Abstract
Inflammatory lung diseases are characterised by increased expression of multiple inflammatory genes that are regulated by proinflammatory transcription factors, such as NF-κ B. Gene expression is regulated by modifications such as acetylation of core histones through the concerted action of coactivators such as CBP (cAMP-response element binding protein (CREB)-binding protein) which have intrinsic histone acetyltransferase (HAT) activity and are able to recruit other HAT enzymes. Conversely gene repression is mediated via histone deacetylases (HDAC) and other corepressors. In biopsies from asthmatic subjects there is an increase in HAT activity and some reduction in HDAC activity. Both of these changes are partially reversed by corticosteroid therapy. Corticosteroids switch off inflammatory genes in asthma through a combination of a direct inhibition of HAT activity and by the recruitment of HDAC2 to the activated NF-κB-stimulated inflammatory gene complex. In chronic obstructive pulmonary disease (COPD), a corticosteroid insensitive disease, there is a reduction in HDAC activity and HDAC2 expression, which may account for the amplified inflammation and resistance to the actions of corticosteroids. The reduction in HDAC2 may be secondary to oxidative and nitrative stress as a result of cigarette smoking and severe inflammation. This may also occur to differing degrees in severe asthma, smoking asthmatic patients and cystic fibrosis. Similar mechanisms may also account for the steroid resistance seen within latent adenovirus infections. The reduction in HDAC activity induced by oxidative stress can be restored by theophylline, acting through specific kinases, which may be able to reverse steroid resistance in COPD and other inflammatory lung diseases. The modulation of HAT/HDAC activity may lead to the development of novel anti-inflammatory approaches to inflammatory lung diseases that are currently difficult to treat.
INTRODUCTION
Inflammation is a major component of the most common lung diseases, including asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis, interstitial lung disease, and acute respiratory distress syndrome. The coordinated expression of cytokines, chemokines, enzymes that synthesise inflammatory mediators, inflammatory mediator receptors, and adhesion molecules, results in an activation of both infiltrating and resident inflammatory cells. Many of these inflammatory genes are regulated by proinflammatory transcription factors, including nuclear factor-κ B (NF-κ B) and activator protein-1 (AP-1). These transcription factors orchestrate, amplify, and perpetuate the inflammatory response and form the molecular basis of chronic inflammation [Citation[1]]. Recently we have learned much more about the molecular mechanisms that enhance inflammatory gene expression.
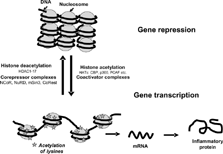
Since the 1960s it has been recognised that acetylation and methylation of histones and remodelling of the tightly packed chromatin structure is associated with gene induction [Citation[2], Citation[3]]. However, only in the last 8–10 years have the molecular mechanisms whereby inflammatory genes are switched on by transcription factors become much better understood with the demonstration that transcriptional co-activators possessed intrinsic histone acetyltransferase (HAT) activity [Citation[4]]. Acetylated histones are generally associated with increased gene transcription. In contrast, histone deacetylases (HDAC), which remove the ε -acetyl groups from hyperacetylated histones counteract the effects of HATs and return histone to its basal state, with the concomitant suppression of gene transcription (). This review focuses on the critical role of histone acetylation/deacetylation in the regulation of inflammatory genes. Understanding the role of histone deacetylation is now giving important insights into the mechanism of action of corticosteroids in treating inflammatory diseases, such as asthma, and the mechanisms of resistance to steroids in COPD and other severe inflammatory lung diseases. These new concepts are now also pointing the way towards the development of novel therapeutic approaches.
Figure 1 Gene activation and repression are regulated by acetylation of core histones. Histone acetylation is mediated by transcription factor-mediated recruitment of coactivators which have intrinsic histone acetyltransferase (HAT) activity. Changes in histone acetylation and recruitment of co-activators such as CBP (CREB binding protein), p300 and PCAF (p300/CBP associated factor) leads to an opening up of the chromatin structure allowing binding of large protein complexes such as RNA polymerase II that were unable to bind DNA in the closed chromatin configuration. This is reversed by corepressor complexes such as NuRD (nucleosome remodeling and deacetylase), Sin3 (Switch insensitive 3) and Co-REST (Co-repressor of REST (RE1 silencing transcription factor)), that include histone deacetylases (HDACs) and other associated proteins such as NCoR (nuclear receptor co-repressor) which reverse this acetylation, thereby causing gene silencing.
Corticosteroids and histone acetylation
Corticosteroids are by far the most effective therapy available for asthma and inhaled steroids have revolutionized its management in the last two decades [Citation[5]]. Inhaled steroids are not only effective in virtually all patients with asthma, but they are almost free of systemic side effects. In view of the complexity of inflammation in asthma, it has been difficult to understand how small doses of corticosteroids could be so effective in suppressing this inflammation, as so many inflammatory cells and mediators are involved in the pathophysiology of asthma. It is now becoming clear that the inflammation in asthma is largely driven by the increased expression of multiple inflammatory genes via the activation of proinflammatory transcription factors, such as AP-1 and NF-κ B. This results in acetylation of core histones as discussed above. Corticosteroids appear to suppress inflammation in asthma by switching off these inflammatory genes by targeting these transcription factors and their ability to induce histone modifications and subsequent gene expression [Citation[6], Citation[7]].
Gene Activation by Corticosteroids
Corticosteroids cross the cell membrane and bind to glucocorticoid receptors (GR) in the cytoplasm, which rapidly translocate to the nucleus, where the activated GR may bind to glucocorticoid recognition elements (GRE) in the promoter region of steroid-sensitive genes. This results in switching on of certain genes, including several anti-inflammatory genes. The activation of genes by relatively high concentrations of corticosteroids is associated with a selective acetylation of N-terminal tail lysine residues (lysines 5 and 16) on histone-H4, resulting in increased gene transcription [Citation[8]]. A more detailed description of these events is given later in this review. This is a different histone acetylation pattern (lysines 8 and 12) seen with inflammatory stimuli such as interleukin (IL)-1. This acetylation pattern is correlated with increased secretion of the antiprotease, anti-inflammatory protein secretory leukoprotease inhibitor (SLPI) in response to corticosteroids in an epithelial cell line. Activated GR may bind to coactivator molecules, such as CBP (cAMP-response element binding protein (CREB)-binding protein) or PCAF (p300/CREB associated factor), as well as steroid-receptor coactivator-1 (SRC-1) and glucocorticoid receptor interacting protein 1 (GRIP-1), which all possess HAT activity [Citation[9], Citation[10]].
Corticosteroids may suppress inflammation by increasing the synthesis of anti-inflammatory proteins, such as annexin-1 (lipocortin-1), SLPI (secretory leukocyte protease inhibitor), interleukin(IL)-10, the inhibitor of NF-κB, IκB-α, glucocorticoid-induced leucine zipper protein (GILZ), which inhibits both NF-κ B and AP-1 (11) and MAP kinase phosphatase-1 that inhibits p38 MAP kinase [Citation[12]]. However, it seems unlikely that the widespread anti-inflammatory actions of corticosteroids could be explained by increased transcription of small numbers of anti-inflammatory genes, particularly as high concentrations of corticosteroids are usually required for these responses, whereas in clinical practice corticosteroids are able to suppress inflammation at low concentrations. It is also likely that many of the side effects of corticosteroids are mediated by gene-activation mechanisms, as mice expressing a mutant form of GR that cannot dimerize and thus bind DNA lose the ability of corticosteroids to affect many metabolic effects of corticosteroids, whilst retaining their anti-inflammatory effects [Citation[13]].
Gene Suppression by Corticosteroids
Most of the inflammatory genes that are activated in asthma do not have GRE sites in their promoter regions, yet are potently repressed by corticosteroids. It has become clear that most of the anti-inflammatory actions of corticosteroids are due to suppression of the actions of pro-inflammatory transcription factors, such as AP-1 and NF-κ B, that regulate the expression of genes that code for inflammatory proteins, such as cytokines, inflammatory enzymes, adhesion molecules and inflammatory receptors [Citation[1]]. The activated GR can interact directly with activated transcription factors by a protein-protein interaction, which in many cases does not alter DNA binding. Thus, treatment of asthmatic patients with high doses of inhaled corticosteroids that suppress airway inflammation is not associated with any reduction in NF-κ B binding to DNA [Citation[14]]. This suggests that corticosteroids are more likely to be acting downstream of the binding of proinflammatory transcription factors to DNA and attention has now focused on their effects on downstream effects such as histone modifications.
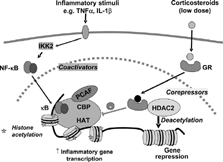
Activated GR may bind to CBP or other coactivators directly to inhibit their HAT activity [Citation[8]], thus preventing the subsequent histone acetylation and chromatin remodelling. More importantly, particularly at low concentrations that are likely to be relevant therapeutically, activated GR recruits co-repressor complexes containing HDAC2 to the activated inflammatory gene transcriptional complex, resulting in deacetylation of histones, and thus a decrease in inflammatory gene transcription [Citation[8]] (). This mechanism may account for the clinical efficacy of corticosteroids in asthma and GR could potentially recruit HDAC2 to all inflammatory gene promoters that have been acetylated activated by NF-κ B and possibly other proinflammatory transcription factors. Using a chromatin immunoprecipitation (ChIP) assay we have demonstrated that corticosteroids reverse the acetylation of the promoter of inflammatory genes such as GM-CSF [Citation[8]]. Other genes are not recognised through this mechanism, so corticosteroids do not switch off genes involved in basal cell functions, proliferation or survival. Furthermore this explains why corticosteroids are relatively safe, as side effects may be mediated mainly by gene activation mechanisms, which requite higher concentrations of corticosteroids, rather than via gene repression and HDAC recruitment.
Figure 2 Inflammatory gene suppression by corticosteroids. Inflammatory genes are activated by inflammatory stimuli, such as interleukin-1β (IL-1β) or tumour necrosis factor-α (TNF-α), resulting in activation of IKK2 (inhibitor of I-κ B kinase-2), which activates the transcription factor nuclear factor κ B (NF-κ B). NF-κ B translocates to the nucleus, binds to specific κ B recognition sites and recruits coactivators, such as CREB-binding protein (CBP) or p300/CBP-activating factor (PCAF), which have intrinsic histone acetyltransferase (HAT) activity. This results in acetylation of lysines in core histones resulting in increased expression of genes encoding inflammatory proteins, such as granulocyte-macrophage colony-stimulating factor (GM-CSF). Glucocorticoid receptors (GR) after activation by corticosteroids translocate to the nucleus and bind to NF-κ B-associated coactivators to inhibit their HAT activity either directly or by recruiting histone deacetylases (HDAC)2, which functionally reverses histone acetylation leading in suppression of inflammatory genes.
HATS/HDAC s IN AIRWAY DISEASES
Asthma
In bronchial biopsies from patients with asthma there is a marked increase in HAT and a small reduction in HDAC activity compared to normal airways, thus favouring increased inflammatory gene expression [Citation[15]]. Similar changes are found in alveolar macrophages obtained by bronchoalveolar lavage from patients with asthma [Citation[16]]. There is a small reduction in the expression of HDAC1, but expression of HDAC2 and 3 is normal in these cells. Peripheral blood mononuclear cells (lymphocytes and monocytes) appear to have normal HAT and HDAC activity, indicating that these changes occur locally in the airways of asthmatic patients. Interestingly, in patients with asthma who smoke there is a significantly greater reduction of HDAC activity in bronchial biopsies than in non-smoking asthmatic patients (unpublished observations) and this may account for why these smoking asthmatics have more severe asthma and resistance to steroids [Citation[17]].
COPD
In contrast to asthma, in COPD there is no change in HAT activity but a marked reduction in HDAC activity in the lung parenchyma and this decrease is correlated with disease severity [Citation[18]]. The reduction in HDACs in peripheral lung is selective with a marked reduction in HDAC2, with lesser reduction in HDAC5 and HDAC8 expression, but normal expression of the other class 1 and 2 HDACs. Furthermore, HDAC5 expression is predominantly cytoplasmic rather than nuclear in patients with COPD. In patients with very severe (GOLD stage 4) disease there is a > 95% reduction in expression of HDAC2. The reduction in HDAC activity is also related to the intensity of inflammation, as measured by expression of IL-8 and the number of inflammatory cells in small airways [Citation[19]]. This does not correspond to an increase in tissue HAT activity [Citation[18]].
Although inhaled corticosteroids are highly effective in asthma, they provide relatively little therapeutic benefit in COPD, despite the fact that active airway and lung inflammation is present. This may reflect that the inflammation in COPD is not suppressed by corticosteroids, with no reduction in inflammatory cells, cytokines or proteases in induced sputum even with oral corticosteroids [Citation[20], Citation[21], Citation[22]]. Furthermore, histological analysis of peripheral airways of patients with severe COPD shows an intense inflammatory response, despite treatment with high doses of inhaled corticosteroids [Citation[19]]. There is some evidence that an active steroid resistance mechanism exists in COPD as corticosteroids fail to inhibit cytokines (such as IL-8 and TNF-α) that they normally suppress [Citation[20], Citation[21]]. In vitro studies show that cytokine release from alveolar macrophages is markedly resistant to the anti-inflammatory effects of corticosteroids, compared to cells from normal smokers and these in turn are more resistant than alveolar macrophages from non-smokers [Citation[23]]. This lack of response to corticosteroids may be explained, at least in part, by an inhibitory effect of cigarette smoking and oxidative stress on HDAC function, thus interfering with the critical anti-inflammatory action of corticosteroids [Citation[24]]. Indeed there is a correlation between HDAC activity and the suppressive effects of a corticosteroid on cytokine release.
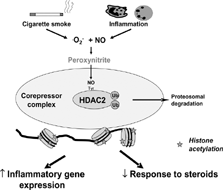
It is possible that oxidative and nitrative stress in COPD specifically impairs HDAC2 expression and activity, as discussed next, resulting in corticosteroid resistance [Citation[25]] (). Although this is seen in all stages of COPD it is most marked in the patients with the most severe disease [Citation[18]]. Even in patients with COPD who have stopped smoking the steroid resistance persists [Citation[20], Citation[21]] and these patients are known to have continuing oxidative stress [Citation[26]]. As oxidative stress is also increased in patients with severe asthma and during exacerbations [Citation[27], Citation[28], Citation[29]], a reduction in HDAC may also account for the reduced responsiveness to corticosteroids in these patients.
Figure 3 Possible mechanism of reduction in histone deacetylase 2 (HDAC2). HDAC2 is inactivated by peroxynitrite, generated by an interaction of nitric oxide (NO) generated by inducible NO synthase (iNOS) and cigarette smoke and superoxide anions (O2−). Peroxynitrite nitrates tyrosine (Tyr) residues on HDAC2 and this may block enzymatic activity and also mark the enzyme for ubiquitination (Ub) and destruction by the proteasome. Tyrosine nitration may also lead to disassembly of the co-repressor complex. The loss of HDAC2 function leads to amplification of the inflammatory response and resistance to corticosteroids.
Asthmatic patients who smoke have more severe disease and are also resistant to the anti-inflammatory effects of corticosteroids [Citation[17], Citation[30]]. Alveolar macrophages from normal smokers show a reduction in HDAC activity and expression of HDAC2 and this is correlated with an increase in release of TNF-α and IL-8 in response to an inflammatory stimulus [Citation[24]]. A combined effect of asthma and cigarette smoking on HAT/HDAC activity may explain the reduced corticosteroid sensitivity seen in these subjects.
Virus Infections
Adenovirus infection increases the expression of inflammatory genes in epithelial cells in vitro and this appears to be mediated via the adenoviral E1A protein, which is capable of interacting with HAT-containing coactivators such as CBP [Citation[31]]. In COPD lungs there is evidence for latent adenovirus infection and increased expression of E1A protein, so that this may be a mechanism for amplification of inflammation in COPD patients [Citation[32], Citation[33]]. Interestingly, adenovirus infection in guinea pigs amplifies the inflammatory response to allergen [Citation[34]] and is associated with a significant reduction in HDAC activity in the lungs in ovalbumin-sensitized animals [Citation[35]]. Thus the amplifying effects of adenovirus may be due to an inhibitory effect on HDAC activity and it is possible that there is a molecular interaction between HDAC and E1A protein within the nucleus. Persistence of adenovirus infections has also been implicated in steroid-resistance in children with asthma [Citation[36]]. Other virus infections may also impair the action of HDAC2 and thus induce steroid resistance, but this still needs to be explored. Thus, increased gene transcription in inflammatory diseases may be due to increased HAT, decreased HDAC or a combination of both.
Other Diseases
In cystic fibrosis and interstitial lung disease the high level of oxidative stress [Citation[26], Citation[37], Citation[38], Citation[39]] may impair HAT/HDAC ratios and this may potentially also account for the poor response to corticosteroid in these diseases [Citation[40], Citation[41]] although data is needed in these diseases.
Effects of oxidative and nitrative stress on HDAC activity
Oxidative stress results in increased acetylation of histone-H4 in epithelial cell lines such as A549 and BEAS-B (Adenovirus-12/SV40 virus immortalized human bronchial epithelial cells), resulting in increased release of inflammatory proteins such as GM-CSF and IL-8 [Citation[42], Citation[43]]. This involves the activation of the transcription factor NF-κ B, which has long been known to be activated by oxidative stress. Oxidative stress may also increase the association between the p65 component of NF-κ B and CBP [Citation[44], Citation[45]] thereby increasing promoter-specific HAT activity. In addition, oxidative stress such as hydrogen peroxide and cigarette smoke markedly reduce HDAC activity and HDAC2 expression in epithelial cell lines in vitro [Citation[46]] which will also result in promoter acetylation. Peroxynitrite, generated through an interaction of superoxide anions and nitric oxide, may be involved in HDAC down-regulation through nitration of tyrosine residues since a peroxynitrite-generating compound SIN-1 causes marked reduction in HDAC activity in epithelial cells in vitro [Citation[46]]. The mechanism whereby tyrosine nitration leads to inactivation of HDAC2 is not yet certain. Nitration of proteins by peroxynitrite has been documented to reduce prostacyclin synthase enzymatic activity [Citation[47]]. It is possible that either a tyrosine in the catalytic region of HDAC2 becomes nitrated, thus interfering with its enzymatic efficiency or, as with phosphorylation, nitration may affect co-factor association. Furthermore, nitration of proteins appears to make HDAC2 more susceptible to proteasomal degradation [Citation[48]], which may explain the marked reduction in HDAC2 protein expression in severe COPD [Citation[18]].
The effects of oxidative stress are mimicked by cigarette smoke extract and its effects are blocked by the antioxidant N-acetylcysteine [Citation[49]]. This suggests that cigarette smoking may be one of the mechanisms inducing HDAC deficiency in COPD patients. There is considerable evidence for increased oxidative stress in the respiratory tract of patients with COPD, including increased concentrations of ethane, and 8-isoprostane, markers of oxidative stress, in exhaled breath and 4-hydroxynonenal in peripheral lungs of COPD patients [Citation[26], Citation[38], Citation[50], Citation[51]]. These markers are increased to a greater extend in COPD patients than normal smokers and are related to disease severity. Importantly, in an animal model of COPD, HDAC2 expression and activity correlate with cigarette smoking, changes in oxidative stress and reduced corticosteroid sensitivity [Citation[52]].
Effects of drugs on HDAC activity
Corticosteroids
As indicated above, corticosteroids inhibit p65-associated HAT activity and enables HDAC activity to be recruited to NF-κB, thus switching off activated inflammatory genes. These actions of corticosteroids do not involve a direct enhancement of HDAC activity but direct or indirect binding of the GR monomer with CBP and HDAC [Citation[53]]. Corticosteroid-induced gene induction, in contrast, requires the GR homodimer recruiting a CBP-HAT complex. This suggests that it may be possible to develop dissociated steroids that discriminate between GR DNA binding, co-activator recruitment of GR and interaction with inflammatory gene complexes which utilises co-repressor recruitment [Citation[54]]. Several dissociated steroids are in development with a view to reducing systemic side effects (that are largely due to DNA binding) and anti-inflammatory effects (that are due to protein-protein interaction) [Citation[55]].
Theophylline
Theophylline has been used to treat asthma for many years, but its mechanism of action has been difficult to elucidate. Originally theophylline was used as a bronchodilator and relaxes airway smooth muscle by inhibiting phosphodiesterases (PDE). There is accumulating evidence that at lower doses theophylline has anti-inflammatory effects, but it is unlikely that these are mediated by PDE inhibition, as the inhibition of these enzymes is trivial at low plasma concentrations that are clinically effective [Citation[56]]. We have shown that the anti-inflammatory effects of theophylline may be mediated via activation of HDAC and that this effect is independent of PDE inhibition [Citation[57]]. Low doses of theophylline significantly increase HDAC activity in bronchial biopsies from asthmatic patients and the increase in HDAC activity is correlated with the reduction in airway eosinophils [Citation[57]]. Theophylline is active in low concentrations (10−7–10−5 M) on nuclear extracts, indicating that it works within the nucleus and does not require surface receptors. This appears to be a novel mechanism of theophylline and is not mimicked by PDE inhibitors or by adenosine receptor antagonists [Citation[57]]. This is of particular importance as the major side effects of theophylline are mediated via PDE inhibition (nausea headaches) and adenosine receptor (A1) antagonism (cardiac arrhythmias, seizures). Theophylline appears to preferentially activate class I HDACs, including HDAC2 [Citation[23]]. However, the exact mechanism whereby theophylline activates HDAC is not yet certain, but is likely to be through signal transduction pathways, probably kinases, that regulate HDAC activity or co-factor association. The effects of theophylline on HDAC appear to be enhanced under conditions of oxidative stress, making it more efficient as a regulator of inflammatory genes [Citation[23]]. This means that the dose of theophylline does not have to be increased as the disease becomes more severe as the increase in oxidative stress would increase drug activity.
This predicts that theophylline will enhance the anti-inflammatory actions of corticosteroids, as the HDAC2 recruited to the activated p65 complex at the promoters of inflammatory genes will be more effective at switching off these genes. Indeed, therapeutic concentrations of theophylline markedly potentiate the anti-inflammatory effects of corticosteroids in vitro [Citation[23]]. This may explain why adding a low dose of theophylline is more effective than increasing the dose of inhaled corticosteroids in patients who are not controlled adequately [Citation[58], Citation[59], Citation[60]].
In BAL macrophages obtained from COPD patients HDAC activity reduced and associated with increased inflammatory gene expression and steroid resistance. Low concentrations of theophylline are able to restore HDAC activity in these macrophages and this results in increased steroid responsiveness in these cells [Citation[23]]. These in vitro studies suggest that low doses of theophylline may have the capacity to reverse steroid resistance in COPD patients and trials are now underway to test this idea. Indeed, in COPD patients low-dose theophylline has anti-inflammatory effects, in contrast to the lack of response to high doses of corticosteroids [Citation[61]], and it is possible that there is an interaction between theophylline and endogenous cortisol. Furthermore, since similar mechanisms of steroid resistance may also apply in severe asthma and smoking asthmatics theophylline may also be useful in these patients and this may explain why theophylline appears to be useful as an add-on therapy to inhaled steroids particularly in patients with severe disease [Citation[62]].
Future directions
The recognition that histone acetylation status regulates inflammatory gene expression has improved our understanding of chronic inflammatory lung diseases and the discovery that oxidative and nitrative stress can lead to amplified inflammation and corticosteroid resistance in COPD and probably severe asthma and smoking asthmatic patients may have important clinical implications.
New Theophylline Derivatives
Activation of HDAC2 may have therapeutic potential in COPD and theophylline represents the first drug that has been shown to have this property, resulting in marked potentiation of the anti-inflammatory effects of corticosteroids. It may be possible to discover other drugs in this class that could form the basis of a new class of anti-inflammatory drugs without the side effects that limit the use of theophylline [Citation[56]]. While low concentrations of theophylline activate HDAC, higher concentrations (> 10−4 M) inhibit HDAC. This may be an indication that theophylline is a partial agonist and a search for fuller agonists might lead to more effective HDAC activators. Novel HDAC activators might be discovered by high throughput screening using HDAC activation, particularly under conditions of oxidative stress. Kinases, or phosphatases, that regulate HDAC2 activity/complex association in a similar manner to theophylline may also prove effective add-on therapies to corticosteroids.
Antioxidants
Since oxidative stress is a mechanism that may impair HDAC activity and expression as seen in COPD and severe asthma, antioxidants have the potential to increase HDAC activity, switch off inflammatory genes and restore steroid responsiveness [Citation[63]]. Currently available antioxidants, such as N-acetyl cysteine, are not very potent and may not sufficiently reduce oxidative stress in the lungs. New more potent antioxidants are needed in the future and there are several drugs in development, including new glutathione and superoxide dismutase analogues [Citation[64]].
iNOS Inhibitors
Peroxynitrite, formed by an interaction of superoxide anions and NO, may lead to a reduction in HDAC activity and expression. NO is derived predominantly from inducible NO synthase (iNOS) and this suggests that inhibition of iNOS may block the formation of peroxynitrite and reverse steroid resistance. Several selective iNOS inhibitors are now in development and one of these has been shown to markedly reduce NO formation in asthmatic patients [Citation[65]].
Histone acetylation and gene expression
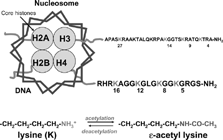
Alterations in the structure of chromatin are critical to the regulation of gene expression. Chromatin is made up of nucleosomes that are particles consisting of 146bp of DNA wrapped around an octomer of two molecules each of the core histone proteins (H2A, H2B, H3, and H4). In the resting cell DNA is wound tightly around these basic core histones, presenting an impenetrable barrier to large protein complexes such as RNA polymerase II, which produce unspliced primary messenger RNA transcripts. This conformation of the chromatin structure is described as closed (heterochromatin) and is associated with suppression of gene expression. Gene transcription occurs when the chromatin structure is opened up, with loosening of the tight nucleosomal structure allowing RNA polymerase II and basal transcription complexes to interact with DNA and initiate transcription. When proinflammatory transcription factors, such as NF-κB, are activated they bind to specific recognition sequences in DNA and subsequently recruit large coactivator proteins, such as cAMP-response element binding protein (CREB)-binding protein (CBP), p300 and PCAF (p300-CBP associated factor) and other complexes to the site of gene expression (). These coactivator molecules act as the molecular switches that control gene transcription and all have intrinsic HAT activity [Citation[66], Citation[67]].
The N-terminal tails of the histone molecules protrude through and beyond the DNA coil presenting accessible targets for post-translational modifications of selective amino acid residues (). Thus, lysine residues in the tails of histone H3 and H4 may be acetylated, thus changing the electrostatic attraction between DNA and histones and also forming bromodomains enabling the association of other co-activators such as TATA box binding protein (TBP), TBP-associated factors, chromatin modifying engines and RNA polymerase II [Citation[67], Citation[68]]. This molecular mechanism is common to all genes, including those involved in differentiation, proliferation and activation of cells. Just as acetylation of histones is associated with gene induction, removal of acetyl groups by HDACs is generally associated with re-packing of chromatin and a lack of gene expression or gene silencing [Citation[69]]. The story is a little more complex since it is now clear that this is not an all-or-nothing event since the basal level of histone acetylation is just below the threshold for gene induction. Recruitment of HAT activity, by NF-κB for example, increases the local acetylation level above the threshold enabling gene expression to occur [Citation[70]].
Figure 4 The structure of nucleosomes. DNA is wound around the histone core, which is composed of 8 histone molecules with two copies of histones 2A, 2B, 3 and 4. Each histone molecule has a long N-terminal tail rich in lysine residues (K), which protrudes through and beyond the DNA helix. These lysine residues are the sites of acetylation resulting in a loss of positively charged histones thereby reducing the electrostatic attraction with negatively charged DNA. This surface also forms a bromodomain structure which enables recruitment of other transcriptional complexes.
Recently these fundamental mechanisms have been applied to understanding the regulation of inflammatory genes in airway diseases. In a human epithelial cell line activation of NF-κ B induced by IL-1β results in acetylation of specific lysine residues on histone-H4 and to a lesser extent on H3 and that this is correlated with increased expression of inflammatory genes, such as granulocyte-macrophage colony stimulating factor (GM-CSF) [Citation[8]]. There is a specific pattern of acetylation of histone-H4 after NF-κ B activation in A549 cells with preferential acetylation of lysines 8 and 12 and a relative sparing of lysines 5 and lysine 16 [Citation[8]].
Histone deacetylases
HDACs play a critical role in the suppression of gene expression by reversing the hyperacetylation of core histones. Lysine acetylation is reversible and is controlled by the opposing actions of HATs and HDACs in vivo. Since histones were thought to be the major cellular proteins modified by lysine acetylation, most lysine HATs and HDACs were initially identified as histone acetyltransferases and HDACs.
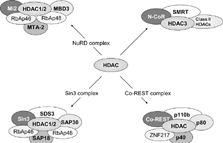
The lysine deacetylases, mammalian HDAC1 and yeast Hda1, were first identified in 1996 [Citation[71], Citation[72]]. According to phylogenetic analyses as well as sequence homology to yeast Rpd3 (reduced potassium dependency 3), Hda1 (histone deacetylase 1), and Sir2 (silent information regulator 2), HDACs are divided into four classes: I (HDAC1, -2, -3, and -8; homologous to Rpd3), II (HDAC4, -5, -6, -7, -9, and -10; related to Hda1), III (Sirt1, -2, -3, -4, -5, -6, and -7; similar to Sir2) and IV (HDAC11) (73–75). Class I HDACs display some sequence homology to members of classes II and IV but not to those of class III. In agreement with this, class I, II, and IV HDACs are zinc-dependent enzymes, whereas the deacetylase activity of class III members is NAD+ dependent. In addition, the widely expressed class I HDACs are exclusively localized to the nucleus whereas the more restricted class II HDACs shuttle between the nucleus and cytoplasm. There is evidence that these different HDACs target different patterns of acetylation and regulate different genes [Citation[76]]. The different HDACs are also likely to be regulated differently. HDACs interact with corepressor molecules, such as nuclear receptor corepressor (NCoR), ligand-dependent corepressor (LCoR), NuRD (nucleosomes remodelling and deacetylase) and mSin3 (Switch independent 3), all of which aid HDACs in gene repression and may provide specificity by selecting which genes are switched off by HDAC [Citation[77], Citation[78]] ().
Composition of HDAC repressor complexes. HDACs lack intrinsic repressor activity and require co-factors for optimal HDAC activity. The co-repressor proteins involved in the major HDAC complexes NuRD (nucleosome remodeling and deacetylase), Sin3 (Switch insensitive 3), Co-REST (Co-repressor of REST (RE1 silencing transcription factor)) and N-CoR and SMRT complexes are shown. NuRD and sin3 complexes share the retinoblastoma associated protein (RbAp)46 and 48 proteins and also contain distinct sets of proteins. Abbreviations: Co-REST, Co-repressor of REST (RE1 silencing transcription factor); MBD3, Methyl CpG binding domain 3; Mi2, Mi2 autoantigen; MTA-2, Metastasis-associated gene family, member 2; N-CoR, Nuclear receptor co-repressor; NuRD, Nucleosome remodelling and deacetylating; RbAp46, Retinoblastoma associated protein of 46 kDa; SAP18, Sin3 associated protein of 18kDa; SDS3, Suppressor of defective silencing 3; Sin3, Switch insensitive 3; SMRT, Silencing mediator for retinoid and thyroid receptors; ZNF217, Zn finger factor 217 kDa.
Trichostatin A is a non-selective inhibitor of class I, II and IV HDACs [Citation[79]] and in alveolar macrophages and airway epithelial cell lines it leads to increased expression of inflammatory genes, such as GM-CSF and interleukin (IL)-8, after activation with inflammatory stimuli [Citation[8], Citation[23]]. This suggests that HDACs normally act to repress the expression of inflammatory genes in these cells. However, acetylated lysine residues are now known to be present in at least 80 other proteins, including approximately 40 sequence-specific transcription factors, approximately 10 transcriptional coregulators, several viral proteins, α -tubulin, acetyl-CoA synthase, Ku70, and Hsp90 [Citation[73], Citation[75]]. Transcription factors, such as the GATA sequence binding protein GATA3, Forkhead box protein O1 (FOXO1), p53 and the p65 component of NF-κB, are targets for acetylation and deacetylation, which thereby modulates their transcriptional activity. Thus, HDACs are also associated with inactive p65 and play a role in the regulation of NF-κ B-mediated gene transcription without altering DNA binding [Citation[80], Citation[81], Citation[82]]. CBP acetylates specific lysine residues on p65, increasing its binding to DNA and causing transcriptional activation. HDACs reverse this process. HDAC1 and HDAC2 are able to deacetylate NF-κ B and promote its association with the inhibitor Iκ B-α within the nucleus to promote export into the cytoplasm and thus terminate the activity of NF-κ B [Citation[80]]. Inhibition of these HDACs by TSA results in increased activation of NF-κB and increased expression of inflammatory genes, such as IL-8. Furthermore, changes in the phosphorylation status can switch p65 from interacting with a corepressor (HDAC) to a coactivator CBP (HAT) [Citation[82]].
Class 3 HDACs are the atypical nicotinamide adenosine dinucleotide (NAD)-dependent sirtuins. These proteins are principally thought to deacetylate non-histone proteins and to play a role in programmed cell death in mononuclear cells [Citation[83], Citation[84]]. For example, SIRT1 can deacetylate p53, thereby inactivating p53-mediated transcription and apoptosis, regulate Bax-induced apoptosis by deacetylating Ku70 and prevent Forkhead-mediated cell death [Citation[85]]. Recently, SIRT1 has also been reported to deacetylate p65 and control apoptosis in a lung cancer cell line [Citation[85]]. Selective inhibitors of class III HDACs such as sirtinol and splitomycin have been described [Citation[75]] and in addition, resveratrol has been reported as a selective activator of sirtuin activity.
Regulation of HDAC activity
Co-Factor Association and DNA Binding
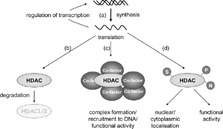
The activities of most if not all HDACs are regulated by protein-protein interactions. In addition, many HDACs are regulated by post-translational modifications as well as by subcellular localization. HDACs generally exist as a component of stable large multi-subunit complexes, and most, if not all, HDACs interact with other cellular proteins. With the exception of mammalian HDAC8, most purified recombinant HDACs are enzymatically inactive [Citation[86]]. Any protein that associates with HDACs, therefore, has the potential to activate or inhibit the enzymatic activity of HDACs. Likewise, HDACs, in general, have no DNA binding activity, therefore, any DNA-binding protein that targets HDACs to DNA or to histones potentially can affect HDAC function ().
Regulation of HDAC activity. HDAC expression is regulated by several processes including (a) inflammatory mediator effects on HDAC gene expression and autoregulation by HDAC isoenzymes; (b) proteasomal degradation of HDAC isoenzymes; (c) complex formation and subsequent changes in functional HDAC activity and/or recruitment to DNA and (d) post-translational modifications. Post-translational modifications such as phosphorylation (P), sumoylation (S) and nitration (N) can affect HDAC subcellular localization and/or functional HDAC activity.
Human HDAC1 and HDAC2 exist together in at least three distinct multi-protein complexes called the Sin3, the NuRD, and the Corepressor of REST (RE1 silencing transcription factor, CoREST) complexes [Citation[86]] (). Sin3 and NuRD complexes share a core comprised of four proteins: HDAC1, HDAC2, retinoblastoma associated protein (RbAp)46, and RbAp48. In addition, each complex contains unique polypeptides (Sin3, sin3 associated protein (SAP)18, and SAP30 in the Sin3 complex; Mi2, metastasis-associated gene family (MTA)-2, and methyl CpG binding domain (MBD)3 in the NuRD complex) which are essential for HDAC activity and function [Citation[87], Citation[88]]. Thus the NuRD complex may link acetylation and methylation in the regulation of gene expression [Citation[86]]. Similar results are seen for HDAC activity within the CoREST complex [Citation[89]]. Furthermore, HDAC3 activity is dependent upon silencing mediator of retinoid and thyroid receptor (SMRT) and nuclear receptor corepressor (N-CoR) association [Citation[86]].
Unlike HDAC3, the class II HDACs cannot be activated by SMRT/N-CoR alone. Instead, the enzymatic activity of HDAC4, 5, and 7 is dependent on the association with the HDAC3/ SMRT/N-CoR complex [Citation[86]]. These studies suggest that HDAC4, 5, and 7 are not active deacetylases but recruit preexisting enzymatically active SMRT/N-CoR complexes containing HDAC3 [Citation[90]] ().
Phosphorylation and Other Modifications
All mammalian HDACs possess potential phosphorylation sites and many of them have been found to be phosphorylated in vitro and in vivo. HDAC1 phosphorylation may either alter its conformation into a more favourable enzymatic active form or affect the ability of HDAC1 to interact with proteins, such as MTA2 and SDS3, which can subsequently stimulate its activity and consequently enhance its enzymatic activity [Citation[86]]. Similarly, HDAC2 phosphorylation is necessary for both enzymatic activity and association with the corepressors mSin3 and Mi2 [Citation[86]]. The activity of class II HDACs may also be regulated by phosphorylation via modulating their subcellular localization [Citation[86]]. This suggests that phosphatases may also be important in regulating HDAC activity and although less well studied, PP1 (protein phosphatase 1) and PP4, may also play an important role in regulating/modulating HDAC activity [Citation[91]]. Interestingly, addition of the small ubiquitin-like peptide Sumo to HDAC1 and HDAC4 can also affect repressor and HDAC activity [Citation[86]] ().
HDACs must reside in the nucleus in order to deacetylate histones and to repress transcription, therefore, signals that enhance HDAC nuclear localization must affect HDAC activity. HDAC1, 2, and 8 are predominantly nuclear proteins but in contrast, HDAC3 can be found both in the nucleus and cytoplasm and the nuclear/cytoplasmic ratio depends on cell types and stimuli [Citation[86]]. Thus, in response to IL-1β stimulation, the N-CoR/TAB2/HDAC3 corepressor complex undergoes nuclear to cytoplasmic translocation, resulting in derepression of a specific subset of NF-κ B-regulated genes [Citation[92]].
In contrast, experiments in cardiac myocytes shows that class II HDACs shuttle between the nucleus and the cytoplasm where they associate with 14-3-3 proteins [Citation[93], Citation[94]]. The binding of class II HDACs to 14-3-3 is absolutely dependent on phosphorylation of conserved N-terminal serine residues and this association results in sequestration of HDACs to the cytoplasm [Citation[93], Citation[94]]. Furthermore, CaMK-mediated phosphorylation of HDACs 4, 5, 7, and 9 promotes their association with 14-3-3 proteins resulting in increased retention of HDACs in the cytoplasm. Binding of 14-3-3 has been suggested to mask an N-terminal nuclear localization signal [Citation[93], Citation[94]].
Expression
Interestingly, HDACs can autoregulate their own expression by feedback mechanisms utilising the DNA binding actions of transcription factors such as NF-Y (nuclear factor Y) and Sp1. Furthermore, some degree of cross-talk in this regulation must also occur as changes in HDAC1 expression can also affect the expression of other class I HDACs (86; ). Recent evidence [Citation[43]] has shown that nitration of HDAC2 can lead to protein degradation. Proteasomal degradation appears to be a major mechanism of regulation of HDAC function [Citation[86]].
SUMMARY
In COPD, a corticosteroid insensitive disease, there is a reduction in HDAC2 function and activity and HDAC2 expression, which may account for the amplified inflammation and resistance to the actions of corticosteroids. The reduction in HDAC2 may be secondary to oxidative and nitrative stress as a result of cigarette smoking and severe inflammation. The reduction in HDAC2 complex activity induced by oxidative stress can be restored by theophylline, acting through specific kinases, which may be able to reverse steroid resistance in COPD and other inflammatory lung diseases. The modulation of HAT/HDAC activity may lead to the development of novel anti-inflammatory approaches to inflammatory lung diseases that are currently difficult to treat.
GLOSSARY
AP-1 = Activator protein-1
CBP = CREB binding protein
CREB = cAMP response element binding protein
Co-REST = Co-repressor of REST (RE1 silencing transcription factor)
FOXO1 = Forkhead box protein O1
HAT = Histone acetyltransferase
Hda1 = Histone deacetylase 1 (yeast)
HDAC = Histone deacetylase
LCoR = Ligand dependent corepressor
MBD3 = Methyl CpG binding domain 3
Mi2 = Mi2 autoantigen
MTA-2 = Metastasis-associated gene family, member 2
N-CoR = Nuclear receptor co-repressor
NF-kB = Nuclear factor kB
NuRD = Nucleosome remodelling and deacetylating
RbAp46 = Retinoblastoma associated protein of 46 kDa
Rpd3 = Reduced potassium dependency 3
SAP18 = Sin3 associated protein of 18kDa
SDS3 = Suppressor of defective silencing 3
Sin3 = Switch insensitive 3
Sir2 = Silent information regulator 2
SMRT = Silencing mediator for retinoid and thyroid receptors
REFERENCES
- Caramori G, Ito K, Adcock I M. Transcription factors in asthma and COPD. IDrugs 2004; 7(8)764–770
- Allfrey V G, Faulkner R, Mirsky A E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA 1964; 51: 786–794, may[CSA]
- Littau V C, Burdick C J, Allfrey V G, Mirsky S A. The role of histones in the maintenance of chromatin structure. Proc Natl Acad Sci USA 1965; 54(4)1204–1212, [CSA]
- Bannister A J, Kouzarides T. The C BP co-activator is a histone acetyltransferase. Nature 1996; 384(6610)641–643, [CSA]
- Barnes P J, Pedersen S, Busse W W. Efficacy and safety of inhaled corticosteroids. New developments. Am J Respir Crit Care Med 1998; 157(3 Pt 2)S1–53, [CSA]
- Barnes P J, Adcock I M. How do corticosteroids work in asthma?. Ann Intern Med 2003; 139(5 Pt 1)359–370, [CSA]
- Leung D Y, Bloom J W. Update on glucocorticoid action and resistance. J Allergy Clin Immunol 2003; 111(1)3–22, [CSA]
- Ito K, Barnes P J, Adcock I M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20(18)6891–6903, [CSA]
- Yao T P, Ku G, Zhou N, Scully R, Livingston D M. The nuclear hormone receptor coactivator SRC-1 is a specific target of p300. Proc Natl Acad Sci USA 1996; 93(20)10626–10631, [CSA]
- Kurihara I, Shibata H, Suzuki T, Ando T, Kobayashi S, Hayashi M, Saito I, Surata T. Expression and regulation of nuclear receptor coactivators in glucocorticoid action. Mol Cell Endocrinol 2002; 189(1–2)181–189, [CSA]
- Mittelstadt P R, Ashwell J D. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem 2001; 276(31)29603–29610, [CSA]
- Lasa M, Abraham S M, Boucheron C, Saklatvala J, Clark A R. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol 2002; 22(22)7802–7811, [CSA]
- Reichardt H M, Tuckermann J P, Gottlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schultz G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J 2001; 20(24)7168–7173, [CSA]
- Hart L, Lim S, Adcock I, Barnes P J, Chung K F. Effects of inhaled corticosteroid therapy on expression and DNA-binding activity of nuclear factor κB in asthma. Am J Respir Crit Care Med 2000; 161(1)224–231, [CSA]
- Ito K, Caramori G, Lim S, Oates T, Chung K F, Barnes P J. Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med 2002; 166(3)392–396, [CSA]
- Cosio B G, Mann B, Ito K, Jazrawi E, Barnes P J, Chung K F, Adcock I M. Histone acetylase and deacetylase activity in alveolar macrophages and blood mononocytes in asthma. Am J Respir Crit Care Med 2004; 170: 141–147, [CSA]
- Chaudhuri R, Livingston E, McMahon A D, Thomson L, Borland W, Thomson N C. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. Am J Respir Crit Care Med 2003; 168(11)1308–1311, [CSA]
- Ito K, Ito M, Eliott W M, Cosio B, Caramori G, Kon O M, Barczyk B, Hayashi S, Adcock I M, Hogg J C, Barnes P J. Decreased histone deacetylase activity in chronic obstructive pulmonary disease: relationship to disease severity. N Engl J Med 2005; 352: 1967–1976, [CSA]
- Hogg J C, Chu F, Utokaparch S, Woods R, Elliott W M, Buzatu L, Cherniak R M, Rogers R M, Sciurba F C, Coxson H O, Pare P D. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350(26)2645–2653, [CSA]
- Keatings V M, Jatakanon A, Worsdell Y M, Barnes P J. Effects of inhaled and oral glucocorticoids on inflammatory indices in asthma and COPD. Am J Respir Crit Care Med 1997; 155(2)542–548, [CSA]
- Culpitt S V, Maziak W, Loukidis S, Nightingale J A, Matthews J L, Barnes P J. Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 160(5 Pt 1)1635–1639, [CSA]
- Loppow D, Schleiss M B, Kanniess F, Taube C, Jorres R A, Magnussen H. In patients with chronic bronchitis a four week trial with inhaled steroids does not attenuate airway inflammation. Respir Med 2001; 95(2)115–121, [CSA]
- Cosio B G, Tsaprouni L, Ito K, Jazrawi E, Adcock I M, Barnes P J. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med 2004; 200(5)689–695, [CSA]
- Ito K, Lim S, Caramori G, Chung K F, Barnes P J, Adcock I M. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J 2001; 15(6)1110–1112, [CSA]
- Barnes P J, Ito K, Adcock I M. Corticosteroid resistance in chronic obstructive pulmonary disease: inactivation of histone deacetylase. Lancet 2004; 363(9410)731–733, [CSA]
- Montuschi P, Collins J V, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov S A, Barnes P J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am J Respir Crit Care Med 2000; 162(3 Pt 1)1175–1177, [CSA]
- Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov S A, Barnes P J. Increased 8-isoprostane, a marker of oxidative stress, in exhaled condensate of asthma patients. Am J Respir Crit Care Med 1999; 160(1)216–220, [CSA]
- Baraldi E, Ghiro L, Piovan V, Carraro S, Ciabattoni G, Barnes P J, Montuschi P. Increased exhaled 8-isoprostane in childhood asthma. Chest 2003; 124(1)25–31, [CSA]
- Caramori G, Papi A. Oxidants and asthma. Thorax 2004; 59(2)170–173, [CSA]
- Chalmers G W, Macleod K J, Little S A, Thomson L J, McSharry C P, Thomson N C. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax 2002; 57(3)226–230, [CSA]
- Higashimoto Y, Elliott W M, Behzad A R, Sedgwick E G, Takei T, Hogg J C, Hayashi S. Inflammatory mediator mRNA expression by adenovirus E1A-transfected bronchial epithelial cells. Am J Respir Crit Care Med 2002; 166(2)200–207, [CSA]
- Retamales I, Elliott W M, Meshi B, Coxson H O, Pare P D, Sciurba F C, Rogers R M, Hayashi S, Hogg J C. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med 2001; 164(3)469–473, [CSA]
- Hogg J C. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med 2001; 164(10 Pt 2)S71–S75, [CSA]
- Yamada K, Elliott W M, Brattsand R, Valeur A, Hogg J C, Hayashi S. Molecular mechanisms of decreased steroid responsiveness induced by latent adenoviral infection in allergic lung inflammation. J Allergy Clin Immunol 2002; 109(1)35–42, [CSA]
- Ito M, Yamada K, Vitalis T Z, Elliott W M, To Y, Hayashi S, Adcock I M, Hogg J C, Barnes P J, Ito K. Latent adenovirus infection decreases histone deacetylase activity in the lungs of ovalbumin-sensitized guinea pigs. Am J Respir Crit Care Med 2004; 169: A78, [CSA]
- Macek V, Sorli J, Kopriva S, Marin J. Persistent adenoviral infection and chronic airway obstruction in children. Am J Respir Crit Care Med 1994; 150(1)7–10, [CSA]
- Montuschi P, Ciabattoni G, Paredi P, Pantelidis P, DuBois R M, Kharitonov S A, Barnes P G. 8-Isoprostane as a biomarker of oxidative stress in interstitial lung diseases. Am J Respir Crit Care Med 1998; 158(5 Pt 1)1524–1527, [CSA]
- Paredi P, Kharitonov S A, Leak D, Ward S, Cramer D, Barnes P J. Exhaled ethane, a marker of lipid peroxidation, is elevated in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 162(2 Pt 1)369–373, [CSA]
- Antuni J D, Kharitonov S A, Hughes D, Hodson M E, Barnes P J. Increase in exhaled carbon monoxide during exacerbations of cystic fibrosis. Thorax 2000; 55(2)138–142, [CSA]
- Dezateux C, Walters S, Balfour-Lynn I. Inhaled corticosteroids for cystic fibrosis. Cochrane Database Syst Rev 2000; 2: CD001915, [CSA]
- Richeldi L, Davies H R, Ferrara G, Franco F. Corticosteroids for idiopathic pulmonary fibrosis. Cochrane Database Syst Rev 2003, 3: CD002880, [CSA]
- Rahman I, Gilmour P S, Jimenez L A, Macnee W. Oxidative stress and TNF-alpha induce histone acetylation and NF-κB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem 2002; 234–235(1–2)239–248, [CSA]
- Tomita K, Barnes P J, Adcock I M. The effect of oxidative stress on histone acetylation and IL-8 release. Biochem Biophys Res Commun 2003; 301(2)572–577, [CSA]
- Marwick J A, Kirkham P A, Stevenson C S, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 2004; 31(6)633–642, [CSA]
- Moodie F M, Marwick J A, Anderson C S, Szulakowski P, Biswas S K, Bauter M R, Kilty I, Rahman I. Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-κB activation and proinflammatory cytokine release in alveolar epithelial cells. FASEB J 2004; 18(15)1897–1899, [CSA]
- Ito K, Hanazawa T, Tomita K, Barnes P J, Adcock I M. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun 2004; 315(1)240–245, [CSA]
- Schmidt P, Youhnovski N, Daiber A, Balan A, Arsic M, Bachschmid M, Przybylski M, Ullrich V. Specific nitration at tyrosine 430 revealed by high resolution mass spectrometry as basis for redox regulation of bovine prostacyclin synthase. J Biol Chem 2003; 278(15)12813–12819, [CSA]
- Buchczyk D P, Grune T, Sies H, Klotz L O. Modifications of glyceraldehyde-3-phosphate dehydrogenase induced by increasing concentrations of peroxynitrite: early recognition by 20S proteasome. Biol Chem 2003; 384(2)237–241, [CSA]
- Cosio B, Jazrawi E, Ito K, Barnes P J, Adcock I M. Cigarette smoke decreases steroid responsiveness in monocytes: the role of histone deacetylase. Am J Respir Crit Care Med 2003; 167: A804, [CSA]
- Biernacki W A, Kharitonov S A, Barnes P J. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax 2003; 58(4)294–298, [CSA]
- Rahman I, van Schadewijk A A, Crowther A J, Hiemstra P S, Stolk J, Macnee W, De Boer W I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166(4)490–495, [CSA]
- Marwick J A, Kirkham P A, Stevenson C S, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 2004; 31(6)633–642, [CSA]
- Ito K, Jazrawi E, Cosio B, Barnes P J, Adcock I M. p65-activated histone acetyltransferase activity is repressed by glucocorticoids: mifepristone fails to recruit HDAC2 to the p65-HAT complex. J Biol Chem 2001; 276(32)30208–30215, [CSA]
- Garside H J, Stevens A, Farrow S, Normand C, Houle B, Berry A, Maschera B, Ray D. Glucocorticoid ligands specify different interactions with NF-κB by allosteric effects on the glucocorticoid receptor DNA binding domain. J Biol Chem 2004; 279: 50050–50059, [CSA]
- Schacke H, Schottelius A, Docke W D, Strehlke P, Jaroch S, Schmees N, Rehwinkel I H, Hennekes H, Asdullah K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA 2004; 101(1)227–232, [CSA]
- Barnes P J. Theophylline: new perspectives for an old drug. Am J Respir Crit Care Med 2003; 167(6)813–818, [CSA]
- Ito K, Lim S, Caramori G, Cosio B, Chung K F, Adcock I M, Barnes P J. A molecular mechanism of action of theophylline: induction of histone deacetylase activity to decrease inflammatory gene expression. Proc Natl Acad Sci USA 2002; 99(13)8921–8926, [CSA]
- Evans D J, Taylor D A, Zetterstrom O, Chung K F, O'Connor B J, Barnes P J. A comparison of low-dose inhaled budesonide plus theophylline and high-dose inhaled budesonide for moderate asthma. N Engl J Med 1997; 337(20)1412–1418, [CSA]
- Ukena D, Harnest U, Sakalauskas R, Magyar P, Vetter N, Steffen H, Leichtl S, Rathgeb F, Keller A, Steinijans V W. Comparison of addition of theophylline to inhaled steroid with doubling of the dose of inhaled steroid in asthma. Eur Respir J 1997; 10(12)2754–2760, [CSA]
- Lim S, Jatakanon A, Gordon D, Macdonald C, Chung K F, Barnes P J. Comparison of high dose inhaled steroids, low dose inhaled steroids plus low dose theophylline, and low dose inhaled steroids alone in chronic asthma in general practice. Thorax 2000; 55(10)837–841, [CSA]
- Culpitt S V, De Matos C, Russell R E, Donnelly L E, Rogers D F, Barnes P J. Effect of theophylline on induced sputum inflammatory indices and neutrophil chemotaxis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 165(10)1371–1376, [CSA]
- Barnes P J, Woolcock A J. Difficult asthma. Eur Respir J 1998; 12(5)1209–1218, [CSA]
- Rahman I, Marwick J, Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-κB and pro-inflammatory gene expression. Biochem Pharmacol 2004; 68(6)1255–1267, [CSA]
- Cuzzocrea S, Riley D P, Caputi A P, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev 2001; 53(1)135–159, [CSA]
- Hansel T T, Kharitonov S A, Donnelly L E, Erin E M, Currie M G, Moore W M, Manning P T, Recker D P, Barnes P J. A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics. FASEB J 2003; 17(10)1298–1300, [CSA]
- Ogryzko V V, Schiltz R L, Russanova V, Howard B H, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996; 87(5)953–959, [CSA]
- Roth S Y, Denu J M, Allis C D. Histone acetyltransferases. Annu Rev Biochem 2001; 70: 81–120, [CSA]
- Urnov F D, Wolffe A P. Chromatin remodeling and transcriptional activation: the cast (in order of appearance). Oncogene 2001; 20(24)2991–3006, [CSA]
- Gao L, Cueto M A, Asselbergs F, Atadja P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem 2002; 277(28)25748–25755, [CSA]
- Waterborg J H. Steady-state levels of histone acetylation in Saccharomyces cerevisiae. J Biol Chem 2000; 275(17)13007–13011, [CSA]
- Rundlett S E, Carmen A A, Kobayashi R, Bavykin S, Turner B M, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci USA 1996; 93(25)14503–14508, [CSA]
- Taunton J, Hassig C A, Schreiber S L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p [see comments]. Science 1996; 272(5260)408–411, [CSA]
- de Ruijter A J, van Gennip A H, Caron H N, Kemp S, van Kuilenburg A B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 2003; 370(Pt 3)737–749, [CSA]
- Yang X J, Gregoire S. Class II histone deacetylases: from sequence to function, regulation, and Clinical Implication. Mol Cell Biol 2005; 25(8)2873–2884, [CSA]
- Porcu M, Chiarugi A. The emerging therapeutic potential of sirtuin-interacting drugs: from cell death to lifespan extension. Trends Pharmacol Sci 2005; 26(2)94–103, [CSA]
- Peterson C L. HDAC's at work: everyone doing their part. Mol Cell 2002; 9(5)921–922, [CSA]
- Jones P L, Shi Y B. N-CoR-HDAC corepressor complexes: roles in transcriptional regulation by nuclear hormone receptors. Curr Top Microbiol Immunol 2003; 274: 237–268, [CSA]
- Fernandes I, Bastien Y, Wai T, Nygard K, Lin R, Cormier O, Lee H S, Eng F, Bertos N R, Pelletier N, Mader S, Han V K, Yang X J. Ligand-dependent nuclear receptor corepressor LCoR functions by histone deacetylase-dependent and -independent mechanisms. Mol Cell 2003; 11(1)139–150, [CSA]
- Marks P A, Richon V M, Miller T, Kelly W K. Histone deacetylase inhibitors. Adv Cancer Res 2004; 91: 137–168, [CSA]
- Chen L, Fischle W, Verdin E, Greene W C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science JID-0404511 2001; 293(5535)1653–1657, [CSA]
- Ashburner B P, Westerheide S D, Baldwin A SJ. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol JID-8109087 2001; 21(20)7065–7077, [CSA]
- Zhong H, May M J, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-κ B determines its association with CBP/p300 or HDAC-1. Mol Cell 2002; 9(3)625–636, [CSA]
- Brunet A, Sweeney L B, Sturgill J F, Chua K F, Greer P L, Lin Y, Tran H, Ross S E, Mostoslavsky R, Cohen H Y, Hu L S, Cheng H L, Jedrychowski M P, Gygi S P, Sinclair D A, Alt F W, Greenberg M E. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303(5666)2011–2015, [CSA]
- Cohen H Y, Miller C, Bitterman K J, Wall N R, Hekking B, Kessler B, Howitz K T, Gorospe M, de Cabo R, Sinclair D A. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004; 305(5682)390–392, [CSA]
- Yeung F, Hoberg J E, Ramsey C S, Keller M D, Jones D R, Frye R A, Mayo M W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004; 23(12)2369–2380, [CSA]
- Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem 2004; 93(1)57–67, [CSA]
- Zhang Y, Ng H H, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 1999; 13(15)1924–1935, [CSA]
- Lechner T, Carrozza M J, Yu Y, Grant P A, Eberharter A, Vannier D, Brosch G, Stillman D J, Shore D, Workman J L. Sds3 (suppressor of defective silencing 3) is an integral component of the yeast Sin3[middle dot]Rpd3 histone deacetylase complex and is required for histone deacetylase activity. J Biol Chem 2000; 275(52)40961–40966, [CSA]
- You A, Tong J K, Grozinger C M, Schreiber S L. CoREST is an integral component of the Co. Proc Natl Acad Sci USA 2001; 98(4)1454–1458, [CSA]
- Fischle W, Kiermer V, Dequiedt F, Verdin E. The emerging role of class II histone deacetylases. Biochem Cell Biol 2001; 79(3)337–348, [CSA]
- Galasinski S C, Resing K A, Goodrich J A, Ahn N G. Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J Biol Chem 2002; 277(22)19618–19626, [CSA]
- Baek S H, Ohgi K A, Rose D W, Koo E H, Glass C K, Rosenfeld M G. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and beta-amyloid precursor protein. Cell 2002; 110(1)55–67, [CSA]
- McKinsey T A, Zhang C L, Olson E N. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 2001; 21(18)6312–6321, [CSA]
- McKinsey T A, Zhang C L, Lu J, Olson E N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000; 408(6808)106–111, [CSA]