a comprehensive list of the factors that co-operate with RB, either directly or indirectly, to cause dispersion.
A chicken and egg problem: dispersion, prolonged cell cycle arrest, and senescence
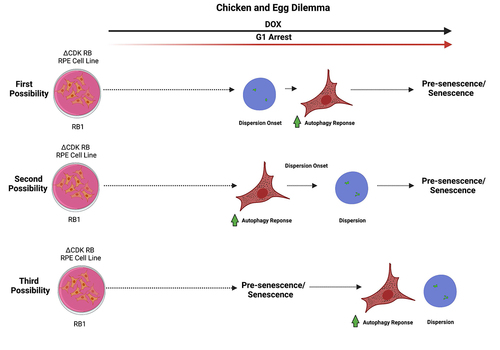
The published data were sufficient, to some degree, to tease dispersion apart from senescence. Nevertheless, the precise chronology of the events leading to dispersion and the cellular changes resulting from dispersion are not completely clear. Here, we discuss some of the potential scenarios. We know that dispersion occurs in the context where active RB induces a prolonged cell cycle arrest and the autophagy program is activated. It is uncertain whether the observed events form a linear pathway, but, even if they do, the data could be explained in at least three different ways. The first possibility is that prolonged expression of active RB causes dispersion onset around 48 h and autophagy response soon after. The second possibility is that sustained expression of ΔCDK RB/high disperser mP-RB causes induction of autophagy response in around 48 h and dispersion onset soon after. In both these scenarios, pre-senescence/senescence occurs after the dispersion onset-increased autophagy response. The third possibility is that prolonged expression of active RB leads cells to enter pre-senescence/senescence state causing increased autophagy response and dispersion onset (). There are two points to keep in mind while considering these possibilities. The first is that the onset of dispersion and the induction of autophagy response were coincident in the published data but may be separable with more time points. The second is that the timing of pre-senescence/senescence is speculative, as we failed to observe senescence markers in RPE1 cells, even after 144 h of ΔCDK RB expression.
Figure 2. Possible chronology of events after the expression of constitutively active ∆CDK RB. The first two scenarios explore the relationship between dispersion and autophagy and its contribution to senescence/presenescence. The third scenario speculates that both dispersion and autophagy response occur as a consequence of presenescence/senescence onset. The solid black arrow represents the DOX-induced expression of active RB. The red arrow shows onset of cell cycle arrest and the prolonging of the arrest. Dashed arrows indicate the progress of time with respect to the major events shown.
Autophagy induction and dispersion
A few studies have shown that the expression of active RB (or the treatment of cells with molecules such as CDK4/6 inhibitors that generate active RB) can, in some cancer cells, induce autophagy [Citation42,Citation43]. Mechanistically, this has been proposed to occur through the inhibition of E2F1. In a previous study [Citation32], when we asked whether the dispersion rankings/scores of the active RB forms correlated with any expression signature, we failed to find any association with canonical E2F transcriptional regulation. Instead, we found a very significant positive correlation between dispersion and autophagy signature. The forms of RB that had the highest dispersion activity showed the biggest change in autophagy gene expression and in autophagy response (measured by LCIIIB punctas). Although 74% of the autophagy genes showing this association had RB binding sites nearby, it is not clear whether active RB was directly involved in all cases, as several of the genes with the tightest correlation lacked RB binding sites in the presumptive regulatory regions.
All of the active forms of RB associated with E2F and repress E2F-dependent transcriptional but the autophagy outcomes were very different, with the low dispersers never inducing an autophagy response. Using forms of RB that did promote markers of autophagy, the response became evident 48 h after DOX induction, whereas cell cycle progression and E2F activity were suppressed rapidly, as soon as 12 h after DOX addition. It seems, then, that the autophagy response is an outcome of RB activation that requires more than just the repression of E2F. Based on the correlation seen between dispersion and autophagy, and their coincident appearance, we speculate that RB may increase the expression of autophagy genes by orchestrating chromatin re-organization, rather than by acting directly on these targets. However, at present, this is just a model and to test this hypothesis rigorously it will be necessary to carefully examine the regulatory sequences in the vicinity of the autophagy genes, especially those that correlated well with the dispersion response. Such experiments are needed to reveal whether RB-mediated stimulation of these genes corresponds to a transient gain or loss of RB-binding sites, with changes in looping between enhancers and promoters, or whether there are changes in other 3D chromatin contacts that correlate with dispersion.
Senescence and dispersion/scatter
Senescence has been associated with widespread changes in chromatin conformation and nuclear organization. Several studies have examined these changes using Hi-C and in different types of senescence. So far, no changes in TAD (Topologically Associated Domains) boundaries have been identified between senescent and proliferating cells, but a common feature seen in all types of senescence is the re-positioning of certain regions to the nuclear lamina. During Oncogene-Induced Senescence (OIS) and Replicative Senescence (RS), there are a loss of local chromatin contacts and a gain of distal contacts [Citation44–46]. In both of these types of senescence, there is compartment switching between active A compartment and repressed B compartment, with OIS showing a loss of A–B contacts and gain of B–B, and RS showing a gain of A–B contacts [Citation47–49]. The movement of genes from one compartment to another affects their transcription/expression.
Imaging studies on RS and OIS have revealed visible changes to chromatin organization. SAD (Senescence-Associated Distension of Satellites) has been observed in RS cells. SAD is formed due to chromatin decondensation and decompaction caused due to altered higher-order genome packing and deposition of inactive histone marks [Citation45,Citation50]. SAHFs are found during OIS but are absent during RS. Recent studies have identified a feature called SAHDs (Senescence-Associated Heterochromatin Domains) in OIS and RS. SAHDs are chromatin regions enriched in H3K9me3 that are highly interactive. It is hypothesized that SAHFs contain SAHDs and are formed because of B–B compartmentalization, and such altered chromatin interactions underlie SAHF formation [Citation47,Citation48]. In a previous study [Citation32], we examined the molecular events that underlie dispersion and scattering using 3C. Although limited in scale, these experiments supported the idea that ΔCDK RB expression does not cause widespread changes in interaction between TAD boundaries, however a genome-wide study of spatial interactions will be needed to obtain a comprehensive picture.
The RB phenotype described in a previous study [Citation32] differs from the changes described above. This phenotype involves visual changes in chromatin organization that include a quantifiable unique compartmentalization (puncta) and extension (oblong shapes and irregular extended shapes) of euchromatin, heterochromatin, and chromosome position. This array of visual changes has not been described before for any of the oncogenes or tumor suppressors that promote senescence. These results are perhaps the clearest evidence that RB has a role in controlling higher-order genome structure, and they provide insight into the cellular context in which this role occurs.
Returning to the question of chronology, does active RB directly cause dispersion, or does it promote senescence and/or autophagy and chromatin dispersion occur as a consequence? The fact that active forms of RB induce dispersion in RPE1 cells without induction of the senescence signature or any markers of senescence indicates that these RB-induced chromatin changes are not a consequence of senescence. Moreover, the fact that the CDK4/6 inhibitor palbociclib fails to induce dispersion in the absence of RB strongly suggests that RB is a necessary intermediate for dispersion. Evidence shows that autophagy promotes the remodeling of cellular constituents during senescence, and thus dispersion-induced autophagy may be part of the processes that help to remodel the cell in preparation for senescence [Citation51]. As such, RB-induced dispersion, by increasing autophagy, may help to prepare cells for senescence. While further experiments are needed to precisely separate events, we favor the notion that RB-induced dispersion reflects a reorganization of chromatin that occurs after cell cycle exit and prior to activation of a full senescent response, that it is associated with specific transcriptional changes, and these include an increased expression of autophagy-related genes.
Summary and future directions
It is now clear that RB activation triggers changes in chromatin and chromosomal organization that are visible under the microscope. These changes extend beyond the regulation of E2F transcriptional activity and are independent of the RB-mediated G1-arrest. RB-induced chromatin dispersion describes an extended chromatin arrangement that affects both euchromatin and heterochromatin regions and impacts entire chromosomes. These changes may be irreversible, and they appear in cells that undergo senescence, but RB-induced chromatin dispersion is not equivalent to cellular senescence. Investigating the molecular mechanisms underlining this phenomenon may provide new insights into the RB’s mechanism of action and explain the consequences of RB inactivation that are not directly related to E2F regulation.
Previously, RB has been linked to the activity of chromatin remodeling complexes (such as NuRD, SWI/SNF, TOPIIA, and TOPIIB) [Citation22,Citation36,Citation38,Citation52,Citation53], epigenetic regulators (such as HDACs, EZH2, KDMs, DNMT1, HP1, Suv39h1) [Citation21,Citation22,Citation34,Citation37,Citation38,Citation54–56], cohesin and condensin II complexes [Citation25,Citation28]. Very recently, RB has been found to associate with the CTCF-bound chromatin insulators [Citation31]. Krishnan et al. [Citation32] show that the time point when active RB arrest cells in G1 can be readily distinguished from the time point where it induces visible changes in chromosome organization. A systematic analysis of the activity of RB-associated chromatin modifiers is required to better understand RB-induced chromatin dispersion, and the temporal window can be used to identify RB-associated chromatin regulators and enzymatic activities that are specifically needed for the dispersion phenotype. In addition, high-throughput HiC-based interaction studies can link the RB-mediated chromosome organization to specific chromatin interactions and possible regulation of the insulation activity of the CTCF sites. Differences in the Hi-C profiles between RB wild-type and RB knockout cells will uncover the RB-dependent chromatin interactions. In parallel, Hi-C differences between cells expressing active forms of RB that are high or low dispersers will reveal whether the RB-induced chromatin dispersion associates with differential regulation in chromatin interactions.
It is fascinating that the chromatin dispersion induced by active RB so clearly extends beyond RB-bound loci, even affecting heterochromatin regions that completely lack RB and E2F-binding sites. This raises the question of whether the RB-induced chromosome rearrangements should be considered as structural changes that are linked to permanent cell cycle exit or whether they should be viewed as events that modify the program of gene expression that is controlled by RB. The correlation of the dispersion phenotype with the expression of the autophagy expression program indicates that the second scenario is certainly possible. In addition to E2F-bound promoters, RB associates with cell-type-specific enhancers [Citation31]. Moreover, the activation of RB controls the activity of cell-type-specific enhancers [Citation24,Citation31]. In combination with expression analysis of the epigenetic markers of enhancer activity, Hi-C chromatin interaction analysis may uncover novel mechanisms of RB-dependent transcription regulation associated with chromosome dispersion. These studies may help to explain aspects of RB-loss transcription signatures that cannot currently be explained via RB-mediated regulation of E2F.
In conclusion, the discovery that RB directly orchestrates genome-wide changes in 3D chromatin organization paves a new direction for studies of RB’s mechanism of action.
Acknowledgments
We would like to thank MGH Cancer Center and Molecular Pathology Confocal Core. This work was supported by NIH grants to NJD (GM117413 and CA236538). NJD is the recipient of the Mary B Saltonstall Chair in Oncology.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bickmore WA. The spatial organization of the human genome. Annu Rev Genomics Hum Genet. 2013;14:67–84.
- Gonzalez-Sandoval A, Gasser SM. On TADs and LADs: spatial control over gene expression. Trends Genet. 2016;32:485–495.
- Corces MR, Corces VG. The three-dimensional cancer genome. Curr Opin Genet Dev. 2016;36:1–7.
- Johnstone SE, Reyes A, Qi Y, et al. Large-scale topological changes restrain malignant progression in colorectal cancer. Cell. 2020;182:1474–1489 e1423.
- Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4:677–687.
- Akdemir KC, Le VT, Chandran S, et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat Genet. 2020;52:294–305.
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682.
- Dick FA, Goodrich DW, Sage J, et al. Non-canonical functions of the RB protein in cancer. Nat Rev Cancer. 2018;18:442–451.
- Dyson N. The regulation of E2F by Prb-family proteins. Genes Dev. 1998;12:2245–2262.
- Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 2016;30:1492–1502.
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70.
- Chicas A, Wang X, Zhang C, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–387.
- Narita M, Nunez S, Heard E, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716.
- Calo E, Quintero-Estades JA, Danielian PS, et al. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010;466:1110–1114.
- Thomas DM, Carty SA, Piscopo DM, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8:303–316.
- Kareta MS, Gorges LL, Hafeez S, et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell. 2015;16:39–50.
- Ku SY, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83.
- Mu P, Zhang Z, Benelli M, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88.
- Walter DM, Yates TJ, Ruiz-Torres M, et al. RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature. 2019;569:423–427.
- Zhang J, Benavente J CA, McEvoy J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334.
- Nielsen SJ, Schneider R, Bauer UM, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565.
- Ishak CA, Marshall AE, Passos DT, et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell. 2016;64:1074–1087.
- Kotake Y, Cao R, Viatour P, et al. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21:49–54.
- Watt AC, Cejas P, DeCristo MJ, et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nat Cancer. 2021;2:34–48.
- Longworth MS, Herr A, Ji JY, et al. RBF1 promotes chromatin condensation through a conserved interaction with the Condensin II protein Dcap-D3. Genes Dev. 2008;22:1011–1024.
- Marshall AE, Ishak CA, Dick FA. An RB-Condensin II complex mediates long-range chromosome interactions and influences expression at divergently paired genes. Mol Cell Biol. 2020;40. DOI:10.1128/MCB.00452-19
- Manning AL, Benes C, Dyson NJ. Whole chromosome instability resulting from the synergistic effects of pRB and p53 inactivation. Oncogene. 2014a;33:2487–2494.
- Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010;24:1364–1376.
- Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer. 2012;12:220–226.
- Manning AL, Yazinski SA, Nicolay B, et al. Suppression of genome instability in Prb-deficient cells by enhancement of chromosome cohesion. Mol Cell. 2014b;53:993–1004.
- Sanidas I, Lee H, Rumde PH, et al. Chromatin-bound RB targets promoters, enhancers, and CTCF-bound loci and is redistributed by cell-cycle progression. Mol Cell. 2022;82:3333–3349 e3339.
- Krishnan B, Yasuhara T, Rumde P, et al., Active RB causes visible changes in nuclear organization. J Cell Bio. 2022;221. DOI:10.1083/jcb.202102144
- Narasimha AM, Kaulich M, Shapiro GS, et al. Cyclin D activates the Rb tumor suppressor by monophosphorylation. Elife. 2014;3. DOI:10.7554/eLife.02872
- Sanidas I, Morris R, Fella KA, et al. A code of monophosphorylation modulates the function of RB. Mol Cell. 2019;73:985–1000 e1006.
- Goodrich DW. The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene. 2006;25:5233–5243.
- Xiao H, Goodrich DW. The retinoblastoma tumor suppressor protein is required for efficient processing and repair of trapped topoisomerase IIDNA-cleavable complexes. Oncogene. 2005;24:8105–8113.
- Brehm A, Miska EA, McCance DJ, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601.
- Zhang HS, Gavin A M, Dahiya A, et al. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-Hswi/SNF/SNF and Rb-Hswi/SNF/SNF. Cell. 2000;101:79–89.
- Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413.
- Nitiss JL. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer. 2009;9:327–337.
- Brown CR, Kennedy CJ, Delmar VA, et al. Global histone acetylation induces functional genomic reorganization at mammalian nuclear pore complexes. Genes Dev. 2008;22:627–639.
- Jiang H, Martin V, Gomez-Manzano C, et al. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010;70:7882–7893.
- Vijayaraghavan S, Karakas C, Doostan I, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916.
- Chandra T, Ewels PA, Schoenfelder S, et al. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015;10:471–483.
- Criscione SW, De Cecco M, Siranosian B, et al. Reorganization of chromosome architecture in replicative cellular senescence. Sci Adv. 2016a;2:e1500882.
- Criscione SW, Teo YV, Neretti N. The chromatin landscape of cellular senescence. Trends Genet. 2016b;32:751–761.
- Rocha A, Dalgarno A, Neretti N. The functional impact of nuclear reorganization in cellular senescence. Brief Funct Genomics. 2022;21:24–34.
- Sati S, Bonev B, Szabo Q, et al. 4D genome rewiring during oncogene-induced and replicative senescence. Mol Cell. 2020;78:522–538 e529.
- Zirkel A, Nikolic M, Sofiadis K, et al. HMGB2 loss upon senescence entry disrupts genomic organization and induces CTCF clustering across cell types. Mol Cell. 2018;70:730–744 e736.
- Swanson EC, Manning B, Zhang H, et al. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Bio. 2013;203:929–942.
- White E, Lowe SW. Eating to exit: autophagy-enabled senescence revealed. Genes Dev. 2009;23:784–787.
- Bhat UG, Raychaudhuri P, Beck WT. Functional interaction between human topoisomerase IIalpha and retinoblastoma protein. Proc Natl Acad Sci U S A. 1999;96:7859–7864.
- Montoya-Durango DE, Ramos KA, Bojang P, et al. LINE-1 silencing by retinoblastoma proteins is effected through the nucleosomal and remodeling deacetylase multiprotein complex. BMC Cancer. 2016;16:38.
- Benevolenskaya EV, Murray HL, Branton P, et al. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 2005;18:623–635.
- Fattaey AR, Helin K, Dembski MS, et al. Characterization of the retinoblastoma binding proteins RBP1 and RBP2. Oncogene. 1993;8:3149–3156.
- Robertson KD, Ait-Si-Ali S, Yokochi T, et al. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–342.