
ABSTRACT
Lung adenocarcinoma (LUAD) is the most common type of lung cancer. Tripartite motif 13 (TRIM13) is a member of TRIM protein family and is downregulated in multiple cancers, especially non-small cell lung cancers (NSCLC). In this study, we investigated anti-tumor mechanism of TRIM13 in non-small cell lung cancer tissues and cell lines. First, the mRNA and protein levels of TRIM13 in LUAD tissue and cells were measured. TRIM13 was overexpressed on LUAD cells to investigate the effects on cell proliferation, apoptosis, oxidative stress, p62 ubiquitination, and autophagy activation. Finally, mechanistic role of TRIM13 in regulating the Keap1/Nrf2 pathway was investigated. Results indicated that low level of TRIM13 mRNA and protein expression was found in LUAD tissue and cells. Overexpression of TRIM13 in LUAD cancer cells suppressed their proliferation, increased apoptosis, and oxidative stress, ubiquitinated p62, and activated autophagy via the RING finger domain of TRIM13. Furthermore, TRIM13 showed interaction with p62 and mediated its ubiquitination and degradation in LUAD cells. Mechanistically, TRIM13 exerted the tumor suppressor functions in LUAD cells by negatively regulating Nrf2 signaling and downstream antioxidants, which was further confirmed by in vivo data from xenografts. In conclusion, TRIM13 behaves like a tumor suppressor and triggers autophagy in LUAD cells by mediating p62 ubiquitination via KEAP1/Nrf2 pathway. Our findings provide a novel insight into targeted therapy plans for LUAD.
1. Introduction
Lung cancer (LC) is ranked as one of the leading causes of death worldwide and causes almost one-quarter of all deaths in both sexes [Citation1]. Histologically, LC can be divided into two main types with different treatment strategies, small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). The most common histological subtype of NSCLC is lung adenocarcinoma (LUAD). Although significant advances have been made to treat LUAD such as surgery, radiotherapy, chemotherapy, targeted therapy, and immunotherapy, yet survival rate of these patients remains low [Citation2]. Currently, imaging examination is effective in screening patients, but it is ineffective for early-stage detection of adenocarcinoma [Citation3,Citation4]. Furthermore, the prognosis remains poor in the advanced stage of NSCLC in patients treated with standard chemotherapy [Citation5]. Hence, understanding disease mechanisms and the development of new therapeutic targets is urgently warranted.
Autophagy is a conserved physiological process that is activated under cellular stress or nutrient depletion condition [Citation6,Citation7]. It is a double-edged sword with both tumor suppressor and tumor promoter activities. Autophagy mediates tumor suppression via many protective mechanisms such as inhibiting ROS accumulation and DNA damage, preventing genomic instability, and clearing oncogenic proteins [Citation8] that increase cell survival and its prolonged activation results in autophagy-mediated cell death [Citation9]. Notably, abnormalities in autophagy have been associated with an enhanced risk of malignancy. Tumor suppressor proteins such as PTEN and Liver kinase B1 are activators of autophagy [Citation10], conversely, oncoproteins such as PI3K and Akt1 are potential inhibitors of this pathway [Citation11]. Additionally, controlled activation of autophagy inhibits cell proliferation and stimulates cell death in Huh7 HCC cells [Citation12]. Atg4C is a protease that senses ROS level in autophagic machinery and enables the formation of autophagosome via LC3/Atg8 [Citation13]. Defects in autophagy have been reported to be associated with the development of neurodegenerative diseases of nervous system [Citation14,Citation15] and cancers [Citation16–18]. Thus, autophagy acts as a tumor suppression mechanism and limits cell growth and genomic instability [Citation19]. However, some findings indicate that autophagy also promotes oncogenesis. For instance, increased autophagy level is observed in tumor cells with activated Ras oncogene [Citation20]. Moreover, autophagy breakdowns cytoplasm and provides nutrients to cancer cells. It also promotes the survival of metastatic cells during detachment [Citation21]. Multiple other studies indicate autophagy as a mechanism to facilitate tumors [Citation17,Citation22]. Taken together, the role of autophagy in cancer is context-dependent and may act as a tumor suppressor or tumor promoter.
TRIM13 is a member of the tripartite motif (TRIM) family, and its gene is located on the 13q14 chromosome. Members of the TRIM family have been reported to regulate multiple processes such as cell proliferation [Citation23,Citation24], cell metabolism [Citation25], autophagy [Citation26] and regulate gene expression [Citation27,Citation28]. Importantly, most of the members of this family are E3 ubiquitin ligase. Several target oncogenic proteins have been identified for ubiquitination by TRIM13 such as TARF6 (Tumor Necrosis Factor Receptor-Associated Factor 6), Akt, and MDM2 [Citation26,Citation29]. When TRIM13 is overexpressed, it leads to the ubiquitination and proteasomal degradation of MDM2 and AKT, resulting in increased p53 stability and decreased AKT kinase activity. Ultimately, this enhances the level of apoptosis induced by irradiation [Citation29]. TRIM13 also downregulates NF-κB, which is essential for negative regulation of clonogenic ability of the cells [Citation30]. It also plays a crucial role in protein degradation (ERAD) during ER stress via the ubiquitin-proteasome system [Citation31]. Autophagy is a well-known mitigator of ER stress and facilitates the degradation of proteins, and even cell death during persistent ER stress [Citation32]. TRIM13 cooperates with p62, a ubiquitin-binding adaptor protein, to regulate the ubiquitination mechanism [Citation26]. Ubiquitin-proteasome pathway (UPP) is a crucial path for protein degradation and its dysfunction results in the development of multiple malignancies. A recent study showed that TRIM13 is downregulated in NSCLC and its overexpression was found to inhibit NF-κB [Citation33]. Importantly, overexpression of TRIM13 in NSCLC cell lines induced apoptosis and hampered tumor growth in the xenograft model suggesting its tumor-suppressive role [Citation33]. Here, we demonstrate that TRIM13 is a tumor suppressor that induces autophagy in lung adenocarcinoma by regulating the Nrf2 pathway and mediating SQSTM1 (p62) ubiquitination. These new findings may open novel avenues for targeted therapy of LAUD.
2. Materials and methods
2.1. Patient specimens
A total of 20 fresh LUAD tissues and 20 matched normal lung samples were supplied from the Department of Thoracic Surgery, The General Hospital of Ningxia Medical University. The study was approved by research ethics Committee of the General Hospital of Ningxia Medical University and in accordance with the Basic & Clinical Pharmacology & Toxicology policy for experimental and clinical studies [Citation34].
2.2. Cell lines and cell culture
Three LUAD cell lines (A549, NCI-H1650, and HCC827) and one normal human bronchial epithelial cell line (BEAS-2B) were purchased from ATCC (MD, USA). BEAS-2B cell line was cultured in BEGM growth medium, A549 cell line was grown in F-2K medium, while NCI-H1650 and HCC827 cell lines were supplemented with RPMI-1640 medium. All the cells were cultured with appropriate media and supplemented with 10% FBS,100 U/ml penicillin, and 100 µg/mL streptomycin. All cells were maintained in a humidified 5% CO2 incubator at 37 degrees. A549 cells received treatment with or without 10 mM of MG132, a protein degradation inhibitor, for 6 h. These cells also received treatment with 50 μg/mL cycloheximide (CHX), a protein synthesis inhibitor, at different time points.
2.3. RNA extraction and RT-qPCR
RNA from cells and tissues was extracted with GENEzolTM reagent (Geneaid) with the standard method. Briefly, samples were homogenized with GENEzol and separated using chloroform. Isopropanol was added to the separate aqueous phase, which is then centrifuged to obtain tight RNA pellets. These pellets were washed with 70% ethanol and finally resuspended in 50uL of RNAse-free water. The RNA extracts were obtained with concentration greater than 500 ng per µL and RNA, 1.98 A260/A280 ratio. cDNA was prepared using reverse transcription kit (QuantiNova), and qPCR was performed on samples using Bio-Rad CFX96 machine according to the recommended protocol. The relative gene expression was analyzed by 2–∆∆Ct method that normalized to the internal control β-actin. The primers used to amplify TRIM13 were: Forward ATTGAAGATGCCTACGCTCG and Reverse: GTGAGTAACTGGAGGGCTTTC. P62 was: Forward GGTGAAGAAACTGCTGTACAAAG and Reverse: CACACTTGGGCACTGAGAG. NQO1 was: Forward: TGAAGAAGAGAGGATGGGAGG and Reverse: GATGACTCGGAAGGATACTGAAAG. For normalization, expression of β-actin was examined with the primer pair of forward: ACCTTCTACAATGAGCTGCG and Reverse: CTGGATGGCTACGTACATGG.
2.4. Immunochemistry (IHC)
Human LUAD tissue specimens (n = 3), human normal lung tissue specimens (n = 3), xenograft specimens from mice in the oe-NC, oe-TRIM13, and oe-TRIM13+oe-NRF2 groups (n = 5 per group) were used in the IHC staining assay. For each specimen, at least three slices were used. PFA-fixed tissues were utilized to prepare paraffin-embedded blocks. A microtome machine was used to prepare 5um tissue sections on charged slides which were deparaffinized and rehydrated using xylene and alcohol. Tissue sections were boiled in the microwave at 95 degrees using 0.01 sodium citrate buffer (pH 6.5) to achieve antigen retrieval. Slides were stained using Novolink Polymer Detection System, which was then counterstained with hematoxylin, dehydrated, mounted, and finally covered with cover-slips. The primary antibodies included anti-TRIM13 (1:100, cat. no. Ab234847; Abcam, Cambridge, MA, USA), anti-Ki67 (Novus, NB500–170), and anti-p62 (1:1000, cat. no. ab207305, Abcam). For each slide, at least five randomly selected fields were photographed under an OLYMPUS B×51 microscope (Olympus, Tokyo, Japan). The staining results were analyzed by two independent pathologists using the ImageJ software to quantify the frequency of positive cells.
2.5. Western blot
Protein from cells and tissues was extracted using RIPA buffer containing protease and phosphatase inhibitors. Protein was then separated using SDS-PAGE and transferred to PVDF membranes. After blocking, membranes were incubated overnight in a cold chamber (4 degrees) with primary antibodies including anti-Bax (Abcam, 1/1000, ab32503), anti-Bcl-2 (Abcam, 1/1000, ab32124), anti-Cyclin A1 (Abcam, 1/1000, ab270940), anti-Cyclin D1 (Abcam, 1/100, ab16663), anti-HA (Abcam, ab9110), anti-Flag (Sigma-Aldrich, F1804), anti-Ub (Santa Cruz Biotechnology, sc8017), anti-TRIM13 (1:100, cat. no. Ab234847), anti-p62 (1:50, sc-48,402), anti-β-actin (1:50 sc-47,778), anti-KEAP1 (Sigma, AV38981), anti-tubulin (Novus, NB100–690), and anti-H3 (Abcam, 1:1000, ab176842). The next day, membranes were washed with washing buffer and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Finally, bands were visualized and detected with an enhanced chemiluminescence kit (Abcam), and protein intensity was analyzed by the ImageJ software. Whole WB membranes have been provided in supplementary material, S1.
2.6. Cell transfection
The oe-TRIM13 and oe-TRIM13+oe-NRF2 lentiviral vectors and a non-targeting control vector oe-NC were provided by Hanbio Therapeutics (Shanghai, China). The oe-TRIM13-Flag, and oe-Nrf2 vectors were constructed using pcDNA3.1 following methodology of standard subcloning. TRIM13 mutant version with depletion of ΔRING (TRIM13-ΔRING) was constructed using CRISPR/Cas9. The sgRNAs were designed by the CRISPR sgRNA design web tool, and the annealed oligonucleotides were integrated into the pSpCas9n(BB)-2A-GFP vector (Addgene 48,140) [Citation35]. The empty vector (oe-NC) was used as a negative control. Upon reaching 70% confluence A549 and HCC287 cells were transfected using Lipofectamine® 2000 at a final concentration of 20 nmol/l (Invitrogen Life Technologies, Carlsbad, CA, USA). Finally, cells were harvested after 48 hours.
2.7. Colony formation
A total of 2 × 103 A549 and HCC287 cells were seeded into 6-well plates and culture for 2 wk. Colonies were fixed with 4% PFA and subsequently stained with 0.1% crystal violet for 15 min at room temperature. The colonies (>50 cells) were visualized using an OLYMPUS B×51 microscope and quantified with the ImageJ software.
2.8. Cell Counting Kit-8 (CCK-8)
Cell proliferation and viability were assessed by CCK-8 assay (Sigma: 96992) according to protocol. Briefly, A549 and HCC287 cells were plated in a 96-well tissue culture plate at a density of 1 × 104 cells/well and maintained for 24 hours. Cell viability was evaluated by adding 100 µL of CCK-8 solution (Dojindo, Japan) added to each well for 24 hours. The absorbance was measured at 450 nm using an ELISA microplate reader.
2.9. Apoptotic assay by flow cytometry
Apoptosis in cells was evaluated using an apoptosis staining kit (Beyotime, Shanghai, China) by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). Briefly, A549 and HCC287 cells were seeded in 6-well culture plates at the density of 2 × 103 cells/mL overnight. After digestion, cells were washed thrice with PBS and resuspended in a binding buffer. Cells were then incubated in darkness at room temperature in FITC-labeled Annexin V and PI for 15 min. The percentage of apoptotic cells was finally assessed by flow cytometry.
2.10. Transmission electron microscopy (TEM)
A549 cells were trypsinized, washed thrice with PBS, and fixed in 2.5% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.2) overnight at 4°C. Cells were washed on the next day with 0.1 mol/L phosphate buffer and fixed in 1% aqueous osmium. After that, they were dehydrated with increasing concentrations of ethanol and finally embedded in araldite. Ultrathin sections were prepared with a microtome and mounted on copper grids. As the last step, samples were stained with 2% uranyl acetate and lead citrate. They were then observed in a transmission electron microscope (TEM; Jeol, Japan) and the number of autophagosomes was manually counted.
2.11. Tunel
Apoptosis in xenografts from mice in the oe-NC, oe-TRIM13, and oe-TRIM13+oe-NRF2 groups was assessed using a TUNEL Assay Kit-Alexa Fluor™ 488 (C10617, Invitrogen). Specimens (n = 5/group) were cut into 5 μm sections, paraffin-embedded, hydrated with an ethanol gradient (100%, 95%, 85%, 70%, and 50%), fixed with 4% formaldehyde solution, and incubated with proteinase K for 20 min at room temperature. For each specimen, at least three slices were used. To block endogenous peroxidases, 3% hydrogen peroxide was used. TUNEL reaction solution was prepared temporarily according to the manufacturer’s protocols. After washing with PBS, the slices were counterstained with DAPI for nuclear staining. Apoptotic signals were captured under five randomly selected fields with an OLYMPUS B×51 microscope and quantified with the ImageJ software.
2.12. DCFH-DA staining
DCFH-DA staining was used to evaluate the intracellular level of ROS. Briefly, A549 and HCC287 cells were incubated with 10uM DCFH-DA (Beyotime) under the manufacturer’s guidance. Cells were incubated at 37°C for 30 min and subsequently washed 1 to 2 times with cell culture medium (serum-free). Intracellular ROS was observed via an OLYMPUS B×51-FL microscopy (Olympus, Japan) and fluorescence intensity was quantified with the ImageJ software.
2.13. Immunofluorescence
Existence of LC3 and co-localization of TRIM13 and p62 were assessed using immunofluorescence. A549 and HCC287 cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. After washing with PBS, slides were blocked with 2% BSA in PBS for 1.5 h at room temperature, followed by overnight incubation with primary antibodies, anti-LC3 (1:500, cat. no. Ab192890), anti-TRIM13 (1:100, cat. no. Ab234847) and anti-p62 (1:50, sc-48,402).
The next day, cells were washed thrice with PBS and incubated with fluorochrome-conjugated secondary goat anti-rabbit antibody at room temperature for 1 h. Nuclei were stained with DAPI (Beyotime) at 0.1 g/mL for 5 min and images were analyzed via a confocal microscopy (Zeiss LSM 510 confocal). Immunofluorescence intensity was analyzed using the ImageJ software.
2.14. Co-immunoprecipitation (Co-IP)
A549, HCC827 cell lines were tagged with Flag-TRIM13 and HA-62. After 48 h, cells were harvested and lysed in lysate buffer (containing 1% NP-40, Jiangsu, China). The extracts were incubated with anti-Flag and HA antibodies at 4°C for 1 h with slow rotation. IP buffer was used five times to wash away unbound proteins. The immunoprecipitated products were visualized and measured using western blot.
2.15. Immunoprecipitation (IP)
A549 cell lysate was extracted on ice for 2 h, centrifuged, and incubated with antibody-conjugated beads at 4°C overnight. Antibody-bead complexes were washed 5 or 6 times with cold lysis buffer. SDS-PAGE loading buffer was utilized to release the precipitated proteins from the resins by boiling them for 15 min. The boiled immune complexes were placed on ice for 2 min and proceeded for SDS-PAGE electrophoresis.
2.16. Animal models
Animal experiments were conducted following institutional guidelines set forth by the ethics board. The female nude mice (5 wk, 18–20 g, n = 5/group) were injected subcutaneously (SC) with 5 × 105 A549 cells transfected with oe-NC, oe-TRIM13, and oe-TRIM13+oe-NRF2 into the right flank of a nude mouse. Tumor growth was examined weekly for at least 4 wk. After 28 d, the mice were euthanized, and tumor volumes were calculated using the formula: V = (ab2)/2 where a = tumor length and b = tumor width.
2.17. Statistical analysis
Data were expressed as the mean ± standard deviation of three independent assays. Statistical analysis was conducted using GraphPad Prism 7 software. Shapiro–Wilk test was performed to assess normality, and all data passed the normality test. Comparisons between two groups were assessed with two-tailed Student’s t-test and comparisons among multiple groups were assessed with one-way ANOVA followed by Tukey’s post hoc test. The five-year overall survival curve was generated by Kaplan-Meier Plotter online database [Citation36]. Association of TRIM13 expression and clinicopathological parameters in LUAD was analyzed by the chi-square test. A p-value less than 0.05 (P < 0.05) in our results indicates statistical significance.
3. Results
3.1. Downregulation of TRIM13 in LUAD tissues
The expression of TRIM13 in 20 cases of matched LUAD and noncancerous lung tissue samples was detected by RT-PCR and Western blot assays. As shown in , the average TRIM13 expression was significantly lower in LUAD tissues than in normal tissue (P < 0.001). IHC analysis also demonstrated the down-regulation of the TRIM13 in LAUD tissues as compared to normal counterparts as well as the subcellular location of TRIM13 in both cytoplasm and nucleus in LUAD (). The association of TRIM13 expression and clinicopathological parameters in LUAD was analyzed. The results indicated that TRIM13 expression had no association with age, gender, TNM stage, smoking, but had significant association with pathology stage ().
Figure 1. Downregulation of TRIM13 in LUAD tissues.
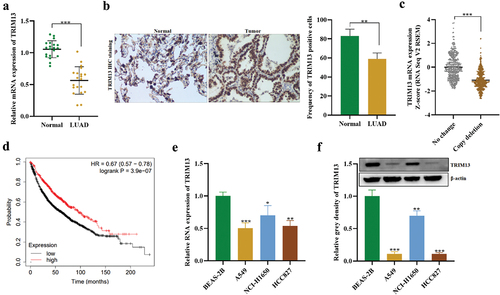
Table 1. Association of TRIM13 expression and clinicopathological parameters in LUAD.
Bioinformatic analysis using cbioportal subset of Metastatic Non-Small Cell Lung Cancer, Nature Medicine 2022 (https://www.cbioportal.org/) on 1249 LUAD samples also indicated reduced mRNA expression and copy deletion in 652 samples showing a correlation between TRIM3 copy deletion and mRNA expression (). Kaplan–Meier curve showed that LAUD patients with low-level TRIM13 had a poor prognosis and reduced survival probability (). Finally, lower mRNA () and protein () of TRIM13 were observed in LUAD cell lines (A549, NCI-H1650, and HCC827) as compared to the control cell line (BEAS-2B). A549 and HCC827 showed relatively lower TRIM13 expression and will be used for experiments. Collectively, TRIM13 shows depletion in LUAD tissue and cells and indicates poor prognosis of LUAD patients.
3.2. TRIM13 suppresses LUAD cell proliferation and facilitates cell apoptosis
We have verified that the basal level of TRIM13 is low in LUAD cell lines. A549 and HCC827 cell lines were generated that ectopically overexpressed TRIM13 (hereafter referred to as oe-TRIM13-A549 and oe-TRIM13-HCC827), while oe-NC without TRIM13 expression was used as a control (oe-NC). TRIM13 expression in the transfected cells was detected by RT-PCR analysis (). Reverse transcription analysis revealed that expression of TRIM13 at mRNA level was increased in both LUAD cells compared with the control. The above result confirmed the expression of recombinant plasmids in transfected cells with TRIM13, indicating that the exogenous TRIM13 was successfully transfected into the LUAD cells.
Figure 2. Ectopic expression of TRIM13 suppressed LUAD cells proliferation and facilitated cell apoptosis. LUAD cells were transfected with TRIM13 and its mutated version.
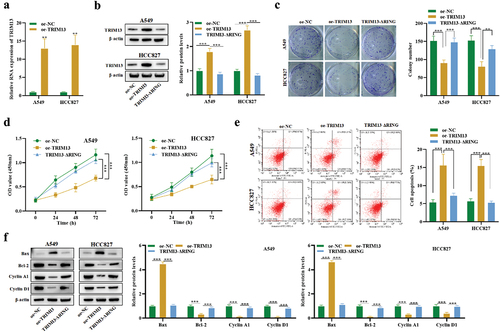
TRIM proteins belong to the RNF protein family that contains the N-terminal RING domain. TRIM13 performs its biological functions using the RING finger domain [Citation37]. Thus, we constructed plasmids of TRIM13 with a mutated RING finger domain (hereafter referred to as TRIM13-ΔRING). The relative proportion of TRIM13 protein in A549 and HCC827 cell lines transfected with wild-type oe-TRIM13 or TRIM13-ΔRING was examined. Interestingly, the mutation in RING finger domain of TRIM13 reduced TRIM13 expression in both cell lines, demonstrating that the TRIM13 RING domain depletion vector is successfully constructed ().
To test whether TRIM13 may function as a tumor suppressor, a clonogenic assay was performed to analyze the effect of TRIM13 upregulation on in vitro growth of LUAD cell lines. Compared with oe-NC and TRIM13-ΔRING, the colony number decreased by oe-TRIM13 by about 50% (p < 0.001) in both cell lines (). To further testify an antiproliferative effect of TRIM13 on the viability of LUAD cells, CCK8 assay was performed. As shown in , oe-TRIM13-A549 and oe-TRIM13-HCC827 cells grew more slowly than oe-NC and oe-TRIM13-ΔRING cells.
To understand if apoptosis was associated with growth restriction, we performed flow cytometry to analyze the apoptosis of LUAD cells with or without the exogenous TRIM13 gene and its mutated version. Annexin V FITC/PI staining of cells was analyzed by flow cytometry, and a substantial increase in the level of apoptotic cells was detected (). Moreover, western blot was conducted to assess apoptosis-associated proteins (Bax and Bcl-2) and cyclins (Cyclin A1 and Cyclin D1). oe-TRIM13-A549 and oe-TRIM13-HCC827 cells showed a significant increase in Bax protein level and a significant increase in Bcl-2, Cyclin A1, and Cyclin D1 protein levels (). Taken together, these results suggest that ectopic expression of TRIM13 can trigger apoptosis in LUAD cells, which was dependent on the RING domain.
3.3. TRIM13 facilitates LUAD cell autophagy and oxidative stress
The above data demonstrated that TRIM13 facilitates the induction of apoptosis in LUAD cells, it was unclear whether autophagy was activated. Therefore, to investigate its involvement, we first assessed the level of autophagy in LAUD cell lines. We characterized it using two classical assays: (1) TEM analysis of the cytoplasmic accumulation of autophagosomes, a direct way to detect autophagy activation, and immunofluorescence staining of LC3, an autophagy-specific marker. TEM Images () showed marked elevation of autophagosome number in A549 and HCC827 cells overexpressed with TRIM13. However, mutation of the RING domain in TRIM13 resulted in a significantly decreased number of the autophagosomes compared with the wild-type oe-TRIM13 group. To further corroborate that TRIM13 influences the autophagosome formation, we directly assessed LC3 expression using immunofluorescence.
Figure 3. TRIM13 facilitated LUAD cell autophagy and oxidative stress.
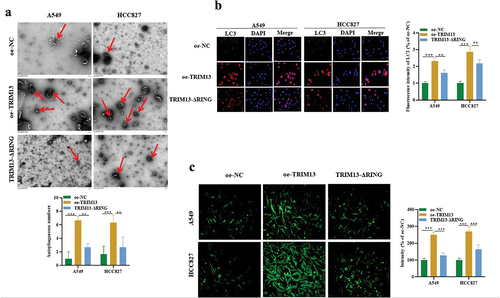
Notably, TRIM13 overexpression dramatically increased LC3 expression in A549, HCC827 cells, whereas mutation in the RING domain significantly decreased the percentage of LC3 positive cells compared with the oe-TRIM13 group (), further proving that TRIM13 affects the autophagy process. Together, our results indicate that TRIM13 is associated with increased autophagosome formation and autophagy, and it requires the cooperation of the RING finger domain. Several reports indicated the contribution of reactive oxygen species (ROS) in the induction of autophagy and apoptosis [Citation38,Citation39]. Therefore, we decided to analyze the concentration of ROS accumulation in the cells. For this purpose, cells were stained with CM-H2-DCFH-DA, a fluorescence dye that reacts to a broad spectrum of ROS. The fluorescence intensity was analyzed using the ImageJ software. As shown in , ROS level was increased significantly by overexpression of TRIM13 in A549 and HCC827 cells. Interestingly, this increased ROS generation was efficiently attenuated in the presence of a mutated RING domain. Collectively, TRIM13 promotes the induction of autophagy and oxidative stress in lung cancer cells through the involvement of the RING finger domain.
3.4. TRIM13 interacts with p62 protein in LUAD cells
Next, we explored the downstream gene of TRIM13 regulating the LUAD cell malignant behaviors. String and BioGrid databases were searched for protein–protein interaction (PPI) of TRIM13. Interestingly, autophagy-related marker p62 (SQSTM1) showed a strong correlation with TRIM13 in both databases () which were further validated with RT-qPCR and western blot. Results showed the reduced protein level of p62 in TRIM13 overexpressed cells. However, this effect was only observed at the protein level and mRNA level of p62 was not influenced (). Finally, TRIM13 co-localized with p62 in the cytoplasm (), and oe-TRIM13 decreased p62 expression in LUAD cells, suggesting that TRIM13 regulates p62 at a post-transcriptional level.
Figure 4. TRIM13 interacts with p62 protein in LUAD cells.
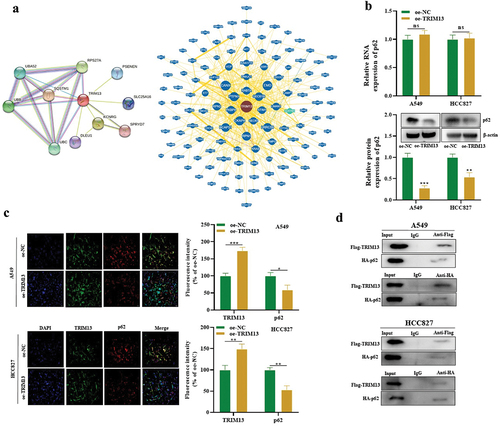
Finally, fusion proteins Flag-TRIM13 and HA-p62 were constructed to validate the interaction of both proteins using Co-IP. The results confirmed that both Flag-TRIM13 and HA-p62 in A549 and HCC827 cells were co-immunoprecipitated with anti-Flag or anti-HA rather than IgG (). Taken together, TRIM13 has been shown to interact with p62 in LUAD cells.
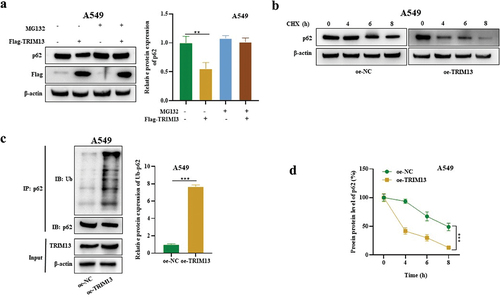
3.5. TRIM13 mediates p62 ubiquitination in LUAD cells
Ubiquitination is a post-transcriptional modification, where ubiquitin moiety is covalently attached to the substrate protein and indicates autophagic degradation. TRIM13 is an E3 ligase and mediates the protein ubiquitination, whereas p62 is an autophagy substrate. Findings from our previous results demonstrate that TRIM13 suppresses p62 protein expression, we speculated that TRIM13 may mediate the process via p62 ubiquitination. Western blot results indicated that the inhibitory effect of TRIM13 on protein p62 in A549 cells was abrogated by the addition of 10 mM of a protease inhibitor, MG132 () confirming that TRIM13 degrades p62 protein. We next treated oe-TRIM13-A549 cells and oe-NC-A549 cells by CHX for the indicated time (4,6,8 h) to examine protein abundance. CHX caused a gradual and time-dependent decline of the p62 protein level. The oe-TRMP13-A549 cells had a more significant decline of p62 protein level than the oe-NC-A549 cells, indicating the role of TRIM13 in degrading the p62 protein (). Finally, IP and WB further confirmed the ubiquitination and decreased protein level of p62 in oe-TRIM13-A549 cells (). These data suggest that p62 is targeted by TRIM13 for ubiquitination in LUAD cells.
Figure 5. TRIM13 mediated SQSTM1 ubiquitination in LUAD cells.
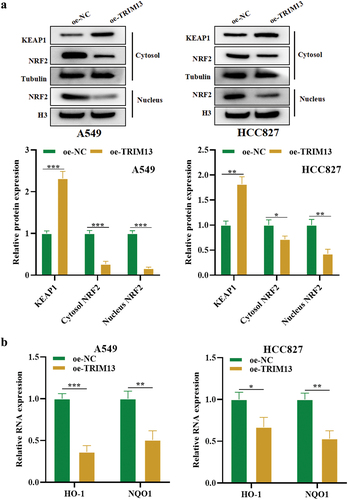
3.6. TRIM13 regulates the Keap1/Nrf2 pathway in LUAD cells
The Keap1-Nrf2 pathway is the primary protective action of the cell against oxidative stress. Under normal circumstances, Keap1, a vital part of an E3 ubiquitin ligase, targets NRF2 for proteasome-dependent degradation [Citation40]. However, in stress, Keap1 allows Nrf2 to escape ubiquitination and translocate to the nucleus, where it can initiate its antioxidant mechanism. To explore whether TRIM13 regulates the Nrf2/Keap1 pathway, the cytoplasmic level of Keap1, whereas the cytoplasmic and nuclear levels of Nrf2 were measured in A549 and HCC827 cells. The western blot result showed that TRIM13 overexpression in these LAUD cells increased cytosolic Keap1 whereas reduced the cytoplasmic and nuclear expression of Nrf2, indicating the inactivation of the Nrf2 pathway (). Nrf2 is a transcriptional regulator of cellular antioxidant genes. Therefore, the mRNA level of downstream antioxidant enzymes, HO-1 and NQO1, was measured in LUAD cells. Accordingly, the mRNA levels of antioxidant HMOX1 and NQO1 were much lower in A549 and HCC827 cells overexpressed with TRIM13 (). The collective results indicate that TRIM13 exerts tumor suppressor functions in lung cancer cells by negatively regulating Nrf2 signaling and downstream antioxidants.
Figure 6. TRIM13 regulates the Keap1/Nrf2 pathway in LUAD cells.
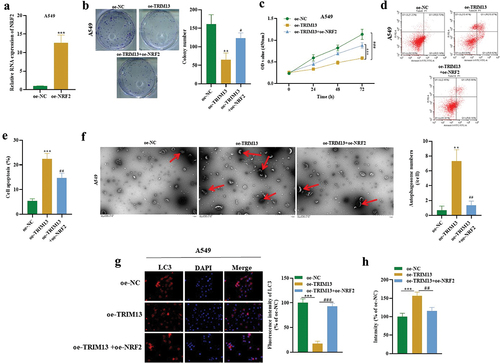
3.7. TRIM13 regulates LUAD cell malignancy through Nrf2
Next, A549 cells were transfected with Nrf2 overexpression vector (oe-NRF2) to investigate whether Nrf2 regulates the functions of LUAD cells. shows successful transfection in these cells by RT-PCR. Interestingly, oe-Nrf2 increased cell proliferation (), enhanced cell viability (), and reduced apoptotic rate (), autophagosome number (), LC3 expression () and oxidative stress () thus countering the effects of TRIM13 to some extent (). It suggests that TRIM13 suppresses LUAD cell malignancy through controlling NRF2.
Figure 7. TRIM13 regulated LUAD cell malignancy through NRF2.
3.8. TRIM13 suppresses LUAD tumor growth through NRF2 in vivo
Based on in vitro data, we sought to determine whether TRIM13 exerts an antitumor effect via Nrf2 in vivo. 5 × 105 A549 cells expressing oe-NC, oe-TRIM13, and oe-TRIM13+oe-Nrf2 were subcutaneously injected into the flank of nude mice. Tumor volume was monitored for 28 d. Mice were killed after 28 d of implantation, and xenografts were weighed. The results revealed that injecting oe-TRIM13-A549 cells reduced the tumor volume, size, and weight in the xenografts. Interestingly, tumor volume, size, and weight from xenograft established by injecting the oe-TRIM13-oe-NRF2-A549 cells were bigger than oe-TRIM13-A549 tumors (, b). IHC analysis revealed a significant decrease in Ki67 (proliferation marker) in xenograft sections from oe-TRIM13-A549 group when compared to sections from oe-TRIM13+oe-Nrf2 and the control group (, d). This suggests that TRIM13 is a tumor suppressor and controls excessive cell proliferation. Ki67 data from Oe-TRIM13+oe-Nrf2 ×enograft tissue sections revealed that cells can proliferate at a higher rate in the presence of Nrf2, but this proliferation was controlled by TRIM13 and thus proliferation was lower than the control group. Moreover, there was a significant increase in TUNEL positive cells and a significant decrease in p62 positive cells in xenograft sections from oe-TRIM13-A549 group when compared to sections from oe-TRIM13+oe-Nrf2 and the control group (, f). These findings revealed that TRIM13 induces apoptosis and autophagy in vivo dependent on Nrf2 depletion.
Figure 8. TRIM13 suppressed LUAD tumor growth through NRF2 in vivo.
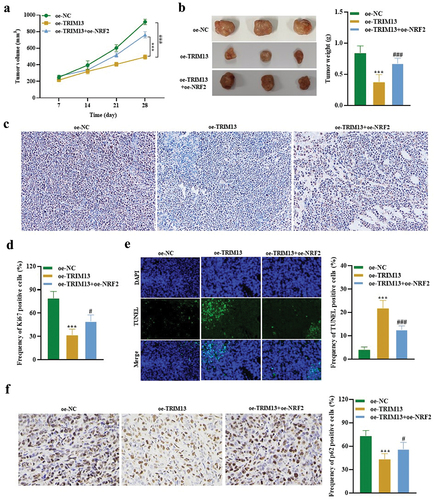
These findings provide compelling evidence that TRIM13 overexpression reduces tumor development in the xenograft mice model. Altogether, these results provide the first in vivo evidence that TRIM13 negatively regulates tumor growth in mice models via Nrf2 downregulation and provides a novel insight for seeking targeted therapy of LUAD.
4. Discussion
Though multiple treatment strategies and diagnostic techniques have been developed for LC, the survival rate of LC remains low, causing great distress to the physical and mental health of patients. Thus, it is of great significance to investigate the mechanism behind LC progression to develop appropriate therapeutic targets.
TRIM13 is an important member of the TRIM family and plays a diverse role in cell death [Citation29] and endoplasmic reticulum-associated degradation [Citation31]. It facilitates apoptosis via protein kinase B [Citation29] and regulates autophagy in endoplasmic reticulum stress [Citation26]. Further studies have shown that Trim13 ubiquitinates Akt, L-type channels, and caspase-8 [Citation41–43]. Bioinformatics analysis revealed lower TRIM13 expression in breast cancer. TRIM13 has been shown to inhibit cell migration and invasion in renal cell carcinoma cell lines of 786-O [Citation42]. A recent report showed that TRIM13 is downregulated in NSCLC cells and regulates NF-KB signaling in these cells [Citation33]. These studies suggest the tumor suppressor function of TRIM13. Here, we found the reduced expression of TRIM13 in human LUAD tissues as compared to non-cancerous tissues, which is further confirmed by cbioportal data analysis. Additionally, TRIM13 overexpression in LUAD cell lines hampered cell proliferation and stimulated apoptosis in LUAD cells. However, this effect is reversed by a mutation in the RING domain of TRIM13 suggesting that tumor suppressor activity of TRIM13 depends on its RING finger domain.
Autophagy is a cellular protective mechanism against pathological conditions like infection, cancer, etc. [Citation26]. The relationship between autophagy and apoptosis is controversial and highly dependent on the context. In some circumstances, autophagy counteracts apoptosis, whereas in other situations, autophagy acts synergistically with apoptosis [Citation44]. Previous studies suggested that TRIM13 regulates autophagy and oxidative stress [Citation45]. Autophagy has been closely associated with ROS [Citation46,Citation47]. ROS has been copiously reported as early inducers of autophagy upon nutrient deprivation [Citation48].
In the present study, we found activation of autophagy and enhanced ROS accumulation in LUAD cells overexpressed with TRIM13, suggesting TRIM13 facilitated activation of autophagy and enhanced the oxidative stress in these cells. Ubiquitination plays an important role in the degradation of proteins or defective organelle either through proteasome or autophagy [Citation26]. Different autophagy receptors interact with autophagy machinery [Citation49]. p62 is a best-studied autophagy marker that can interact with ubiquitinated cargo [Citation50]. This interaction leads to the degradation of the p62 level, which is associated with the activated autophagy process. LC3 is also often employed as a surrogate marker for autophagic vesicles [Citation51].
Our study indicates a strong correlation of TRIM13 with p62 using String and BioGrid pathway, which was further validated experimentally. Interestingly, TRIM13 overexpression reduces the p62 level only at the protein level but not the mRNA value, indicating that TRIM13 degrades p62. We also found increased staining of LC3 in TRIM13-overexpressed LUAD cells suggesting autophagy activation in these cancer cells. This reduced protein level of an autophagy substrate, p62 in oe-TRIM13-A549 cells is caused by ubiquitination of TRIM13-mediated ubiquitination of p62, which was reversed by protease inhibitor, MG132. The RING finger domain can bind to ubiquitinase and substrate simultaneously and mediate ubiquitination. We demonstrated that ubiquitination of p62 by TRIM13 in LUAD cells takes place through its RING finger domain.
p62 is involved in multiple signal transduction pathways, including the Keap1–Nrf2 pathway [Citation52]. It is a major signaling pathway that responds to increased oxidative stress [Citation53]. Keap1 normally functions as an adaptor protein for the E3 ubiquitin ligase complex and sequesters Nrf2 in the cytoplasm at a low level [Citation54]. Genomic data of humans have demonstrated that deregulation of this pathway is reported in multiple disorders. Mutations in Keap1-Nrf2 in lung cancer are associated with therapeutic resistance and poor prognosis [Citation55]. Blake et al. showed malignant lung cancer in Keap Knock-out mice when exposed to additional proliferative stimuli [Citation56]. Nrf2 inhibitors in advanced NSCLC patients harboring Keap1-Nrf2 mutation appear to be a promising treatment [Citation57]. Substantial evidence has shown that p62 links autophagy to the Keap1-Nrf2 pathway [Citation53,Citation58].
In the p62-Keap1-Nrf2 interaction, the Keap1-interacting region of p62 binds to Keap1, thereby restricting Keap1 from trapping Nrf2. It results in stabilization and finally activation of Nrf2 [Citation59]. The p62 gene is a target of Nrf2 [Citation60], suggesting a positive-feedback loop within the p62-Keap1-Nrf2 axis. Autophagy is directly involved in the whole process. p62 is degraded by autophagy [Citation61], and Keap1 is also degraded by autophagy in a p62 interaction manner [Citation62]. We found the reduced cell apoptosis and increased cell proliferation and viability of LUAD cell line by transfection with oe-Nrf2, suggesting that the negative impact of TRIM13 on tumor growth was through Nrf2 regulation. Accordingly, we found big tumors in xenografts injected with control cells, but with TRIM13 overexpression in these injected cells, the tumor remained smaller. Nrf2 in the presence of TRIM13 showed bigger tumors than TRIM13 overexpression ones, indicating that TRM13 is a tumor suppressor and regresses tumor growth via the Nrf2 pathway.
In conclusion, TRIM13 triggers apoptosis and autophagy in LUAD cells through mediating p62 ubiquitination via KEAP1/NRF2 pathway, providing a novel insight for seeking targeted therapy for LUAD patients.
Authorship
Bo Yu and Yu Zhou planned, executed the experiments, and wrote the initial manuscript. Jinxi He performed statistical analysis. All authors revised the manuscript and approved for the final version.
Supplemental Material
Download MS Word (2.8 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2023.2216504
Additional information
Funding
References
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. Ca A Cancer J Clinicians. 2021;71(1):7–33.
- Al-Dherasi A, Huang QT, Liao Y, et al. A seven-gene prognostic signature predicts overall survival of patients with lung adenocarcinoma (LUAD). Cancer Cell Int. 2021;21(1):294. Online First: Epub Date]|.
- Ariozzi I, Paladini I, Gnetti L, et al. Computed tomography-histologic correlations in lung cancer. Pathologica. 2013;105(6):329–336.
- Kozielski J, Kaczmarczyk G, Porębska I, et al. Lung cancer in patients under the age of 40 years. Contemp Oncol (Pozn). 2012;16(5):413–415. Online First: Epub Date]|
- Cataldo VD, Gibbons DL, Pérez-Soler R, et al. Treatment of non-small-cell lung cancer with erlotinib or gefitinib. Online First: Epub Date]| N Engl J Med. 2011;364(10):947–955.
- Bagherniya M, Butler AE, Barreto GE, et al. The effect of fasting or calorie restriction on autophagy induction: a review of the literature. Ageing Res Rev. 2018;47:183–197. Online First: Epub Date]|.
- He L, Zhang J, Zhao J, et al. Autophagy: the Last Defense against Cellular Nutritional Stress. Advances In Nutrition (Bethesda, MD). 2018;9(4):493–504. Online First: Epub Date]|.
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015;34(7):856–880. Online First: Epub Date]|.
- Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Diff. 2005;12 Suppl 2(S2):1528–1534. Online First: Epub Date]|.
- Liang J, Shao SH, Xu ZX, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9(2):218–224. Online First: Epub Date]|.
- Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. Online First: Epub Date]|.
- Li P, Du Q, Cao Z, et al. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012;314(2):213–222. Online First: Epub Date]|.
- Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Online First: Epub Date]| Embo J. 2007;26(7):1749–1760.
- Seranova E, Connolly KJ, Zatyka M, et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017;61(6):733–749. Online First: Epub Date]|.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nature Med. 2013;19(8):983–997. Online First: Epub Date]|
- Di Fazio P, Matrood S. Targeting autophagy in liver cancer. Translational gastroenterology and hepatology. 2018;3:39. Online First: Epub Date]|.
- Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Investig. 2003;112(12):1809–1820. Online First: Epub Date]|.
- Cicchini M, Chakrabarti R, Kongara S, et al. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014;10(11):2036–2052. Online First: Epub Date]|.
- Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. Online First: Epub Date]|.
- Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–470. Online First: Epub Date]|.
- Witz IP. Tumor-microenvironment interactions: dangerous liaisons. Advances in cancer research. 2008;100:203–229. Online First: Epub Date]|.
- Yang L, Zhang X, Li H, et al. The long noncoding RNA HOTAIR activates autophagy by upregulating ATG3 and ATG7 in hepatocellular carcinoma. Online First: Epub Date]| Mol Biosyst. 2016;12(8):2605–2612.
- Lv D, Li Y, Zhang W, et al. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat Commun. 2017;8(1):1454. Online First: Epub Date]|.
- Czerwińska P, Mazurek S, Wiznerowicz M. The complexity of TRIM28 contribution to cancer. Online First: Epub Date]| J Biomed Sci. 2017;24(1):63.
- Cambiaghi V, Giuliani V, Lombardi S, et al. TRIM proteins in cancer. Adv Exp Med Biol. 2012;770:77–91. Online First: Epub Date]|.
- Tomar D, Singh R, Singh AK, et al. TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Online First: Epub Date]| Biochim Biophys Acta. 2012;1823(2):316–326.
- Cheng B, Ren X, Kerppola TK. KAP1 represses differentiation-inducible genes in embryonic stem cells through cooperative binding with PRC1 and derepresses pluripotency-associated genes. Online First: Epub Date]| Mol Cell Biol. 2014;34(11):2075–2091.
- Oleksiewicz U, Gładych M, Raman AT, et al. TRIM28 and Interacting KRAB-ZNFs Control Self-Renewal of Human Pluripotent Stem Cells through Epigenetic Repression of Pro-differentiation Genes. Stem Cell Rep. 2017;9(6):2065–2080. Online First: Epub Date]|.
- Joo HM, Kim JY, Jeong JB, et al. Ret finger protein 2 enhances ionizing radiation-induced apoptosis via degradation of AKT and MDM2. Eur J Cell Biol. 2011;90(5):420–431. Online First: Epub Date]|.
- Tomar D, Singh R. TRIM13 regulates ubiquitination and turnover of NEMO to suppress TNF induced NF-κB activation. Online First: Epub Date]| Cell Signal. 2014;26(12):2606–2613.
- Lerner M, Corcoran M, Cepeda D, et al. The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Molecular biology of the cell. Online First: Epub Date]|. 2007;18(5):1670–1682.
- Verfaillie T, Salazar M, Velasco G, et al. Linking ER Stress to Autophagy: potential Implications for Cancer Therapy. Int J Cell Biol. 2010;2010:930509. Online First: Epub Date]|.
- Xu L, Wu Q, Zhou X, et al. TRIM13 inhibited cell proliferation and induced cell apoptosis by regulating NF-κB pathway in non-small-cell lung carcinoma cells. Gene. 2019;715:144015. Online First: Epub Date]|.
- Tveden-Nyborg P, Bergmann TK, Jessen N, et al. BCPT policy for experimental and clinical studies. Online First: Epub Date]| Basic Clin Pharmacol Toxicol. 2021;128(1):4–8.
- Liu J, Zhang C, Xu D, et al. The ubiquitin ligase TRIM21 regulates mutant p53 accumulation and gain-of-function in cancer. J Clin Investig. 2023;133(6): Online First: Epub Date]|. doi:10.1172/jci164354.
- Győrffy B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput Struct Biotechnol J. 2021;19:4101–4109. Online First: Epub Date]|.
- Cai C, Tang YD, Zhai J, et al. The RING finger protein family in health and disease. Signal transduction and targeted therapy. Signal Transduct Target Ther. 2022;7(1):300. Online First: Epub Date]|.
- Macchioni L, Davidescu M, Sciaccaluga M, et al. Mitochondrial dysfunction and effect of antiglycolytic bromopyruvic acid in GL15 glioblastoma cells. J Bioenerg Biomembr. 2011;43(5):507–518. Online First: Epub Date]|.
- Kaminskyy VO, Piskunova T, Zborovskaya IB, et al. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Online First: Epub Date]| Autophagy. 2012;8(7):1032–1044.
- Baird L, Yamamoto M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol Cell Biol. 2020;40(13): Online First: Epub Date]|. DOI:10.1128/MCB.00099-20.
- Tan P, He L, Cui J, et al. Assembly of the WHIP-TRIM14-PPP6C Mitochondrial Complex Promotes RIG-I-Mediated Antiviral Signaling. Molecular Cell. 2017;68(2):293–307.e5. Online First: Epub Date]|.
- Yang B, Wang J, Wang Y, et al. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J Immun (Baltimore. 2013;190(7):3613–3619. Md. : 1950 Online First: Epub Date]|.
- Castanier C, Zemirli N, Portier A, et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. Online First: Epub Date]|.
- Liu G, Pei F, Yang F, et al. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int J Mol Sci. 2017;18(2):367. Online First: Epub Date]|.
- Tomar D, Prajapati P, Sripada L, et al. TRIM13 regulates caspase-8 ubiquitination, translocation to autophagosomes and activation during ER stress induced cell death. Biochim Biophys Acta. 2013;1833(12):3134–3144. Online First: Epub Date]|.
- Chen Y, McMillan-Ward E, Kong J, et al. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. Online First: Epub Date]| J Cell Sci. 2007;120(Pt 23):4155–4166.
- Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Online First: Epub Date]| Autophagy. 2010;6(7):838–854.
- Filomeni G, Desideri E, Cardaci S, et al. Under the ROS … thiol network is the principal suspect for autophagy commitment. Online First: Epub Date]| Autophagy. 2010;6(7):999–1005.
- Mijaljica D, Nazarko TY, Brumell JH, et al. Receptor protein complexes are in control of autophagy. Autophagy. 2012;8(11):1701–1705. Online First: Epub Date]|.
- Rogov V, Dötsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Molecular Cell. 2014;53(2):167–178. Online First: Epub Date]|
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy 3rd. Autophagy. 2016;12(1):1–222. Online First: Epub Date]|
- Liu WJ, Ye L, Huang WF, et al. P62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21(1):29. Online First: Epub Date]|.
- Kageyama S, Saito T, Obata M, et al. Negative Regulation of the Keap1-Nrf2 Pathway by a p62/Sqstm1 Splicing Variant. Mol Cell Biol. 2018;38(7): Online First: Epub Date]|. DOI:10.1128/MCB.00642-17.
- Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–7139. Online First: Epub Date]|.
- Singh A, Misra V, Thimmulappa RK, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLOS Med. 2006;3(10):e420. Online First: Epub Date]|.
- Best SA, Sutherland KD. “Keaping” a lid on lung cancer: the Keap1-Nrf2 pathway. Cell cycle (Georgetown, Tex. 2018;17(14):1696–1707. Online First: Epub Date]|.
- Barrera-Rodríguez R. Importance of the Keap1-Nrf2 pathway in NSCLC: is it a possible biomarker? Online First: Epub Date]| Biomed Rep. 2018;9(5):375–382.
- Zhang W, Feng C, Jiang H. Novel target for treating Alzheimer’s Diseases: crosstalk between the Nrf2 pathway and autophagy. Ageing Res Rev. 2021;65:101207. Online First: Epub Date]|.
- Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12(3):213–223. Online First: Epub Date]|.
- Jain A, Lamark T, Sjøttem E, et al. P62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–22591. Online First: Epub Date]|.
- Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283(33):22847–22857. Online First: Epub Date]|.
- Taguchi K, Fujikawa N, Komatsu M, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci USA. 2012;109(34):13561–13566. Online First: Epub Date]|.