
ABSTRACT
In chronic liver injury, quiescent hepatic stellate cells (HSCs) transdifferentiate into activated myofibroblast-like cells and produce large amounts of extracellular matrix components, e.g. collagen type 1. Cellular senescence is characterized by irreversible cell-cycle arrest, arrested cell proliferation and the acquisition of the senescence-associated secretory phenotype (SASP) and reversal of HSCs activation. Previous studies reported that H2S prevents induction of senescence via its antioxidant activity. We hypothesized that inhibition of endogenous H2S production induces cellular senescence and reduces activation of HSCs. Rat HSCs were isolated and culture-activated for 7 days. After activation, HSCs treated with H2S slow-releasing donor GYY4137 and/or DL-propargylglycine (DL-PAG), an inhibitor of the H2S-producing enzyme cystathionine γ-lyase (CTH), as well as the PI3K inhibitor LY294002. In our result, CTH expression was significantly increased in fully activated HSCs compared to quiescent HSCs and was also observed in activated stellate cells in a in vivo model of cirrhosis. Inhibition of CTH reduced proliferation and expression of fibrotic markers Col1a1 and Acta2 in HSCs. Concomitantly, DL-PAG increased the cell-cycle arrest markers Cdkn1a (p21), p53 and the SASP marker Il6. Additionally, the number of β-galactosidase positive senescent HSCs was increased. GYY4137 partially restored the proliferation of senescent HSCs and attenuated the DL-PAG-induced senescent phenotype. Inhibition of PI3K partially reversed the senescence phenotype of HSCs induced by DL-PAG. Inhibition of endogenous H2S production reduces HSCs activation via induction of cellular senescence in a PI3K-Akt dependent manner. Our results show that cell-specific inhibition of H2S could be a novel target for anti-fibrotic therapy via induced cell senescence.
1. Introduction
Liver fibrosis is characterized by the excessive deposition of extracellular matrix (ECM) in the liver. Activated hepatic stellate cells (HSCs) are the main producers of ECM in liver fibrogenesis. In physiological conditions, quiescent HSCs (qHSCs), located in the space of Disse, store 50–80% of whole-body vitamin A content in the form of lipid droplets. In chronic inflammatory liver diseases, qHSCs transdifferentiate into myofibroblast-like cells, termed activated HSCs (aHSCs). During activation, HSCs lose their vitamin A droplets, start to proliferate and produce excessive amounts of ECM, including collagen type 1 (Col1α1) and α-smooth muscle actin (Acta2) [Citation1,Citation2]. In addition, the expression of the quiescence markers lecithin:retinol acyltransferase (Lrat), a retinol esterifying enzyme, and the transcription factor peroxisome proliferator-activated receptor gamma (Pparγ) is strongly reduced upon activation of HSCs [Citation3]. The key fibrogenic cytokine, transforming growth factor-beta (TGFβ) is released and increases the production of ECM by HSCs [Citation4]. Reversal of activated HSCs into the quiescent stage and/or induction of apoptosis of activated HSCs are considered as potential strategies to cure liver fibrosis [Citation5,Citation6].
Cell senescence is defined as irreversible cell cycle arrest accompanied by increased cytokine secretion, in particular Il6, termed the Senescence-Associated Secretory Phenotype (SASP) [Citation7]. P21Cip1 (mRNA: Cdkn1a) is an essential cell cycle checkpoint regulator that arrests cell proliferation at the G1 phase and initiates senescence [Citation8]. In general, the PI3K-AKT signaling pathway plays an important role in the cellular response to growth factors, including cell proliferation, metabolic rate and cell migration. More recently, a role of PI3K-AKT signaling has also been proposed in cellular senescence by modulating various pathways including P53/P21Cip1 [Citation9,Citation10]. In the absence of Akt kinase activity, P21Cip1 is unable to arrest the cell cycle and initiate senescence [Citation11]. It has been reported that induction of senescence in activated HSCs reverses the fibrogenic phenotype of activated HSCs [Citation12,Citation13]. Therefore, recent interest has been focused on inducing cellular senescence as a new mechanism for the resolution of liver fibrosis [Citation14]. It has been reported that some bioactive anti-fibrotic compounds induce cellular senescence via P53 and YAP or via increased Natural Killer cell activation [Citation5,Citation15]. In addition, some anti-fibrotic proteins and cytokines, e.g. the matricellular protein CCN1 and the cytokines IL-10 and IL-22 have been reported to induce senescence via an integrin-dependent mechanism, via the generation of ROS or via the activation of p53 and/or p21cip1 [Citation16–18].
Hydrogen sulfide (H2S) is one of the gaseous signaling molecules along with nitric oxide and carbon monoxide. The liver is an essential organ for H2S production and clearance. H2S is an established regulator of cellular redox homeostasis [Citation19]. Hepatic H2S is involved in mitochondrial biogenesis and bioenergetics, insulin sensitivity, lipoprotein synthesis and glucose metabolism [Citation20]. Endogenous H2S is synthesized by the enzymes cystathionine γ-lyase (CTH), cystathionine β-synthase (CBS) and 3-mercaptopyruvate sulfur transferase (MPST) [Citation1]. We previously reported that CTH expression and the generation of H2S are increased during HSCs activation and that H2S promotes activation of HSCs: inhibition of CTH decreased HSCs proliferation and showed anti-fibrotic effects [Citation21]. It has been demonstrated that H2S has potent anti-senescence effects on endothelial cells via the induction of splicing factors HNRNPD, SRSF2 and via the PI3K/Akt signaling pathway [Citation22,Citation23]. Therefore, we hypothesized that the inhibition of endogenous production of H2S in activated HSCs is anti-fibrogenic via the induction of senescence.
2. Materials and methods
2.1. Hepatic stellate cell isolation and culture
Primary hepatic stellate cells (HSCs) were isolated from specified pathogen-free male Wistar rats (400–500 g; Charles River, Wilmington, MA, USA). Animals were housed under standard conditions on a 12 h light–dark cycle with free access to chow and water. All experiments were approved by the Committee for Care and Use of laboratory animals of the University of Groningen. In order to isolate the HSCs, rats were anesthetized and perfused via the portal vein with Pronase-E (Merck, Amsterdam, the Netherlands) and Collagenase-P (Roche, Almere, the Netherlands). After perfusion, the digested liver was removed and the cell suspension centrifuged on 13% w/v Nycodenz (Axis-Shield POC, Oslo, Norway) to obtain HSCs. Following isolation, HSCs were cultured in Iscove’s Modified Dulbecco’s Medium supplemented with Glutamax (Thermo Fisher Scientific, Waltham, MA, USA), 20% heat inactivated fetal calf serum (Thermo Fisher Scientific), 1% MEM Non Essential Amino Acids (Thermo Fisher Scientific), 1% Sodium Pyruvate (Thermo Fisher Scientific) and antibiotics: 50 µg/mL gentamycin (Thermo Fisher Scientific), 100 U/mL penicillin (Lonza, Verviers, Belgium), 10 µg/mL streptomycin (Lonza) and 250 ng/mL fungizone (Lonza) in an incubator containing 5% CO2 at a 37°C. HSCs were expanded and culture-activated for seven days on tissue culture plastic. Human LX-2 cells (SCC064, Merck) were cultured as described for primary activated rat stellate cells [Citation24].
2.2. Induction of liver fibrosis in rats
Fifteen 6-to 8-week-old male Wistar rats (200-400 g) were maintained on a light/dark cycle (12:12) and provided Purina Rodent Chow and water ad libitum, with acclimation period of one week before starting the induction process. The experiment began with two groups: (i) the non-treated (control) group (n = 5) and (ii) the cirrhosis group (n = 10). Fibrosis was induced by the intraperitoneal injection of CCl4, 0.8 ml/kg, as a 1:1 mixture with petrolatum, 2 times per week for 4 weeks. Each animal was weighed once per week throughout the experiment, after the induction of fibrosis, rats were sacrificed with pentobarbital overdose (MAVER Laboratories), liver was collected and cut into pieces and fixed in neutral formalin (4%) for subsequent histological analysis. All animal experiments were approved by the Animal Welfare and Research Ethics Committee of the Autonomous University of Aguascalientes and conducted according to the institutional and national regulation (NOM-062-ZOO-1999). Several signs related to pain and suffering resulting from the induction of liver fibrosis, were analyzed, including decreased mobility, withdrawal, weight loss, reduced food and water intake, self-mutilation, and changes in behavior such as aggressiveness.
2.3. Experimental design
Activated HSCs (aHSCs) or LX-2 cells were seeded at a density resulting in a confluency of around 90% at the end of each treatment. Depending on the assay, the duration of the treatments was 48 to 72 h. Unless otherwise stated, all treatments were performed in fresh medium containing 20% fetal calf serum (FCS) (v/v) and other supplements as described above. The experiments with TGFβ1 were performed at reduced (1%) serum concentrations to avoid interference with HSC activating factors present in FCS. Hydrogen sulfide (H2S) slow-releasing donor GYY4137 (kind gift of prof. Matt Whiteman, University of Exeter, United Kingdom) and the pharmacological inhibitor of CTH DL-propargylglycine (DL-PAG; Sigma-Aldrich, Zwijndrecht, the Netherlands) were freshly prepared prior to administration. GYY4137 and DL-PAG were used at concentrations that were previously shown to be nontoxic to HSCs [Citation20]. The PI3K inhibitor LY294002 (Calbiochem, Darmstadt, Germany) was dissolved in DMSO to prepare a stock solution. The antioxidant N-acetylcysteine was used at a concentration of 5 mmol/L. Each experimental condition was performed in duplicate wells and repeated at least 3 times. A complete set of experiments was always performed with HSCs from one isolation. All experiments were repeated at least three times using cells from different isolations.
2.4. Quantitative real-time polymerase chain reaction
Cellular mRNA was isolated by Tri-reagent (Sigma Aldrich) according to manufacturer’s protocol. Total RNA concentration was determined by Nano-Drop 2000c (Thermo Fisher Scientific). 0.5 to 2.5 µg of RNA was used for preparation of cDNA using MLV reverse transcriptase and RNase Out (Thermo). cDNA was diluted in RNAse-free water and used for real-time polymerase chain reaction on the QuantStudioTM 3 system (Thermo Fisher Scientific). All samples were analyzed in duplicate using 36b4 as housekeeping gene. The mRNA levels of Cth, Cbs, Mpst (Thermo Fisher Scientific) were quantified using SYBR Green (Applied Biosystems), other genes were quantified by TaqMan probes and primers. Relative gene expression was calculated via the 2−ΔΔCt method. The primers and probes are listed in .
Table 1. Primer and probe sequences.
2.5. Senescence-associated β-galactosidase staining
Culture-activated HSCs were treated as described before. Cellular senescence was determined by Senescence-associated β-galactosidase staining kit (Cell Signaling Technology, Danvers, Massachusetts, USA) according to manufacturer’s protocol. After fixation, senescent cells were stained by X-gal solution (pH = 5.9–6.1) for 24 hr at 37°C in a dry incubator. Images were captured by digital phase contrast microscopy.
2.6. Cell proliferation assay
Cell proliferation was determined by BrdU incorporation assay (Roche Diagnostic Almere, the Netherlands) and Real-Time xCelligence assay (RTCA DP; ACEA Biosciences, Inc., CA, USA). Cells were seeded in 96 well plates and treated as described. Incorporation of BrdU was detected by chemiluminescence using Synergy-4 (Bio-Tek). For xCelligence, aHSCs were seeded in 16 well E-plates. Real-time cell proliferation was measured as cell index in the xCelligence system.
2.7. Western blot analysis
Cells were seeded and treated as described. Protein lysates were scraped in cell lysis buffer (HEPES 25 mmol/L, KAc 150 mmol/L, EDTA pH 8.0 2 mmol/L, NP-40 0.1%, NaF 10 mmol/L, PMSF 50 mmol/L, aprotinin 1 µg/µL, pepstatin 1 µg/µL, leupeptin 1 µg/µL, DTT 1 mmol/L). The concentration of protein was measured by Bio-Rad protein assay (Bio-Rad; Hercules, CA, USA). 10–20 µg protein was loaded on SDS-PAGE gels. Proteins were transferred to nitrocellulose transfer membranes using Trans-Blot Turbo Blotting System for tank blotting. Proteins were detected using the primary antibodies listed in . Protein band intensities were determined and detected using the Chemidoc MR (Bio-Rad) system.
Table 2. Antibodies used for Western blotting.
2.8. Immunofluorescence microscopy on cultured cells and tissue sections
Cells were cultured on glass coverslips and fixed with 4% paraformaldehyde/PBS. 5 min incubation with 1% Triton X-100 was used to permeabilize cells. Nonspecific antibody binding sited were blocked by 0.5% BSA/PBS for 30 min. After blocking, cells were incubated by primary antibody against collagen type 1 (#1310–01, Southern Biotech) 1:400 diluted in 0.5% BSA/PBS for 1 hr at room temperature. Coverslips were washed 3 times by PBS and then incubated with secondary antibody Alexa fluorophore (Molecular Probes) 1:400 diluted in 0.5% BSA/PBS for 1 hr at room temperature. Coverslips were mounted with fluorescence mounting medium containing DAPI (Vectashield, Burlingame, CA, USA). Images were captured by Zeiss 410 inverted laser scanning microscope.
Tissue slides were first deparaffinized in xylene and rehydrated in alcohol. Slides were then incubated in citric acid buffer (10 mmol/L; pH 6.0) for antigen retrieval. Afterward, slides were washed three times with PBS and blocked using a blocking solution (1% bovine serum albumin (BSA)/PBS) for 30 minutes at room temperature. Primary antibodies against ACTA2 and CTH () were used after blocking at 4°C in a humidity chamber for 1 hour. The secondary antibody solution was added to samples and allowed to incubate at room temperature for 30 minutes (goat anti-rabbit Alexa Flour 488). DAPI staining was applied for nuclear staining. Images were captured using slide scanner Nanozoomer (Hamamatsu Photonics K.K, Shizuoka, Japan).
2.9. Immunohistochemistry staining on tissue sections
Hematoxylin-eosin-stained slides were prepared following the standard protocol. Paraffin embedded tissue sections of the liver were cut (4 µm) and placed on Starfrost slides (3054–1, Klinipath, VWR, Breda, The Netherlands), dried, deparaffinized in xylene and rehydrated in alcohol. The endogenous peroxidase was blocked with 0.3% H2O2 in phosphate-buffered solution (PBS, 30 minutes). Subsequently, the slides were blocked with 1% bovine serum albumin (BSA)/PBS for 30 minutes. Then, slides were incubated with primary antibody against CTH (). The secondary and tertiary antibodies, labeled with horseradish peroxidase, were diluted in 1% BSA/PBS supplemented with 1% rat serum 1:50 (Santa Clara, Dako, Agilent, CA, USA; goat anti-rabbit and rabbit anti-mouse respectively) and incubated for 30 minutes. Binding was detected using 3,3-diaminobenzidine and slides were counterstained with hematoxylin. Images were captured using slide scanner Nanozoomer (Hamamatsu Photonics K.K, Shizuoka, Japan).
2.10. Sirius red and fast green staining
Tissue samples were fixed in formalin and embedded in paraffin. 4 micrometer tissue sections were deparaffinized and rehydrated in alcohol to continue with the staining with 0.1% Sirius red and 0.1% Fast green dissolved in water saturated with picric acid for 30 minutes. Slides were then washed with water, dehydrated and mounted using xylol-based mounting media. Images were captured using slide scanner Nanozoomer (Hamamatsu Photonics K.K, Shizuoka, Japan).
2.11. Statistical analysis
Data are presented as mean ± standard deviation (mean ± SD) of at least three independent experiments. Statistical significance was analyzed by Mann–Whitney test between the two groups and one-way ANOVA or Kruskal-Wallis followed by post-hoc Dunn’s test for multiple comparison test. p < 0.05 was considered statistically significant. Analysis was performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA).
3. Results
3.1. TGFβ1 increases cystathionine γ-lyase (CTH) expression in activated hepatic stellate cells
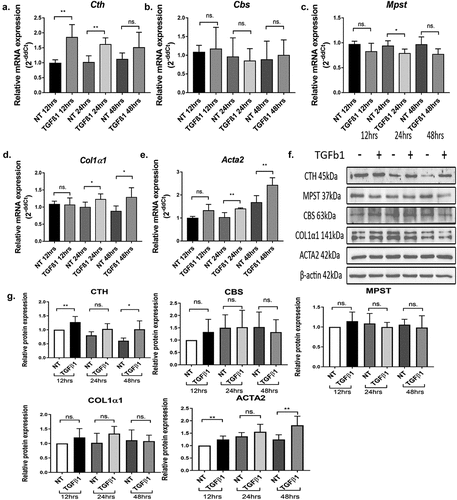
We previously reported that the expression of the endogenous H2S synthesizing enzyme cystathionine γ-lyase (CTH) and the production of H2S (CTH/H2S) were increased during transdifferentiation of quiescent hepatic stellate cells (qHSCs) into activated hepatic stellate cells (aHSCs) [Citation21]. Here, we investigated the effect of the pro-fibrogenic cytokine TGFβ1 on the expression of the H2S producing enzymes CTH, CBS, and MPST . The fibrogenic cytokine TGFβ1 significantly increased the mRNA and protein expression of CTH but not of CBS and MPST. As expected, the expression of the fibrogenic markers Col1α1 and Acta2 mRNA were also increased upon treatment of HSCs with TGFβ1 but only modestly, since the HSCs were already activated and displayed already high levels of Col1α1 and Acta2 mRNA compared to quiescent HSCs. In addition, fibrotic and healthy liver tissue from rats was analyzed (Supplemental Figure S1). CTH was detected at high levels in both healthy and cirrhotic tissue due to the high constitutive expression of this enzyme in hepatocytes. In fibrotic rat liver tissue, ACTA2 was detected, but not in healthy tissue, which correlates with the presence of activated hepatic stellate cells. Furthermore, in cirrhotic liver tissue, we observed co-staining of ACTA2 and CTH, which implies that CTH is present in activated hepatic stellate cell.
Figure 1. Panels A-E: CTH mRNA and protein expression are increased by the pro-fibrogenic cytokine TGFβ1 in HSCs cultured in 1% FCS. Relative gene expression of endogenous H2S producing enzymes Cth, Cbs, Mpst and hepatic stellate cell activation markers Col1α1, Acta2 (mean ± SD) were measured at 12, 24 and 48 h in 1% FCS medium with or without treatment with 5 ng/ml TGFβ1. Expression levels were calculated relative to 36b4 housekeeping gene. Panels F,G: Relative protein expression of CTH, CBS, MPST and stellate cell activation markers COL1α1 and ACTA2. β-actin was used as loading control. Results are expressed as mean ± SD; *: p < 0.05, **: p < 0.005.
3.2. CTH inhibition reduces activation of hepatic stellate cells
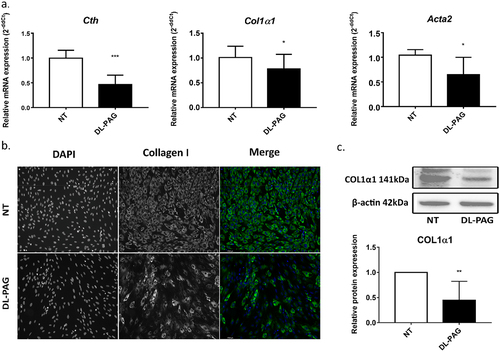
Since CTH expression was increased by TGFβ1, we hypothesized that inhibition of CTH could inhibit TGFβ1-driven transdifferentiation of HSCs. As shown in , inhibition of CTH by DL-PAG reduced the expression of the fibrogenic markers Acta2 and Col1a1. In addition, the protein level of collagen type 1 (COL1α1) was also reduced by DL-PAG as shown by immunofluorescence staining and Western blot .
Figure 2. The CTH inhibitor DL-PAG reverses activation of HSCs. Panel A: mRNA levels of the activation markers Col1α1 and Acta2 were reduced in activated HSCs after 72 h treatment with DL-PAG. Cth expression was also reduced by DL-PAG. 36b4 was used as housekeeping gene. Panel B, C: Immunofluorescence staining and Western blotting of COL1α1 (magnification 200x) in activated HSCs. β-actin was used as loading control. Results are expressed as mean ± SD; *: p<0.05, **: p < 0.005, ***: p < 0.0005.
3.3. CTH inhibition induces cellular senescence in hepatic stellate cells
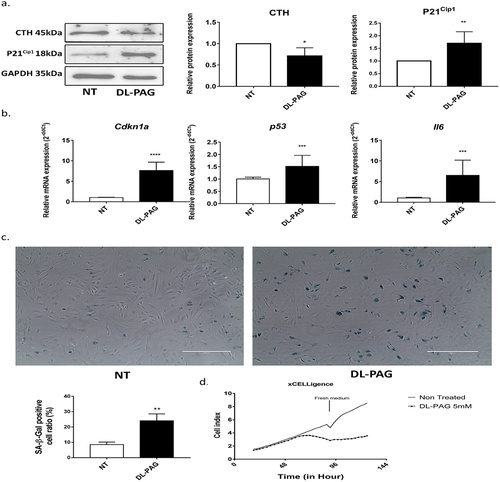
To determine whether the anti-fibrotic effect of CTH inhibition correlated with the induction of senescence, we investigated markers of senescence in DL-PAG treated aHSCs. Inhibition of CTH by DL-PAG increased the protein expression of the senescence marker P21Cip1 ) a cyclin-dependent kinase inhibitor. The mRNA expression of the cell cycle arrest markers Cdkn1α (p21) and p53 as well as the senescence associated secretory phenotype (SASP) marker IL-6 were increased upon CTH inhibition . In addition, inhibition of CTH by DL-PAG increased the number of senescence associated β-galactosidase positive (SA-β-gal) cells . Of note, CTH inhibition did not restore mRNA expression of the quiescence markers PPARγ and Lrat, suggesting that induction of senescence is not equivalent to reversal to the quiescent phenotype in aHSCs (data not shown).
Figure 3. Inhibition of CTH induces cellular senescence. Panel A: Protein expression of H2S synthesizing enzyme CTH is decreased and cell cycle arrest marker P21Cip1 is increased upon CTH inhibition at 24 h. GAPDH is used as loading control. Panel B: mRNA expression of senescence markers Cdkn1α, p53 and SASP marker IL-6 after 72 h treatment of aHscs with DL-PAG. 36b4 was used as housekeeping gene. Panel C: SA-β-gal staining (magnification 200x) and its quantification after 48 h treatment of HSCs with DL-PAG. Panel D: Cell proliferation was measured by the xCelligence system. Cells were treated for 48 h followed by the addition of fresh medium. Cell proliferation is represented as cell index. Results are expressed as mean ± SD; *: p < 0.05, **: p < 0.005.
Since DL-PAG induced cellular senescence in aHSCs, the effect of DL-PAG on primary quiescent rat HSCs (qHSCs) and human LX-2 cells was investigated. DL-PAG showed no effect on the expression of Col1a1 and Acta2 in qHSCs nor in TGFβ1-treated qHSC. DL-PAG also did not affect the quiescence marker Pparg, whereas the expression of the quiescence marker Lrat was increased by DL-PAG. Finally, mRNA expression of the senescence markers Cdkn1a and IL6 was increased by DL-PAG in qHSCs, both in the absence and presence of TGFβ1. Together, these results suggest that the effect of DL-PAG on qHSCs is mainly on senescence markers (both in qHSCs and in aHSCs), partly on quiescence markers, but not on fibrotic markers since the expression of fibrotic markers is very low in qHSCs (Supplemental Figure S2). Next, we investigated whether DL-PAG has similar effects on human LX-2 cells compared to primary rat HSCs. DL-PAG decreased expression of the fibrotic markers Col1a1 and Acta2 mRNA and concomitantly increased markers of cell senescence and markers of the SASP (Cdkn1a (p21) and Il6) as shown in Supplemental Figure S3.
3.4. H2S donor GYY4137 restores proliferation ability of senescent HSCs
Since inhibition of the endogenous H2S synthesizing enzyme CTH showed anti-fibrotic effects via the induction of cellular senescence, we hypothesized that supplementation of exogenous H2S reverses induction of senescence. To test this hypothesis, HSCs were treated with both the inhibitor of CTH, DL-PAG and different concentrations of the H2S slow-releasing donor GYY4137. P21Cip1 expression, SA-β-galactosidase positive cell and mRNA expressions of Cdkn1α, p53 and Il6 were does-dependently reduced by GYY4137 . The impaired proliferation of HSCs treated with DL-PAG was improved by the H2S donor GYY4137 . To investigate whether the pro-senescence effect of DL-PAG may be reversed by additional stimulation with the pro-fibrogenic cytokine TGFβ1, aHSCs were co-treated with TGFβ1 and DL-PAG. As shown in Supplemental , TGFβ1 did not reverse the induction of senescence induced by DL-PAG. Lastly, since pro-oxidants are known to drive HSC activation and antioxidants are known to inhibit and/or reverse HSC activation, we also investigated the effect of the antioxidant N-acetylcysteine (NAC; 5 mmol/L) on DL-PAG-induced senescence. As shown in Supplemental , NAC reduced DL-PAG induced β-galactosidase staining and Cdkn1α, p53 mRNA expression in aHSCs.
Figure 4. H2S donor GYY4137 partially reversed the pro-senescence effect of DL-PAG. Panel A: P21Cip1 expression in HSCs treated with DL-PAG with or without different concentrations of GYY4137 at 24 h. Panel B: Cell proliferation was measured by BrdU incorporation assay. Panel C: mRNA expression of senescence markers Cdkn1α, p53, IL-6. 36b4 was used as housekeeping gene. Panel D: SA-β-Gal staining and its quantification of HSCs treated with DL-PAG with or without GYY4137 (GYY) (magnification 200x). Results are expressed as mean ± SD; *: p < 0.05, **: p < 0.005.
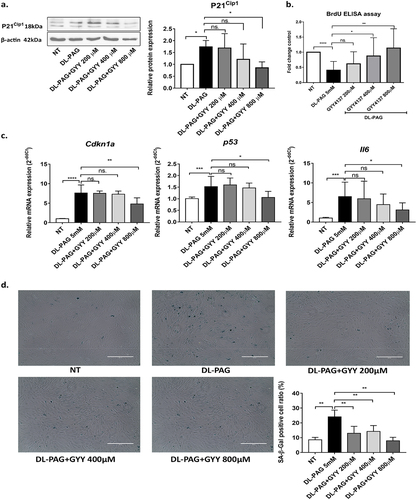
Figure 5. DL-PAG induced cellular senescence is mediated via PI3K-AKT signalling. Panel A: protein expression of Akt phosphorylation at Serine 471, total Akt and senescence marker P21Cip1 at 24 hr. β-Actin was used as loading control. COL1α1 and β-actin protein expression at 72 h. Panel B: mRNA expression of senescence markers Cdkn1α, p53, IL-6, and fibrotic marker Acta2. 36b4 was used as housekeeping gene. Panel C: SA-β-Gal staining of non-treated HSCs and HSCs treated by DL-PAG with (LY29) or without PI3K inhibitor for 48 h (magnification 200x) and its quantification. Panel D: Real-time cell proliferation was measured using the xCelligence system. Results are expressed as mean ± SD; *: p < 0.05, **: p < 0.005.
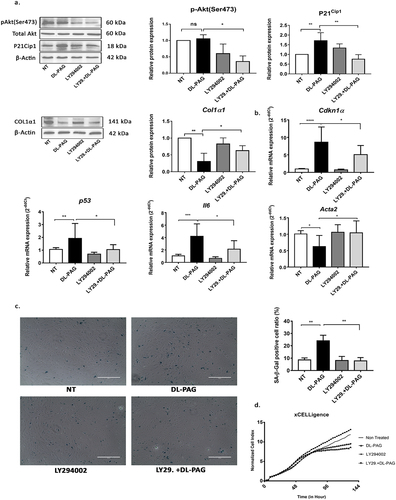
3.5. PI3K-Akt pathway is involved in DL-PAG induced senescence
We next investigated the signaling pathway involved in the anti-senescence effect of H2S. Akt activity is essential for induction of P21Cip1-dependent cellular senescence [Citation7,Citation11,Citation22,Citation25]. To determine the role of Akt in DL-PAG induced senescence, the pan-PI3K inhibitor LY294002 was applied to block Akt activity in HSCs. LY294002 decreased the level of Akt phospho-Ser473 and Akt phospho-Thr308 in HSCs. At the same time, LY294002 attenuated the DL-PAG-induced increased expression of the senescence marker P21cip1 . In addition, the DL-PAG-induced increase in mRNA expression of cell cycle arrest markers Cdkn1α, p53 and SASP marker IL-6 was attenuated by the PI3K inhibitor . The downregulation of CTH expression by DL-PAG was not reversed by LY294002 . The attenuation of DL-PAG-induced expression of P21Cip1 by the PI3K inhibitor was correlated with an attenuation of the DL-PAG-induced increase in number of SA-β-Gal positive cells . Also, the DL-PAG-induced downregulation of the pro-fibrotic marker Acta2, but not Col1a1, was reversed by the PI3K inhibitor LY294002 . Finally, the PI3K inhibitor LY294002 improved the proliferation ability of DL-PAG-treated aHSCs . Taken together, the results demonstrate that senescence induced by CTH/H2S inhibition is mediated via PI3K-Akt signaling. To investigate whether the proliferation arrest induced by DL-PAG was irreversible, we performed wash-out experiments. In the first 48 h, DL-PAG inhibited HSC proliferation. After refreshing the medium (and removing DL-PAG), the proliferation ability remained impaired compared to non-treated HSCs .
4. Discussion
Cellular senescence has been identified as a promising therapeutic strategy for the treatment of liver fibrosis due to its potential to inactivate HSCs [Citation12,Citation24–26]. Senescence can be induced in HSCs in response to a variety of stimuli, such as matricellular protein CCN1 and the cytokines IL-10 and IL-22. In addition, cell cycle arrest marker p53 double knockout (p53−/) mice contain more fibrotic tissue compared to wild-type mice due to impaired cellular senescence [Citation12–14]. However, the specific signaling pathways in HSCs that account for the induction of senescence remain to be fully elucidated. Previously, we have shown that hydrogen sulfide (H2S) promotes stellate cell activation [Citation21]. It has also been shown that H2S hampers senescence in various cell types, including endothelial cells and fibroblasts via the induction of splicing factors like HNRNPD and posttranslational modification of Keap1 [Citation22,Citation27]. In the present study, we demonstrate a causal relationship between the induction of senescence by inhibiting H2S production and the (partial) reversal of stellate cell activation: inhibition of CTH reverses activation of stellate cells and induces a senescent phenotype, whereas H2S donors promote stellate cell activation and (partially) reverse the senescent phenotype. However, inhibition of H2S does not lead to a complete reversal to the quiescent phenotype, for the expression of the quiescence marker PPARγ and the re-appearance of lipid droplets was not restored by H2S inhibition. The effects of inhibiting H2S synthesis in quiescent stellate cells on markers of activation and fibrogenesis were less pronounced compared to activated stellate cells, most likely because the expression of these markers in quiescent stellate cells were already very low. However, the changes in expression of senescence markers were similar in quiescent stellate cells compared to activated stellate cells. Our results could be reproduced in the human stellate cell-line LX-2, confirming the relevance of our findings for future clinical application. We also demonstrated that activated, but not quiescent, HSCs in vivo express CTH, suggesting that CTH may be a valid target for inducing senescence in vivo as well. Moreover, we also demonstrate that antioxidants reverse the senescence phenotype induced by DL-PAG. Since pro-oxidants are known to induce the activated phenotype of HSCs and antioxidants reverse or inhibit HSC activation, the reversal of senescence by antioxidants does not imply a reversal to the activated phenotype. It will be interesting to investigate whether inhibition of H2S production in combination with antioxidants reverses the phenotype of activated HSCs to the quiescent phenotype rather than the senescent phenotype. This would be a major advantage since induction of senescence alone in liver fibrosis may not be sufficient as it will activate the immune system to clear senescent HSCs. We also demonstrate that the induction of the senescent phenotype in HSCs by inhibition of H2S is dependent on PI3K-Akt signaling, since an inhibitor of PI3K is capable to partially reverse the senescent phenotype and restore the fibrogenic phenotype.
TGFβ1 plays a central role in fibrogenesis: it promotes stellate cell activation and increases the production of extracellular matrix (ECM) [Citation4]. We previously reported that expression of CTH/H2S is increased during trans-differentiation of quiescent HSCs into activated stellate cells [Citation21]. In addition, previous studies reported that H2S shows anti-fibrotic effects via pro-apoptotic, antioxidant and anti-inflammatory effects in HSC-T6 cells and in rat primary HSCs [Citation28,Citation29]. However, in these studies a high concentration of the fast-releasing NaHS donor was used, which could be toxic. In another study, the natural compound diallyl trisulfide (DATS) was used as a H2S donor, which could be different from H2S alone [Citation30]. Furthermore, accumulating evidence supports the notion that H2S slow releasing donors better reflect endogenous H2S levels within biological systems over a longer time course [Citation31].
In the present study we demonstrate that TGFβ1 time-dependently increased protein and mRNA expression of CTH in HSCs cultured at low serum concentration. In contrast, TGFβ1 did not change CBS and MPST expression. We did not observe increased production of H2S in response to TGFβ1 treatment, probably because endogenous H2S production was already at a high level in the culture-activated HSCs or the lack of sensitivity of our method. Our observation of increased expression of CTH upon stimulation with TGFβ1 is in line with several reports on the effect of growth factors (PDGF) on fibrogenic markers and proliferation in fibroblasts during skin wound healing and overexpression of CTH in proliferating cancer cells [Citation32–34]. These results suggest that CTH has the potency to regulate cell proliferation and activation via endogenous H2S production in fibroblasts and cancer cells as well as in HSCs.
PI3K-dependent signaling has been demonstrated to be an essential regulator of cellular senescence [Citation7]. Activation of the PI3K-Akt axis permits the induction of P21Cip1 dependent senescence [Citation11,Citation24,Citation35]. As downstream target and substrate of Akt, Ser9-phosphorylated GSK3β stabilizes P21Cip1 and Ser9 phosphorylation of GSKβ is positively correlated with the proportion of senescent cells [Citation36,Citation37]. The PI3K inhibitor LY294002 inhibits Akt kinase activity and phosphorylation of GSK3β and restores the proliferation ability of oncogene Ras-induced senescent fibroblasts [Citation11,Citation37]. In our present study, a similar effect of LY294002 was observed in DL-PAG-treated senescent HSCs. LY294002 downregulated the cell cycle arrest marker Cdkn1a, reduced the number of SA-β-Gal positive cells, suppressed the SASP in DL-PAG treated HSCs and restored proliferation. Furthermore, LY294002 reversed the downregulation of the fibrogenic marker Acta2 in DL-PAG-treated HSCs. These results demonstrate that the inactivation of HSCs results from the induction of HSC senescence. Nevertheless, neither the senescent phenotype and proliferation arrest induced by DL-PAG nor the downregulation of CTH was completely rescued by the PI3K inhibitor. This suggests that the PI3K-Akt signaling pathway accounts for part of the senescent phenotype. It has been stated that activation of PI3K is involved in HSC activation. However, the data are to some extent controversial and we hypothesize that senescence is different from reversal of activation back to quiescence. An association between Akt and P21 has been demonstrated by Kim YY et al. [Citation11]. In addition, the regulation of the quiescence markers Pparg and Lrat was not identical in DL-PAG treated aHSCs (Supplemental Figure S2), which demonstrates that senescent HSCs are not identical to quiescent HSCs. Phosphorylation of Akt is regulated by the mTORC2 complex [Citation38]. Similarly, it has been demonstrated that mTORC2 signaling induces fibroblast senescence concomitantly with phosphorylation of Akt [Citation39]. In this study, p-Akt level as well as CDKN1A expression were reduced in senescent fibroblasts treated with rapamycin or LY294002 [Citation39]. Therefore, the role of PI3K/Akt in activation and senescence of stellate cells may be context-dependent.
In addition to PI3K-Akt signaling, mitochondrial dysfunction may be a driving factor in the induction of cellular senescence [Citation40]. This is in line with our previous observation that H2S improves mitochondrial function and hence, the lack of the H2S may contribute to mitochondrial dysfunction and senescence [Citation22].
To date, several studies have reported that H2S shows anti-senescence and anti-aging effects via splicing factors HNRNPD, SRSF2 and upregulation of SIRT1 in endothelial cells [Citation22,Citation41,Citation42]. In addition, Yang et al., reported that CTH knockout mice display increased senescence, whereas exogenous H2S post-translationally S-sulfhydrates the transcription factor Keap1 to stimulate antioxidant systems and reverse senescence in fibroblasts [Citation27]. Furthermore, the H2S fast-releasing donor NaHS was able to reverse senescence in pulmonary fibrosis via MDM2 mediated p53 degradation in CBS knockout mice [Citation43]. In line with these reports, the DL-PAG-increased senescence markers P21cip1, β-gal staining and decreased HSCs proliferation were dose-dependently and partially reversed by the H2S slow releasing donor GYY4137. Since the senescent phenotype is the integrative effect of both senescent and non-senescent cells, the modulation of single signaling pathways may affect both cell populations and lead to a partially reversed phenotype. The restoration of DNA synthesis by DL-PAG can be attributed to H2S, a phenomenon we reported before [Citation21]. These results demonstrate that H2S has a central role in HSCs senescence and inhibition of H2S signaling shows anti-fibrotic effects via induction of senescence. Senescence is defined as irreversible cell cycle arrest. However, in our study we show that treatment with GYY4137 partially reverses the effect of DL-PAG on senescence. A possible explanation is that dysfunctional mitochondria induce (part of) the SASP, including SASP-related growth arrest [Citation38]. Since H2S supplementation increases bioenergetics of mitochondria, it can be hypothesized that H2S (GYY4137) improves mitochondrial function of senescent HSCs, thus modulating the SASP and SASP-induced senescence and consequently restoring fibrogenic potential of aHSCs.
Our study has some advantages and limitations that need to be addressed. First, we used primary hepatic stellate cells that better reflect the process of activation and we used both quiescent and activated HSCs. This is an important advantage compared to the use of cell lines which have a “fixed” intermediate phenotype. The use of H2S donors and inhibitors in studies on HSC activation have been reported previously by our group, providing important background information on the effects of these compounds on HSC biology. Our study is limited by the lack of extensive in vivo data and knockdown experiments, e.g. using siRNA transfection. Since we investigated only one cell type, the hepatic stellate cell, these additional studies are necessary to extrapolate our findings to intact animals.
In conclusion, our results suggest that CTH and endogenous H2S generation are increased during fibrogenesis [Citation21]. Inhibition of CTH shows anti-fibrotic effects through increased cellular senescence and this cellular senescence is regulated through PI3K-Akt pathways.
Abbreviations
| MPST | = | 3-mercaptopyruvate sulfur transferase |
| CBS | = | cystathionine β-synthase |
| CTH | = | cystathionine γ-lyase |
| DL-PAG | = | DL-propargylglycine |
| ECM | = | extracellular matrix |
| FCS | = | fetal calf serum |
| GYY | = | GYY4137 |
| HSCs | = | hepatic stellate cells |
| LY29 | = | LY294002 |
| SA-β-gal | = | senescence associated β-galactosidase positive |
| SASP | = | senescence associated secretory phenotype |
| TGFβ | = | transforming growth factor-beta |
Author contributions
“Conceptualization, M.Z., T.D. and H.M.; methodology, M.Z., T.D.; validation, M.B.H., H.M.; investigation, M.B,H; resources, H.G.; writing – original draft preparation, T.D., M.Z. S.S.S; writing – review and editing, supervision, H.M., M.B.H., H.G.;. All authors have read and agreed to the published version of the manuscript.”.
Supp Figs.docx
Download MS Word (2.8 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384101.2024.2345477
Additional information
Funding
References
- Kasparek MS, Linden DR, Kreis ME, et al. Gasotransmitters in the gastrointestinal tract. Surgery. 2008;143(4):455–459. doi: 10.1016/j.surg.2007.10.017
- Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21(03):311–336. doi: 10.1055/s-2001-17550
- Trivedi P, Wang S, Friedman SL. The power of plasticity—metabolic regulation of hepatic stellate cells. Cell Metab. 2021;33(2):242–257. doi: 10.1016/j.cmet.2020.10.026
- Dewidar B, Meyer C, Dooley S, et al. TGF-β in hepatic stellate cell activation and liver fibrogenesis—updated 2019. Cells. 2019;8(11):1419. doi: 10.3390/cells8111419
- Jin H, Lian N, Zhang F, et al. Inhibition of YAP signaling contributes to senescence of hepatic stellate cells induced by tetramethylpyrazine. Eur J Pharm Sci. 2017;96:323–333. doi: 10.1016/j.ejps.2016.10.002
- Schrader J, Fallowfield J, Iredale JP, et al. Senescence of activated stellate cells: not just early retirement. Hepatology. 2009;49(3):1045–1047. doi: 10.1002/hep.22832
- Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell. 2019;179(4):813–827. doi: 10.1016/j.cell.2019.10.005
- Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15(7):397–408. doi: 10.1038/nrc3960
- Zhang M, Damba T, Wu Z, et al. Bioactive coumarin-derivative esculetin decreases hepatic stellate cell activation via induction of cellular senescence via the PI3K-Akt-GSK3β pathway. Food Biosci. 2022;50:50. doi: 10.1016/j.fbio.2022.102164
- Liu S, Liu S, Wang X, et al. The PI3K-Akt pathway inhibits senescence and promotes self-renewal of human skin-derived precursors in vitro. Aging Cell. 2011;10(4):661–674. doi: 10.1111/j.1474-9726.2011.00704.x
- Kim YY, Jee HJ, Um JH, et al. Cooperation between p21 and akt is required for p53-dependent cellular senescence. Aging Cell. 2017;16(5):1094–1103. doi: 10.1111/acel.12639
- Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi: 10.1016/j.cell.2008.06.049
- Zhang M, Serna-Salas S, Damba T, et al. Hepatic stellate cell senescence in liver fibrosis: characteristics, mechanisms and perspectives. Mech Ageing Dev. 2021;199:111572. doi: 10.1016/j.mad.2021.111572
- Aravinthan AD, Alexander GJM. Senescence in chronic liver disease: is the future in aging? J Hepatol. 2016;65(4):825–834. doi: 10.1016/j.jhep.2016.05.030
- Jin H, Jia Y, Yao Z, et al. Hepatic stellate cell interferes with NK cell regulation of fibrogenesis via curcumin induced senescence of hepatic stellate cell. Cell Signal. 2017;33:79–85. doi: 10.1016/j.cellsig.2017.02.006
- Kim KH, Chen CC, Monzon RI, et al. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol. 2013;33(10):2078–2090. doi: 10.1128/MCB.00049-13
- Huang YH, Chen MH, Guo QL, et al. Interleukin‑10 promotes primary rat hepatic stellate cell senescence by upregulating the expression levels of p53 and p21. Mol Med Rep. 2018;17:5700–5707. doi: 10.3892/mmr.2018.8592
- Kong X, Feng D, Wang H, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56(3):1150–1159. doi: 10.1002/hep.25744
- Xie ZZ, Liu Y, Bian JS. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid Med Cell Longev. 2016;2016:6043038. doi: 10.1155/2016/6043038
- Wu DD, Wang DY, Li HM, et al. Hydrogen sulfide as a novel regulatory factor in liver health and disease. Oxid Med Cell Longev. 2019;2019:3831713. doi: 10.1155/2019/3831713
- Damba T, Zhang M, Buist-Homan M, et al. Hydrogen sulfide stimulates activation of hepatic stellate cells through increased cellular bio-energetics. Nitric Oxide. 2019;92:26–33. doi: 10.1016/j.niox.2019.08.004
- Latorre E, Torregrossa R, Wood ME, et al. Mitochondria-targeted hydrogen sulfide attenuates endothelial senescence by selective induction of splicing factors HNRNPD and SRSF2. Aging (Albany NY). 2018;10(7):1666–1681. doi: 10.18632/aging.101500
- Niu H, Li J, Liang H, et al. Exogenous hydrogen sulfide activates PI3K/Akt/eNOS pathway to improve replicative senescence in human umbilical vein endothelial cells. Cardiol Res Pract. 2023;2023:1–10. doi: 10.1155/2023/7296874
- Xu L, Hui AY, Albanis E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54(1):142–151. doi: 10.1136/gut.2004.042127
- Huy H, Song HY, Kim MJ, et al. TXNIP regulates AKT-mediated cellular senescence by direct interaction under glucose-mediated metabolic stress. Aging Cell. 2018;17(6):e12836. doi: 10.1111/acel.12836
- Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. doi: 10.1038/nrgastro.2017.38
- Yang G, Zhao K, Ju Y, et al. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18(15):1906–1919. doi: 10.1089/ars.2012.4645
- Fan HN, Wang HJ, Yang-Dan CR, et al. Protective effects of hydrogen sulfide on oxidative stress and fibrosis in hepatic stellate cells. Mol Med Rep. 2013;7(1):247–253. doi: 10.3892/mmr.2012.1153
- Zhang F, Jin H, Wu L, et al. Diallyl trisulfide suppresses oxidative stress-induced activation of hepatic stellate cells through production of hydrogen sulfide. Oxid Med Cell Longev. 2017;2017:1406726. doi: 10.1155/2017/1406726
- Rao PS, Midde NM, Miller DD, et al. Diallyl sulfide: potential use in novel therapeutic interventions in alcohol, drugs, and disease mediated cellular toxicity by targeting cytochrome P450 2E1. Curr Drug Metab. 2015;16(6):486–503. doi: 10.2174/1389200216666150812123554
- Rose P, Dymock BW, Moore PK. GYY4137, a novel water-soluble, H2S-releasing molecule. Methods Enzymol. 2015;554:143–167.
- Hassan MI, Boosen M, Schaefer L, et al. Platelet-derived growth factor-BB induces cystathionine γ-lyase expression in rat mesangial cells via a redox-dependent mechanism. Br J Pharmacol. 2012;166(8):2231–2242. doi: 10.1111/j.1476-5381.2012.01949.x
- Wang YH, Huang JT, Chen WL, et al. Dysregulation of cystathionine γ-lyase promotes prostate cancer progression and metastasis. EMBO Rep. 2019;20(10):e45986. doi: 10.15252/embr.201845986
- Goren I, Köhler Y, Aglan A, et al. Increase of cystathionine-γ-lyase (CSE) during late wound repair: hydrogen sulfide triggers cytokeratin 10 expression in keratinocytes. Nitric Oxide. 2019;87:31–42. doi: 10.1016/j.niox.2019.03.004
- Qiu T, Tian Y, Gao Y, et al. PTEN loss regulates alveolar epithelial cell senescence in pulmonary fibrosis depending on Akt activation. Aging (Albany NY). 2019;11(18):7492–7509. doi: 10.18632/aging.102262
- Iwagami Y, Huang CK, Olsen MJ, et al. Aspartate β-hydroxylase modulates cellular senescence through glycogen synthase kinase 3β in hepatocellular carcinoma. Hepatology. 2016;63(4):1213–1226. doi: 10.1002/hep.28411
- Rössig L, Badorff C, Holzmann Y, et al. Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J Biol Chem. 2002;277(12):9684–9689. doi: 10.1074/jbc.M106157200
- Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi: 10.1126/science.1106148
- Bernard M, Yang B, Migneault F, et al. Autophagy drives fibroblast senescence through MTORC2 regulation. Autophagy. 2020;16(11):2004–2016. doi: 10.1080/15548627.2020.1713640
- Wiley CD, Velarde MC, Lecot P, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23(2):303–314. doi: 10.1016/j.cmet.2015.11.011
- Zheng M, Qiao W, Cui J, et al. Hydrogen sulfide delays nicotinamide-induced premature senescence via upregulation of SIRT1 in human umbilical vein endothelial cells. Mol Cell Biochem. 2014;393(1–2):59–67. doi: 10.1007/s11010-014-2046-y
- Suo R, Zhao ZZ, Tang ZH, et al. Hydrogen sulfide prevents H₂O₂-induced senescence in human umbilical vein endothelial cells through SIRT1 activation. Mol Med Rep. 2013;7(6):1865–1870. doi: 10.3892/mmr.2013.1417
- Wang XL, Xu YT, Zhang SL, et al. Hydrogen sulfide inhibits alveolar type II cell senescence and limits pulmonary fibrosis via promoting MDM2-mediated p53 degradation. Acta Physiol (Oxf). 2023;240(1):e14059. doi: 10.1111/apha.14059