
ABSTRACT
This study aims to investigate how exercise-induced myocardial hypertrophy preconditioning affects cardiac fibroblasts in the context of myocardial fibrosis, a chronic disease that can cause cardiac arrhythmia and heart failure. Heart failure was induced in male C57BL/6 mice via Transverse aortic constriction, and some mice were given swimming exercise before surgery to test the effects of exercise-induced myocardial hypertrophy preconditioning on myocardial fibrosis. Myocardial tissue was evaluated for fibrosis, senescent cells, and apoptotic cells. Myocardial fibroblasts from rats were cultured and treated with norepinephrine to induce fibrosis which were then treated with si-Nrf2 and analyzed for markers of fibrosis, senescence, apoptosis, and cell proliferation. Exercise-induced myocardial hypertrophy preconditioning reduced myocardial fibrosis in mice, as shown by decreased mRNA expression levels of fibrosis-related indicators and increased cell senescence. In vitro data indicated that norepinephrine (NE) treatment increased fibrosis-related markers and reduced apoptotic and senescent cells, and this effect was reversed by pre-conditioning in PRE+NE group. Preconditioning activated Nrf2 and downstream signaling genes, promoting premature senescence in cardiac fibroblasts and tissues isolated from preconditioned mice. Moreover, Nrf2 knockdown reversed proapoptotic effects, restored cell proliferation, reduced senescence-related protein expression, and increased oxidative stress markers and fibrosis-related genes, indicating Nrf2‘s crucial role in regulating oxidative stress response of cardiac fibroblasts. Exercise-induced myocardial hypertrophy preconditioning improves myocardial fibrosis which is Nrf2-dependent, indicating the protective effect of hypertrophy preconditioning. These findings may contribute to the development of therapeutic interventions to prevent or treat myocardial fibrosis.
1. Introduction
Myocardial ischemic diseases, such as coronary heart disease, aortic stenosis, and hypertension, are among the most common causes of myocardial fibrosis [Citation1]. When the heart experiences ischemia, injury, or stress, fibroblasts are activated and transformed into myofibroblasts [Citation2]. Fibrosis is characterized by the accumulation of extracellular collagen without cardiomyocyte necrosis [Citation3]. Fortunately, fibrosis can be repaired with timely and effective treatment. However, chronic pressure overload over time can lead to myocardial cell necrosis and alternative fibrosis which is considered irreversible [Citation4]. Fibrosis restricts the heart’s oxygen supply and nutrition, leading to electrical conduction and structural changes, making patients more susceptible to arrhythmias, heart failure, and ischemia [Citation5]. Consequently, there is an urgent need to explore potential treatments for myocardial fibrosis in early interventions to improve patients’ prognosis.
Studies have shown that transient ischemia can increase the heart’s resistance to subsequent ischemia [Citation6]. This concept of ischemic preconditioning has been observed in many organs, including the brain, liver, and kidney, suggesting that it is a fundamental characteristic among different cell types [Citation7–9]. Similarly, short-term hypertrophic stimulation can make the heart resistant to subsequent hypertrophic stress. This preconditioning phenomenon can be induced by ischemia, hypoxia, hyperbaric oxygen, or certain drugs [Citation10,Citation11]. Our previous research has shown that preconditioning of cardiac hypertrophy delays the onset of heart failure by alleviating cardiac hypertrophy [Citation12]. Whether cardiac hypertrophy inhibits myocardial fibrosis is a promising area for further exploration.
Cell senescence is a natural process that can suppress tumor growth and reverse the impact of damage on healthy cells [Citation13,Citation14]. When senescence occurs, specific factors are activated to block the cell cycle [Citation15]. In addition to its role in preventing tumor progression, studies have also confirmed that cell senescence can inhibit the development of liver and myocardial fibrosis [Citation16,Citation17].
Myocardial damage caused by ischemia is primarily due to oxidative stress, which leads to cell damage [Citation18]. During myocardial reperfusion, the already damaged myocardium undergoes sudden biochemical and metabolic changes, including mitochondrial energization, oxidative stress, intracellular Ca2+ overload, restoration of physiological pH, and inflammation [Citation19]. All these factors work together to mediate cardiomyocyte death. However, the complex antioxidant defense system regulates oxidative stress through multiple pathways, including the activation of Nrf2.
Nrf2 is an important factor that controls the expression of various antioxidants and phase 2 enzymes in cardiomyocytes, protecting them from oxidation and electrophilic stress-induced damage [Citation20]. Upon exposure to ROS or electrophiles, Nrf2 is translocated to the nucleus and binds with the antioxidant response element (ARE) to activate the transcription of its target genes such as HO-1, NQO-1, and GCLC [Citation21]. The Keap1/Nrf2/ARE redox-sensitive signaling system is crucial in regulating cellular homeostasis under conditions of stress, inflammation, carcinogenesis, and pro-apoptosis [Citation22]. The activation of the Nrf2/ARE pathway may also be involved in myocardial hypertrophic preconditioning, although the specific mechanism is still unclear [Citation23]. Several studies have shown the benefits of exercise preconditioning in preventing cardiac myopathy [Citation24,Citation25] and that exercise-induced cardiac hypertrophy preconditioning enhances resistance to pathological stress through anti-hypertrophic effects mediated by Mhrt779/Brg1/Hdac2/p-Akt/p-GSK3β signaling pathway [Citation26].
Given the clinical significance and research value of myocardial hypertrophic preconditioning, considerable effort has been devoted to analyzing its underlying mechanism and potential mediators. In this study, we aimed to investigate the effect of hypertrophic preconditioning on myocardial fibrosis and to elucidate the underlying mechanisms.
2. Materials and methods
2.1. Ethics statement
All procedures related to animal experiments were conducted in strict adherence to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication, 8th edition, 2011). The local animal ethics committee of Nanjing Drum Tower Hospital (No.2020AE01065) reviewed and approved the experimental protocols involving animal subjects.
2.2. Cell treatment
To euthanize the neonatal rats, we utilized 2% isoflurane inhalation followed by cervical dislocation in accordance with established protocols and ethical guidelines. Fibroblasts were then isolated and cultured following previously described methods [Citation27]. Control group cells were cultured in Dulbecco’s modified Eagle’s medium for 48 hours. Cells in the norepinephrine (NE) group were treated with 1 μmol/L NE, which was dissolved in Dulbecco’s modified Eagle’s medium, for 48 hours. For the preconditioning (PRE)+NE group, cells were stimulated with NE for 12 hours, followed by a 12-hour period of culturing without NE, before finally being stimulated with NE again for 48 hours. Fibroblasts were transfected with si-NC and si-Nrf2 (100 ng) obtained from GenePharma (Shanghai, China) using lipofectamine 2000 for 24 hours. The harvested fibroblasts were then analyzed for mRNA and protein expression levels of markers of myocardial fibrosis, cell senescence, apoptosis, and cell proliferation.
2.3. Experimental design
To investigate the effects of Nrf2 knockdown on fibroblasts, we designed five groups as follows: a) Control group, b) NE+NC group: Cardiac fibroblasts stimulated with NE and transfected with si-NC. c) NE+si-Nrf2 group: Cardiac fibroblasts stimulated with NE and transfected with si-Nrf2. d) PRE+NE+NC group: Cardiac fibroblasts subjected to preconditioning and NE stimulation and transfected with si-NC. e) PRE+NE+si-Nrf2 group: Cardiac fibroblasts subjected to preconditioning and NE stimulation and transfected with si-Nrf2. In each group, we assessed the mRNA and protein expression levels of markers related to myocardial fibrosis, cell senescence, apoptosis, and cell proliferation.
2.4. Western blot
The myocardial fibroblasts were washed twice with PBS. Total proteins were extracted from cardiac tissues using RIPA lysis buffer with either protease inhibitor cocktail or phosphatase inhibitor cocktail (Selleckchem, China). Protein concentrations were determined using the Bradford assay (Bio-Rad, USA), and then 20 µg of total proteins were separated using 10% SDS-PAGE and transferred onto a PVDF membrane. After blocking with 5% nonfat milk in TBS at room temperature for 2 hours, the PVDF membranes were incubated with primary antibodies against p16 (1:1000), p21 (1:1000), p53 (1:1000), β-actin (1:1000), procollagen I (1:1000), procollagen III (1:1000), MMP2 (1:1000), MMP9 (1:1000), and TGF-β (1:1000) overnight at 4°C, followed by incubation with an HRP-linked IgG secondary antibody (Sigma-Aldrich, 1:10,000 dilution) at room temperature for 1 hour. The protein bands were visualized using enhanced chemiluminescence (ECL, Merck Millipore, Germany). The band intensities were quantified using ImageJ software (NIH, Bethesda, MD, USA).
2.5. EdU assay
Cells were seeded in 96-well plates at a density of 4 × 103 cells per well during the logarithmic growth stage. After the cells adhered, NE was added. To prepare the appropriate 50 μM EdU medium, the EdU solution (reagent A) was diluted with cell medium at a ratio of 1000:1. The cells were then fixed with 50 μL of 4% paraformaldehyde in PBS per well for 30 min. After incubating the cells with 2 mg/mL glycine for 5 min, they were treated with 100 μL of 0.5% Triton for 10 min. Images were captured after incubating with the Apollo dye reaction solution and DAPI solution.
2.6. Detection of the oxidative stress response
Quantitative assessment of reactive oxygen species (ROS) in cell samples was performed using the DCFDA-Cellular ROS Assay Kit (ab113851, abcam). The activities of SOD and GPx in cell lysates were measured using the Superoxide Dismutase Activity Assay Kit (Colorimetric) (ab65354, abcam) and the Glutathione Peroxidase Assay Kit (Colorimetric) (ab102530, abcam), respectively.
2.7. TAC mouse model
Transverse aortic constriction (TAC) was performed on male C57BL/6 mice (6–8 weeks old, weighing 18–23 g) to induce heart failure. Anesthesia was induced by intraperitoneal injection of ketamine (80 mg/kg), methylthiazide (20 mg/kg), and atropine (0.6 mg/kg). The skin in the middle of the chest was incised to the second rib, followed by the separation of the thymus to expose the aortic arch with micro tweezers. Using a No.5 silk thread and needle, the aorta was ligated 0.3 cm apart from the artery. The chest was then closed layer by layer, and the skin was sutured. Three groups were established: (1) the control group, in which the mice were sham-operated; (2) the TAC group, in which mice were observed for 7 days after the surgery; and (3) the SW+TAC group, in which male C57BL/6 mice underwent swimming training in a tank (25 cm) at a constant temperature of 29–34°C. Swimming exercise started at 10 minutes a day, increasing by 10 minutes each day up to 90 minutes, twice a day with a 6-hour interval, according to previous studies [Citation3,Citation28,Citation29]. After 3 weeks of swimming training, TAC was performed, and myocardial tissues were isolated for subsequent experiments after 7 days of TAC surgery. All mice were euthanized by neck amputation.
2.8. Masson staining
Formalin-fixed tissue was embedded in paraffin, and sections of 5 μm were prepared for staining with Masson’s trichrome reagent. The staining protocol included fixation in Bouin’s or Zenker’s liquor overnight, followed by washing in running water until the yellow color faded. The sections were then stained with Mayer’s hematoxylin for 5 minutes and immersed in 0.5% hydrochloric acid in 70% alcohol for 5 seconds. After washing in running tap water for 30 seconds, the sections were stained with acid ponceau for 5–10 minutes, followed by dissolution in 1% phosphomolybdic acid aqueous solution. Aniline blue or brilliant green was used to stain the sections for 5 minutes, and then dissolved in 1% glacial acetic acid for 5 minutes. The sections were dehydrated in 95% ethyl alcohol several times, followed by anhydrous alcohol. Hyalinization was performed with dimethylbenzene, and the sections were sealed with neutral balsam. The staining protocol enabled collagen fibers to be stained blue, while cytoplasm, muscle fibers, and red blood cells were stained red, and the nuclei were stained black.
2.9. TUNEL assay
The fixed myocardial tissue sections were subjected to a series of treatments. Firstly, they were fixed in 4% paraformaldehyde in PBS for 15 minutes. Next, they were treated with 1 μg/mL proteinase K in PBS for 10–20 minutes, followed by another fixation with 4% paraformaldehyde for 5 minutes. After thorough washing with PBS, the sections were incubated with 100 μL per slide of equilibration buffer for 5–10 minutes. Subsequently, the sections were incubated with a reaction mixture comprising TdT and digoxigenin-conjugated duTP at 37°C for 1 hour. Finally, the labeled DNA was visualized by using peroxidase-conjugated anti-digoxigenin antibody, with 3,3’−diaminobenzidine serving as the chromogen.
2.10. Light microscopy immunohistochemical staining of β-galactosidase assay (SA-β-gal)
To detect activation of SA-β-gal, we used cardiac tissue from sham mice or TAC mice with or without preconditioning. The expression of the SA-β-gal biomarker is independent of DNA synthesis, making it a reliable method for distinguishing between senescent and quiescent cells. This enzymatic activity is distinct from the ubiquitous acidic β-galactosidase and can be detected at pH 6.0 using the chromogenic substrate X-gal. We identified senescent cells using a histochemical staining kit (Sigma, St. Louis, MO), and observed senescent cells using light microscopy.
2.11. Quantitative real-time PCR
We employed quantitative Real-time PCR (qRT-PCR) to analyze the gene expression. The analysis was carried out using a 7500 ABI Real-time PCR system (Applied Biosystems, United States) and SYBR Premix Ex Taq (Takara Bio, Kusatsu, Shiga, Japan). In each 20 μL reaction mixture, 10 μL of SYBR Premix Ex Taq, 2 μL of cDNA, 0.8 μL of forward primer, 0.8 μL of reverse primer, and 6.4 μL of distilled water were added to a 96-well optical plate. The mixture was subjected to amplification at 95°C for 10 s, followed by 40 cycles of 5 s at 95°C and 30 s at 60°C. The melting curve was obtained by heating the PCR product from 65 to 95°C (0.5°C/5 s), and the raw Ct values were recorded. The amplification efficiency of each candidate gene was calculated using the slope of the standard curve, which was generated by serial 10-fold dilutions of the cDNA samples. The primers used for qRT-PCR are listed in . The experiment was performed in technical triplicate to ensure accuracy.
Table 1. List of primers used for Qrt-PCR.
2.12. Statistical analysis
The data is reported as mean ± SD and normality was assessed using the Shapiro-Wilk test. Statistical significance was assessed using unpaired t-tests or one-way ANOVA. A P value of less than 0.05 was considered statistically significant. All experimental results were analyzed using ImageJ and GraphPad Prism 5.0 software (CA, USA).
3. Results
3.1. Exercise-induced myocardial hypertrophy preconditioning alleviated cardiac hypertrophy
To investigate the effect of exercise-induced myocardial hypertrophy preconditioning on myocardial fibrosis, we utilized a TAC mouse model. The results of this study showed that the TAC mice had a higher heart weight to body weight ratio than the control mice. However, the preconditioned TAC mice showed a lower heart weight to body weight ratio than the TAC mice ().
Figure 1. Ratio of heart weight (mg)/weight (g) of mice and cardiac functional and structural changes. Control mice are the sham group. TAC mice received TAC and were observed for 1 week; Sw+TAC: preconditioned mice on which TAC was performed at the end of 21 days of swimming training. We observed a significantly higher heart weight to body weight ratio in TAC mice as compared to control and preconditioned TAC mice (A) (***P < 0.001 vs. control, ##P < 0.01 vs. TAC). Two-dimensional echocardiography was performed to analyze cardiac structure (B), and we monitored time-course changes in several echo-derived parameters including LV mass, LVIDd, LVESD, and EF in control, TCA, and TCA+SW mice. We normalized the mass of LV+ S, LA, RV, and RA to the corresponding tibial length (TL), and also assessed wet and dry lung weights. Statistical analysis was performed with *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control, and #P < 0.05 and ##P < 0.01 vs. TAC. All data were presented as mean ± SD from three independent experiments.
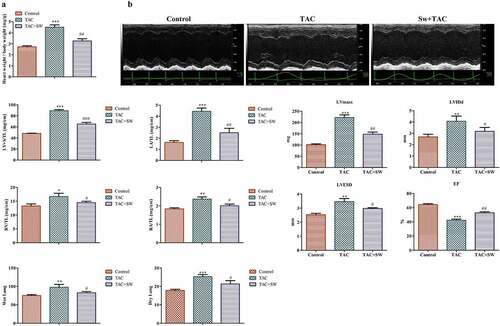
Furthermore, based on the echocardiographic data, the TAC mice showed clear signs of cardiac hypertrophy. This was evident by the increase in left ventricular (LV) mass, left ventricular internal dimension in end-diastole (LVIDd), left ventricular end-diastolic and systolic diameter (LVESD), and the decrease in ejection fraction (EF). However, the preconditioning of TAC mice rescued these changes induced by TAC, as shown in .
Additionally, TAC induction led to an increase in left ventricle + septum (LV + S) and left atrial (LA) masses in the mice, while the right ventricle (RV), right atrial (RA) masses were also elevated. The wet and dry lung weights were increased in the TAC mice. However, these TAC-induced cardiac hypertrophic phenotypes were rescued by preconditioning, as depicted in .
Therefore, our findings suggest that we successfully established a mouse model of TAC-induced myocardial hypertrophy and preconditioning plays a protective role against it.
3.2. Exercise-induced myocardial hypertrophy preconditioning inhibits myocardial fibrosis by promoting premature senescence of fibroblasts in vivo
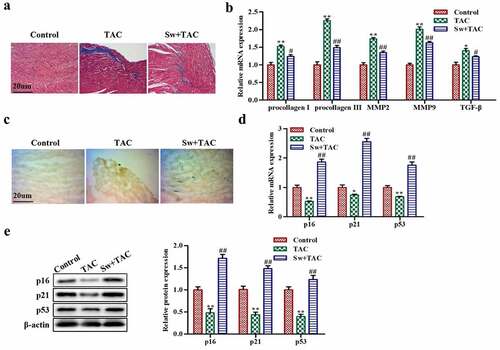
The histological experiment showed that myocardial fibrosis occurred following TAC (blue color) and was significantly inhibited by preconditioning (). Additionally, the degree of fibrosis was evaluated by measuring mRNA expression levels of fibrosis-related genes such as procollagen I, procollagen III, MMP2, MMP9, and TGF-β. As shown in , mice exhibited significantly high expression of these indicators in response to TAC treatment, whereas preconditioning reversed this effect, suggesting that preconditioning significantly inhibited myocardial fibrosis. To explore the underlying cause of the decrease in fibrosis, we conducted SA-β-Gal staining experiment for examining cellular senescence. The results confirmed more positive fibroblasts in preconditioned TAC mice compared to TAC mice () suggesting that preconditioning promoted premature senescence and reduced fibrosis. To support this result, we evaluated mRNA and protein expression of several senescence-related indicators including p21, p16, and p53, which hindered the cell cycle and promoted the occurrence of cell senescence. The results demonstrated that the level of these markers were upregulated in the preconditioning group compared with the TAC group (). In conclusion, exercise-induced myocardial hypertrophy preconditioning inhibited myocardial fibrosis by promoting premature senescence of fibroblasts in vivo.
Figure 2. Exercise-induced myocardial hypertrophy preconditioning inhibits myocardial fibrosis by promoting premature senescence of fibroblasts in vivo.
3.3. Exercise-induced myocardial hypertrophy preconditioning promotes the premature senescence of cardiac fibroblasts in vitro
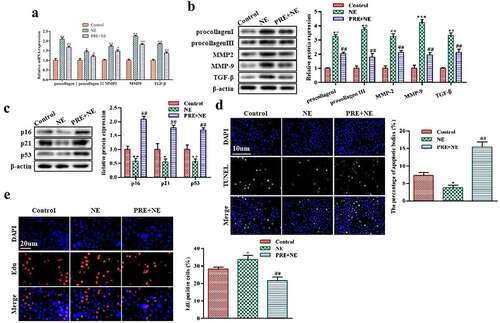
We extracted cardiac fibroblasts to investigate the impact of exercise-induced myocardial hypertrophy preconditioning on fibrosis in vitro. We have shown that norepinephrine (NE) can induce fibrosis in myocardial tissues, but mice that had undergone preconditioning exhibited less myocardial fibrosis than those treated with NE alone (Figure S1). NE treatment increased the mRNA and protein expression levels of fibrosis-related markers such as procollagen I, procollagen III, MMP2, MMP9, and TGF-β in the NE group. However, cells that underwent preconditioning PRE+NE exhibited downregulation of these markers (as shown in ). Furthermore, western blot data demonstrated that protein expression levels of p21, p16, and p53 were upregulated in the PRE+NE group compared to the NE group of cells (). In addition, the TUNEL assay indicated that there was a higher percentage of apoptotic fibroblasts in the PRE+NE group than in cells treated with NE alone (). The EdU experiment revealed that NE stimulation increased cell proliferation, which was rescued by cardiac hypertrophy preconditioning in the PRE+NE group (). Overall, cardiac hypertrophy preconditioning promoted premature senescence, increased apoptosis, and inhibited fibroblast proliferation in vitro.
Figure 3. The effect of myocardial hypertrophy preconditioning on premature senescence of cardiac fibroblasts in vitro. (a,b): The levels of procollagen I, procollagen III, MMP2, MMP9, and TGF-β proteins in cardiac fibroblasts were evaluated using western blot analysis. (c): the protein expression levels of p16, p21, and p53 in cardiac fibroblasts were analyzed using western blotting. (d): the TUNEL assay was performed on cardiac fibroblasts, and TUNEL-positive cells were stained green. (e): to detect proliferation, EdU staining was performed on cardiac fibroblasts. **P < 0.01, *P < 0.05 vs. Control, ##P <0.01, #P <0.05 vs. NE. Data were presented as mean±SD from three independent experiments.
3.4. Exercise-induced myocardial hypertrophy preconditioning promotes the activation of Nrf2 signaling pathway
Nrf2 is known to function as an antioxidant factor, but previous research has shown that its activation in fibroblasts can promote cell senescence [Citation30]. To investigate the role of Nrf2 in myocardial hypertrophy, we examined its expression in both in vitro and in vivo models. Our results demonstrated that the mRNA and protein expression levels of Nrf2 were significantly decreased in fibroblasts after treatment with TAC or NE but were increased after preconditioning ().
Figure 4. The effect of exercise-induced myocardial hypertrophy preconditioning on Nrf2 signaling pathway. (a,b) shows Qrt-PCR and western blot analysis for detecting Nrf2 mRNA and protein levels in cardiac fibroblasts, respectively. GAPDH was used as the housekeeping gene for the RT-qPCR experiments. (c, d) shows the Qrt-PCR and western blot analysis for detecting Nrf2 mRNA and protein levels, respectively, in cardiac tissues of an animal model. (e) indicates the Qrt-PCR analysis of HO-1, NQO-1, and GCLC mRNA levels in cardiac fibroblasts with GAPDH used as the control. (f) shows western blot analysis of HO-1, NQO-1, and GCLC protein levels in cardiac fibroblasts. ***P < 0.01, ##P < 0.05 vs. control, *P < 0.01 vs. NE. Data were presented as mean±SD from three independent experiments.
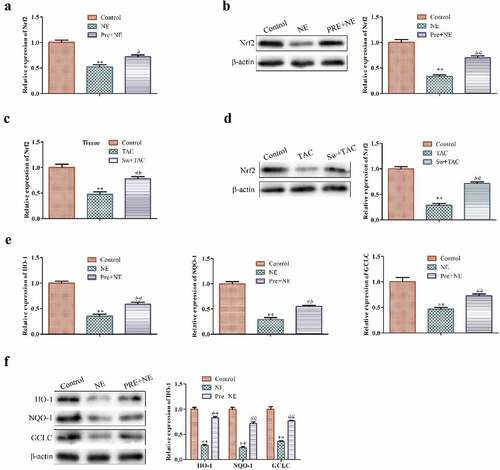
To confirm the activation of the Nrf2 signaling pathway, we further assessed the expression of its downstream target genes in NE and PRE+NE cardiac fibroblasts using qRT-PCR and western blot analysis. We found that NE treatment for 48 hours decreased the mRNA and protein expression levels of HO-1, NQO-1, and GCLC, whereas preconditioning followed by NE treatment upregulated the expression of these target genes ().
Taken together, our findings suggest that preconditioning promotes premature senescence through the activation of the Nrf2 signaling pathway.
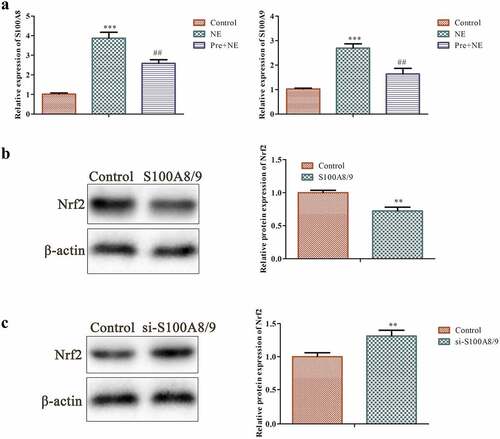
3.5. Effects of S100A8/S100A9 on Nrf2 protein in fibroblasts
We examined the expression of S100A8/S100A9 and its impact on Nrf2 protein in fibroblasts. It has been observed that the release of S100A8/A9 protein complex during inflammation prompts the secretion of cytokines, making it a potential therapeutic target in cardiovascular conditions, including myocardial inflammation and heart failure [Citation31]. The results of our investigation showed that NE stimulation increased the expression of S100A8/S100A9, while preconditioning decreased its expression (as shown in ). Western blot analysis revealed that S100A8/S100A9 had a suppressive effect on Nrf2 protein. Knocking down S100A8/S100A9 through siRNA exerted an opposite effect on Nrf2 protein, resulting in an increase in its level (as demonstrated in ). Nevertheless, the precise mechanism underlying the regulatory effects of S100A8/S100A9 on Nrf2 warrants further exploration.
Figure 5. Effects of S100A8/9 on Nrf2 protein in fibroblasts. (a) Qrt-PCR analysis was performed to determine the expression of S100A8/9 using GAPDH as the housekeeping gene in the control, NE, and Pre + NE groups. (b and c) Western blot analysis of Nrf2 protein in basal fibroblasts, S100A8/9 overexpressing fibroblasts, and S100A8/9 silenced fibroblasts. The data indicate a significant increase in Nrf2 protein expression in S100A8/9 silenced fibroblasts compared to the control (***P < 0.001, ##P < 0.01 vs. control, *P < 0.01 vs. NE). All data presented are the mean±SD from three independent experiments.
3.6. Silencing of Nrf2 attenuates the antifibrosis effects of exercise-induced myocardial hypertrophy preconditioning
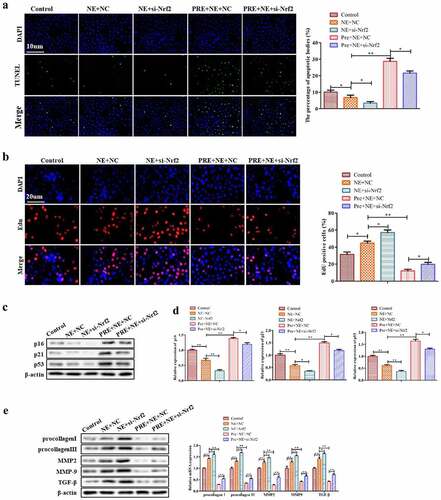
To gain a better understanding of the role of Nrf2 in exercise-induced myocardial hypertrophy preconditioning, we investigated the phenotypic changes of fibroblasts after Nrf2 knockdown in cardiac fibroblasts. Our study showed that Nrf2 knockdown reversed the pro-apoptotic effect of exercise-induced myocardial hypertrophy preconditioning on fibroblasts, as demonstrated by TUNEL assay (). Furthermore, EdU assay results indicated that Nrf2 knockdown restored the decreased cell proliferation induced by preconditioning plus NE (). In addition, Western blot and qRT-PCR analyses revealed that Nrf2 knockdown reversed the preconditioning-induced increase in expression of senescence-related proteins () and the decrease in protein and mRNA expression of procollagen I, procollagen III, MMP2, MMP9 and TGF-β (). Taken together, these findings suggest that exercise-induced myocardial hypertrophy preconditioning may reduce myocardial fibrosis in an Nrf2-dependent manner.
Figure 6. The effect of Nrf2 knockdown on the myocardial fibrosis. (a) Representative pictures of TUNEL assay of cardiac fibroblasts. TUNEL-positive cells were stained green. (b) Representative images of EdU-positive cardiac fibroblasts. (c) Western blots of p16, p21 and p53 proteins in cardiac fibroblasts. (d) Qrt-PCR analysis was conducted to detect procollagen I, procollagen III, MMP2, MMP9, and TGF-β mRNA levels in cardiac fibroblasts using GAPDH as the housekeeping gene. (e) Western blotting of procollagen I, procollagen III, MMP2, MMP9 and TGF-β proteins in cardiac fibroblasts. **P < 0.01, *P < 0.05 vs. control, ##P <0.01, #P <0.05 vs. NE. Data were presented as mean±SD from three independent experiments.
3.7. Silencing of Nrf2 reversed the effects of preconditioning on the oxidative stress response of NE-stimulated fibroblasts
Considering that Nrf2 is a key regulator of antioxidants and oxidative stress plays a critical role in cardiac fibrosis [Citation32], we conducted a study to investigate the effects of Nrf2 on reactive oxygen species (ROS) levels and oxidative stress markers in cardiac fibroblasts. Using DCFDA staining, we observed that NE stimulation increased ROS levels in cardiac fibroblasts, while preconditioning reduced this effect. Moreover, Nrf2 knockdown reversed the protective effects of preconditioning on ROS levels, leading to an increase in ROS levels in fibroblasts ().
Figure 7. The impact of preconditioning and silencing of Nrf2 on the response of fibroblasts to oxidative stress induced by NE stimulation. The panel (a) shows representative fluorescence images of fibroblasts stained with DCFDA (2´,7´-dichlorofluorescein diacetate) for detecting ROS levels in four groups: control, NE, Pre + NE, and Pre + NE + Si-Nrf2. B the levels of SOD and GPx activities in these groups were measured using Superoxide Dismutase Activity Assay Kit and Glutathione Peroxidase Assay Kit, respectively. The control group served as a baseline for comparison, and statistical significance was represented as ***P < 0.001 vs control, ##P < 0.01 vs NE, P < 0.001, $$P < 0.01 vs Pre + NE.
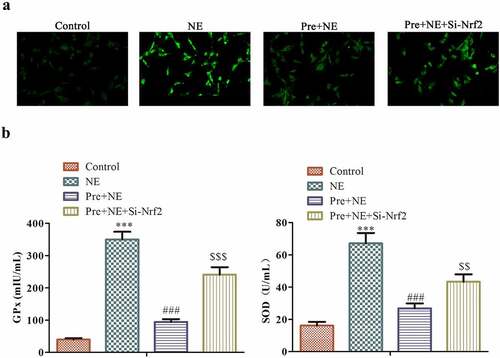
To further investigate the role of Nrf2 in regulating oxidative stress in cardiac fibroblasts, we evaluated the levels of two key oxidative stress markers, SOD and GPx, using specific assays (). Our results showed that NE stimulation led to an increase in both SOD and GPx levels in fibroblasts, which were significantly reduced in the Pre+NE group. However, silencing Nrf2 again raised the levels of both GPx and SOD in fibroblasts, indicating that Nrf2 played a crucial role in regulating the oxidative stress response of cardiac fibroblasts (). In conclusion, our findings suggest that Nrf2 is a critical regulator of ROS levels and oxidative stress markers in cardiac fibroblasts.
Discussion
Recently, studies have shown that targeting Nrf2 to regulate oxidative stress can inhibit the activation and proliferation of cardiac fibroblasts, thereby improving myocardial fibrosis [Citation33,Citation34]. Additionally, the activation of the Nrf2-mediated antioxidant response has been shown to inhibit cardiac hypertrophy [Citation35]. However, whether cardiac hypertrophy preconditioning can enhance fibroblast senescence and improve myocardial fibrosis via the Nrf2 signaling pathway remains unexplored.
This study has successfully confirmed that preconditioning of cardiac hypertrophy has a protective effect on the heart’s response to stress. This was evidenced by decreased proliferation, increased senescence, and apoptosis of fibroblasts, as well as downregulated fibrosis after hypertrophic preconditioning. The study further demonstrated that the inactivation of the Nrf2 signal pathway rescued these phenotypic changes.
Prior research has also substantiated the protective impact of myocardial hypertrophic preconditioning. Studies provide evidence that it exerts an antihypertrophic effect that helps to decelerate the progression of heart failure [Citation12]. Additionally, it has been observed that myocardial hypertrophy is partially alleviated upon release of pressure, indicating possible mechanisms to inhibit cardiac hypertrophy following pressure removal [Citation36–38]. Gaining insight into the mechanism of exercise-induced myocardial hypertrophy preconditioning can aid in identifying protective genes or pathways that could potentially reverse or prevent myocardial hypertrophy. Many diseases with excessive pressure load not only cause cardiac hypertrophy, but also myocardial fibrosis and subsequent functional changes such as heart failure and arrhythmia [Citation39–42]. Thus, it is crucial to investigate whether myocardial hypertrophy preconditioning can exert a beneficial influence on myocardial fibrosis.
Myocardial fibrosis is characterized by the accumulation of collagen in the intercellular matrix, and increased synthesis of type I and type III collagens are observed in the remodeling fibrotic heart [Citation43]. TGF-β is a potent activator of fibroblasts and myocardial fibrosis is usually accompanied by the upregulation of MMP2 and MMP9 [Citation44,Citation45]. We found that after preconditioning, the procollagen I, procollagen III, TGF-β, MMP2, and MMP9 levels were suppressed, indicating that myocardial fibrosis was inhibited. The senescence process is triggered by cell cycle inhibitor protein p21, p53 and p16, which prevent the cell cycle process [Citation15,Citation46–48]. We found that the decrease of cell proliferation and the increase of senescence and apoptosis of fibroblasts can inhibit the process of myocardial fibrosis and all these targets were achieved by myocardial hypertrophic preconditioning.
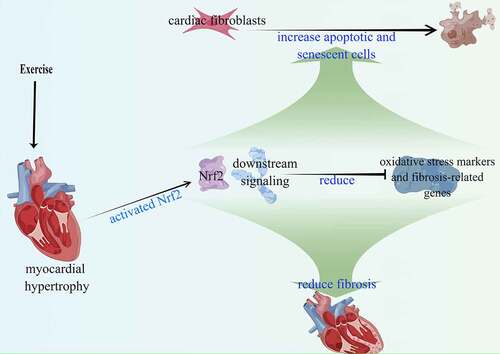
Oxidative stress is involved in cardiac fibrosis [Citation32] and we found that preconditioning reversed the effects of NE on ROS level, SOD, and GPx activities in an Nrf2-dependent way. Nrf2 is a classic gene that fights against oxidative stress and increases the antioxidant capacity of cells [Citation49–51]. Keap1 is the main regulator of Nrf2 [Citation52] which under normal circumstances, promotes ubiquitination and degradation of Nrf2, while under stress condition, Nrf2 is released from Keap1 [Citation49]. It is considered that cellular processes such as cell proliferation and differentiation are controlled by the cellular levels of reactive oxygen species (ROS) [Citation53]. HO-1 and NQO-1, as typical antioxidant enzymes, are downstream target genes of Nrf2 [Citation54,Citation55]. GCLC is the most abundant antioxidant cellular cofactor and the target gene of Nrf2, which reflects the activity of Nrf2 [Citation56]. Our findings indicate that exercise-induced myocardial hypertrophy preconditioning activates the Nrf2 signaling pathway, resulting in a significant upregulation of Nrf2, HO-1, NQO-1, and GCLC. Additionally, the activation leads to downregulation of oxidative stress and fibrosis-related genes. (). This suggests that the anti-fibrosis effect of exercise-induced myocardial hypertrophy preconditioning is Nrf2-dependent.
Figure 8. Exercise-induced myocardial hypertrophy preconditioning activates the Nrf2 signaling pathway. The activation results in increased expression of downstream target genes (HO-1, NQO-1, and GCLC), leading to reduced expression of oxidative stress and fibrosis-related genes.
In conclusion, our study demonstrates that cardiac hypertrophy preconditioning effectively inhibits the development of myocardial fibrosis, and this anti-fibrotic effect is dependent on Nrf2. Our findings suggest that enhancing the expression of Nrf2 and its downstream targets through myocardial hypertrophy preconditioning may be a promising strategy for the prevention and treatment of myocardial fibrosis.
Authors contributors
Xuan Wei, Yajing Mao planned and executed the experiments. Zheng Chen, Lina Kang, Biao Xu performed statistical analysis, and wrote the manuscript. Kun Wang designed the idea of research and designed the experiments. Xuan Wei and Yajing Mao Equal contribution in this work.
Supplemental Material
Download MS Word (888.2 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
SUPPLEMENTARY MATERIAL
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2023.2215081
Additional information
Funding
References
- Gyongyosi M, Winkler, J., Ramos, I. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. 2017;19(2):177–191.
- Espeland T. Myocardial fibrosis. Tidsskr Nor Laegeforen. 2018;138(16).
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040.
- Rathod RH, Powell AJ, Geva T. Myocardial Fibrosis in Congenital Heart Disease. Circ J. 2016;80(6):1300–1307.
- Ma ZG, Yuan, Y.P., Wu, H.M. Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci. 2018;14(12):1645–1657.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136.
- Camara-Lemarroy CR. Remote ischemic preconditioning: lung protection in the time of a pandemic? J Clin Anesth. 2020;66:109920.
- Mukai A, Suehiro, K., Kimura, A. Protective effects of remote ischemic preconditioning against spinal cord ischemia-reperfusion injury in rats. J Thorac Cardiovasc Surg. 2020;163(2):e137–156.
- Kurabayashi A, Iwashita, W., Tanaka, C. Murine remote ischemic preconditioning suppresses diabetic ketoacidosis by enhancing glycolysis and entry into tricarboxylic acid cycle in the liver. Life Sci. 2020;253:117748.
- Minamino T. Cardioprotection from ischemia/reperfusion injury: basic and translational research. Circ J. 2012;76(5):1074–1082.
- Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116(4):674–699.
- Wei X, Wu B, Zhao J. Myocardial hypertrophic preconditioning attenuates cardiomyocyte hypertrophy and slows progression to heart failure through upregulation of S100A8/A9. Circulation. 2015;131(17):1506–1517. discussion 1517.
- Chen Z, Trotman LC, Shaffer D. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–730.
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740.
- Shimizu I, Minamino T. Cellular senescence in cardiac diseases. J Cardiol. 2019;74(4):313–319.
- Krizhanovsky V, Yon M, Dickins RA. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667.
- Meyer K, Hodwin B, Ramanujam D. Essential role for premature senescence of Myofibroblasts in Myocardial Fibrosis. J Am Coll Cardiol. 2016;67(17):2018–2028.
- Kylie MV, David MK. Myocardial Ischemia-Reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr Med Chem. 2007;14(14):1539–1549.
- Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–1135.
- Zhu H, Jia Z, Misra BR. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol. 2008;8(2):71–85.
- Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43.
- Tu W, Wang H, Li S. The anti-inflammatory and anti-oxidant mechanisms of the Keap1/Nrf2/ARE signaling pathway in chronic diseases. Aging Dis. 2019;10(3):637–651.
- Ferdinandy P, Hausenloy DJ, Heusch G. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. 2014;66(4):1142–1174.
- Schmidt C, Bovolini JA, Gonçalves N. Exercise preconditioning prevents left ventricular dysfunction and remodeling in monocrotaline-induced pulmonary hypertension. Porto Biomed J. 2020;5(5):e081.
- Xu T, Tang H, Zhang B. Exercise preconditioning attenuates pressure overload-induced pathological cardiac hypertrophy. Int J Clin Exp Pathol. 2015;8(1):530–540.
- Lin H, Zhu Y, Zheng C. Antihypertrophic memory after regression of exercise-induced physiological Myocardial hypertrophy is mediated by the long noncoding RNA Mhrt779. Circulation. 2021;143(23):2277–2292.
- Tiede K, Melchior-Becker A, Fischer JW. Transcriptional and posttranscriptional regulators of biglycan in cardiac fibroblasts. Basic Res Cardiol. 2010;105(1):99–108.
- Liu X, Xiao J, Zhu H. MiR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21(4):584–595.
- Liu T, Zhang M, Niu H. Astragalus polysaccharide from astragalus melittin ameliorates inflammation via suppressing the activation of TLR-4/NF-κB p65 signal pathway and protects mice from CVB3-induced virus myocarditis. Int j biol macromol. 2019;126:179–186.
- Hiebert P, Wietecha MS, Cangkrama M. Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome. Dev Cell. 2018;46(2):145–161 e10.
- Wang S, Song R, Wang Z. S100A8/A9 in Inflammation. Front Immunol. 2018;9:1298.
- Souza-Neto FV, Jiménez-González S, Delgado-Valero B. The Interplay of mitochondrial oxidative stress and endoplasmic reticulum stress in cardiovascular fibrosis in obese rats. Antioxidants (Basel). 2021;10(8):1274.
- Zhang Q, Wang L, Wang S. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct Target Ther. 2022;7(1):78.
- Cai SA, Hou N, Zhao GJ. Nrf2 is a key regulator on puerarin preventing cardiac fibrosis and upregulating metabolic enzymes UGT1A1 inrats. Front Pharmacol. 2018;9:540.
- Shi X, Zhang B, Chu Z. Wogonin inhibits cardiac hypertrophy by activating Nrf-2-mediated antioxidant responses. Cardiovasc Ther. 2021;2021:9995342.
- Bjornstad JL, Sjaastad I, Nygård S. Collagen isoform shift during the early phase of reverse left ventricular remodelling after relief of pressure overload. Eur Heart J. 2011;32(2):236–245.
- Yang DK, Choi BY, Lee YH. Gene profiling during regression of pressure overload-induced cardiac hypertrophy. Physiol Genomics. 2007;30(1):1–7.
- Miana LA, Assad, RS, Abduch MC. Intermittent systolic overload promotes better myocardial performance in adult animals. Arq Bras Cardiol. 2010;95(3):364–372.
- Zhang QJ, Tran TA, Wang M. Histone lysine dimethyl-demethylase KDM3A controls pathological cardiac hypertrophy and fibrosis. Nat Commun. 2018;9(1):5230.
- Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262.
- Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387–407.
- Lyon RC, Zanella F, Omens JH. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116(8):1462–1476.
- Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71(4):549–574.
- Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–338.
- Yuan H, Xu J, Xu X. Calhex231 alleviates high glucose-induced myocardial fibrosis via inhibiting itch-ubiquitin proteasome pathway in vitro. Biol Pharm Bull. 2019;42(8):1337–1344.
- Schafer MJ, Haak AJ, Tschumperlin DJ. Targeting senescent cells in fibrosis: pathology, paradox, and practical considerations. Curr Rheumatol Rep. 2018;20(1):3.
- Rufini A, Tucci P, Celardo I. Senescence and aging: the critical roles of p53. Oncogene. 2013;32(43):5129–5143.
- He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169(6):1000–1011.
- Bellezza I, Giambanco I, Minelli A. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta, Mol Cell Res. 2018;1865(5):721–733.
- Kerins MJ, Ooi A. The Roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2018;29(17):1756–1773.
- Kitamura H, Motohashi H. NRF2 addiction in cancer cells. Cancer Sci. 2018;109(4):900–911.
- Taguchi K, Yamamoto M. The KEAP1-NRF2 system in cancer. Front Oncol. 2017;7:85.
- Murakami S, Motohashi H. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med. 2015;88(Pt B):168–178.
- Fujiki T, Ando F, Murakami K. Tolvaptan activates the Nrf2/HO-1 antioxidant pathway through PERK phosphorylation. Sci Rep. 2019;9(1):9245.
- Zhao Y, Sun Y, Wang G. Dendrobium officinale polysaccharides protect against MNNG-Induced PLGC in rats via activating the NRF2 and Antioxidant Enzymes HO-1 and NQO-1. Oxid Med Cell Longev. 2019;2019:9310245.
- Beinse G, Just PA, Rance B. The NRF2 transcriptional target NQO1 has low mRNA levels in TP53-mutated endometrial carcinomas. PLoS ONE. 2019;14(3):e0214416.