Abstract
Cigarette smoke has a major impact on health issues worldwide. Although genetics certainly is a factor in the sensitivity to cigarette smoke, other lung environmental factors, such as infection, potentially could interact with cigarette smoke to induce inflammatory changes associated with various diseases. Four groups of BALB/c mice (smoking only; smoking + M. pneumoniae infection; mycoplasma only; saline control) were studied for eight weeks to determine the interactive outcomes of inflammation and structural changes in the smoking plus mycoplasma group. This group did have significantly higher amounts of neutrophil degranulation in the outer airway wall area (smooth muscle to alveolar attachments) (p = 0.03) and mRNA expression of matrix metalloproteinase-9 (p = 0.045). Although there was not a significant difference in alveolar tissue elastin between the groups, the smoking plus mycoplasma group had a level approximately 20% below the other groups. Even in this relatively short duration study, it appears that an infectious process can interact with cigarette smoke to produce a destructive type of inflammatory response (activated neutrophils and metalloproteinase-9) seen in the outer airway wall area.
INTRODUCTION
All cigarette smoke health related diseases are a major and increasing problem worldwide. In particular, chronic obstructive pulmonary disease (COPD) is predicted to rapidly become the fifth most common cause of disability and third commonest cause of mortality [Citation[1]].
Although chronic lung (especially small airway) inflammation and associated elastolytic enzymes (e.g., matrix metalloproteinases) have been proposed to contribute to lung disease, the cellular and molecular mechanisms behind this remain unclear [Citation[2]]. It has been stated that smoking is implicated in greater than 90% of COPD patients [Citation[3]]. However, the question arises if all cigarette smokers get COPD. Perhaps all smokers eventually evolve into COPD, but it is more certain that only 20% of smokers develop COPD at a much more rapid pace [Citation[3]]. Thus, why do these 20% have a “rapid” development of COPD? There is no doubt that a gene-environment interaction occurs in the development of COPD from cigarette smoke, but the question remains what is a key environmental contributor to enhance the process? Perhaps most importantly, what is the environment within the lungs that interacts with cigarette smoke to enhance the development of lung disease? Previous studies have suggested a role of respiratory tract infections in the acute exacerbations of COPD, and also stable COPD. These microorganisms may include bacteria (e.g., Haemophilus influenza) and viruses (e.g., adenovirus) [Citation[4]].
Atypical bacteria (Mycoplasma pneumoniae [Mp] and Chlamydia pneumoniae [Cp]) have been linked with chronic asthma [Citation[5], Citation[6], Citation[7], Citation[8], Citation[9], Citation[10], Citation[11], Citation[12], Citation[13]], another obstructive airway process. In COPD, chronic Cp infection was demonstrated in 46% of mild to moderate and 71% of severe patients [Citation[14]]. In a murine model, Mp respiratory infection produces neutrophilic inflammation and bronchial hyperresponsiveness (BHR) [Citation[15]] and enhances allergen-induced inflammation and BHR [Citation[16]]. Thus, this present study evaluates the interaction of cigarette smoking and Mp respiratory infection for the potential of developing a murine model of increased lung inflammation with the focus on the airway neutrophil [Citation[2]].
MATERIALS AND METHODS
This study was approved by the Institutional Animal Care and Use Committee. Four groups () of 15 BALB/c mice each were studied over an 8-week time interval. M. pneumoniae (strain FH, ATCC 15531) at approximately 1 × 108 colony forming units [Citation[11]] were used to nasally infect specific groups. A similar 50 μl inoculation of saline was given to the control mice.
Figure 1 Four groups of mice were studied over an eight week time interval. Group 1 mice were exposed to cigarette smoke for 6 hours per day, 5 days per week. Group 2 mice had the same cigarette exposure as group 1 and additionally had Mycoplasma pneumoniae (Mp) respiratory infection induced at weeks 2 and 6. Group 3 mice only had Mp inoculations at weeks 2 and 6 while group 4 mice had saline inoculation at these time points. *All mice were sacrificed at week 8 for study analyses.
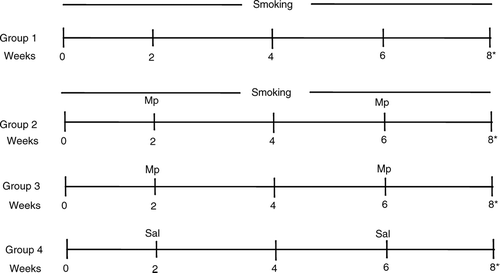
Mice that were exposed to cigarette smoke (2RF4 reference cigarettes, University of Kentucky) were placed in a smoking chamber [Citation[17], Citation[18]] for 6 hours per day, 5 days per week of continual smoking with the total suspended particulate concentration of approximately 75 mg/m3, carbon monoxide concentration of 190 ppm and nicotine concentration of 6 mg/m3.
BAL was performed in one-half of mice (n = 7/group) by using 1 ml of saline with 50 μl used for total white cell count. The remaining BAL was centrifuged for cell cytospin slides for cell differential counts.
One-half of the mice in each group (n = 8) were evaluated for inflammation and structural changes. The lungs were fixed with 4% paraformaldehyde at a hydrostatic pressure of 25 cm H2O for a minimum of 1 hour. The lungs, heart and mediastinal contents were then removed in situ from the chest and stored in 4% paraformaldehyde for 3–4 hours prior to transfer to 70% ethanol to measure total lung volume by displacement, and specific lung volume [Citation[19]]. Thereafter, paraffin embedded H&E stained tissue sections were analyzed for mean linear intercept [Citation[19]], as an index of structural change. The mean linear intercept was randomly measured by dividing the total length of lines (80 to 100 lines/lung) across the lung section by the total number of intercepts.
The number of alveolar attachments in small airways, diameter ≤200 μm, were counted in the outer wall region (smooth muscle to alveolar attachments) without accompanying blood vessels and expressed as number of alveolar attachments per mm of airway epithelial basement membrane [Citation[20]]. At least 5 small airways per mouse were analyzed.
Immunohistochemistry was used to identify neutrophils [clone NIMP-R14, Cell Sciences, Inc., Norwood, MA] and MMP-9 [Chemicon, Temecula, CA] positive cells. Neutrophils in the outer wall of small airways were expressed as number of cells per mm of epithelial basement membrane. MMP-9 positive cells in the alveolar tissue were expressed as volume per surface area of alveolar tissue [Citation[21]]. To measure neutrophil degranulation, the area of NIMP-R14 antibody positively stained extracellular matrix in the outer wall of small airways was measured with an NIH Scion image program. The total area of outer wall was also measured. The percent of outer wall occupied by NIMP-R14 antibody positively stained extracellular matrix (positive area/outer wall area X100) was used to represent the degree of neutrophil degranulation. Elastin was identified using a modified Movat's pentachrome stain and quantified by morphometry with data expressed as volume per surface area of alveolar tissue. MMP-9 mRNA was determined by reverse transcription (RT), followed by real-time PCR [Citation[22]]. MMP-9 mRNA expression levels were obtained using the comparative threshold cycle (CT) method. Zymography was performed to detect MMP-9 protein activity in lung tissue homogenates. Scoring was based on a 0–5 scale.
Normally distributed data are presented as means ± SEM and between group comparisons evaluated by analysis of variance (ANOVA). Non-normally distributed data are expressed as medians with interquartile (25–75%) ranges and the comparisons between groups performed by using the Kruskal–Wallis with Dunn's procedure. A two-tailed p-value < 0.05 is considered statistically significant.
RESULTS
Bronchoalveolar lavage (BAL) cell counts
Groups 1–3 (smoking and/or Mp) had similar and significantly higher total white cell counts compared to control Group 4 (−smoking, −Mp), p = 0.003 (). The two Mp groups (2 and 3) had significantly lower percentage of macrophages and higher lymphocytes than the non-Mp groups (1 and 4), p ≤ 0.009. The percentage of “black” macrophages (those cells engulfing cigarette particulate) were significantly higher in the two smoking groups (1 and 2, p = 0.001). Eosinophils were essentially absent in all groups.
Table 1 Bronchoalveolar lavage cell counts
Morphometric analysis for neutrophils, mean linear intercept and elastin
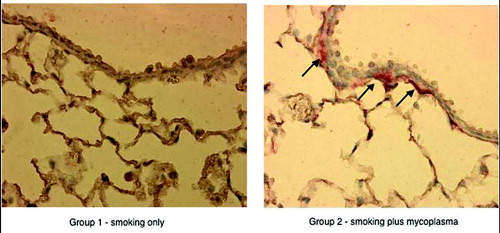
Morphometric analysis for neutrophils in the outer wall (smooth muscle to alveolar attachment) demonstrated that Groups 1 and 2 had higher counts than 3 and 4 (number per mm basement membrane, mean ± SEM: group 1 = 2.2 ± 0.7; group 2 = 1.8 ± 0.6; group 3 = 1.4 ± 0.3; group 4 = 1.2 ± 0.3, p = 0.005. However, group 2 (+smoking, +Mp) had significantly higher neutrophil degranulation of the outer wall area: group 1 = 1.7 ± 0.9%; group 2 = 10.3 ± 4.9%; group 3 = 2.7 ± 1.4%; group 4 = 0.2 ± 0.2%, p = 0.03. demonstrates the neutrophil degranulation in the outer wall of small airways for groups 1 and 2.
Figure 2 There is greater neutrophil degranulation (immunoperoxidase stain, arrows) in the outer wall of a small airway (diameter ≤ 200 μm) for a smoking plus Mp mouse (group 2) compared to a smoking without Mp mouse (group 1).
There were no significant group differences in mean linear intercept (group 1 = 45.8 ± 0.8 μm, group 2 = 43.6 ± 1.8 μm, group 3 = 41.6 ± 1.9 μm, group 4 = 45.2 ± 2.4 μm, overall p = 0.29). Also, the alveolar attachment count was not significantly different (group 1 = 76.8 ± 2.6/mm basement membrane, group 2 = 76.9 ± 3.0, group 3 = 80.8 ± 2.2, group 4 = 77.9 ± 2.3, p = 0.68). The fixed lung volume was significantly increased in groups 1–3 compared to group 4 (p = 0.001), but was similar between groups 1–3 (). The specific lung volume (lung volume/body weight ratio) was significantly greater in both smoking groups (1 and 2) compared to the non-smoking groups (3 and 4), p = 0.001 (). Although, there were no significant group differences found in regard to elastin levels in the alveolar tissue, the +smoking, +Mp group 2 was approximately 20% lower than the other groups (group 1 = 967 ± 108 mm3/mm2, group 2 = 763 ± 71 mm3/mm2, group 3 = 933 ± 155 mm3/mm2, group 4 = 993 ± 145 mm3/mm2).
Table 2 Measurement of fixed and specific lung volume
Matrix metalloproteinase (MMP-9)
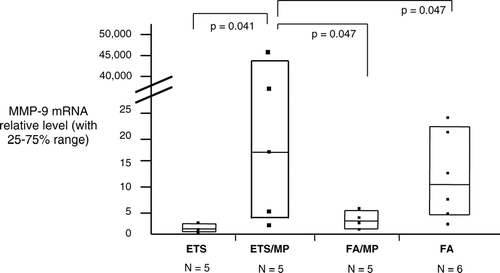
MMP-9 mRNA expression levels were the greatest in group 2 (smoking+, Mp+) (), p < 0.05. Additionally, this group had significantly higher alveolar wall neutrophil staining for MMP-9 protein than the other groups (73 ± 2 mm3/mm2 vs groups 1, 3, and 4, 50 ± 9, 60 ± 12, 25 ± 7, respectively, p = 0.017). MMP-9 protein activity was highest in group 2, 2.7 ± 0.4 compared to group 1, 2.1 ± 0.2; group 3, 2.0 ± 0.7; and group 4, 1.5 ± 0.2; p < 0.05.
Figure 3 Lung tissue relative expression levels of MMP-9 mRNA by real-time RT-PCR. Group 1, smoking only is used as the baseline standard. Group 2, smoking plus Mp has a 10-fold increase in MMP-9 compared to group 1. Group 3 = Mp only and group 4 = saline control. Data expressed are medians with interquartile ranges. Group 2 is significantly increased compared to the other groups. Sample sizes are reduced due to RNA quality variability, only good quality accepted based on housekeeping gene 18sRNA.
DISCUSSION
The results from this relatively short term study demonstrated that Mp respiratory infections appear to interact with cigarette smoke to induce inflammatory changes, perhaps similar to early stages of COPD. The neutrophil, a cell felt to play a major role in the pathogenesis of emphysema [Citation[2]], was significantly increased in the outer wall area, where the alveolar attachments start, in both smoking groups compared to the nonsmoking groups. However, most importantly, neutrophil degranulation was only significantly increased in the smoking plus Mp group in the outer airway wall area. To support that neutrophil degranulation in the outer wall is an active process, we performed terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate-biotin nick end-labeling (TUNEL) staining on lung tissue sections from three mice with ETS + Mp. Although we observed apoptotic cells (e.g., neutrophils) in the alveolar space, we did not see apoptotic cells in small airway outer wall. This suggests that neutrophils in the outer wall are live, and actively degranulate. In concert with this finding, the proteolytic enzyme, MMP-9, expression was ten-fold greater at the mRNA level in the smoking plus Mp group compared to the smoking alone group. Additionally, protein activity in lung homogenates and neutrophil staining for MMP-9 protein was significantly greater in the smoking plus Mp group compared to the other three groups. Other MMPs and elastases need to be evaluated in future studies to develop a clear picture of the involved components.
The histologic index for structural change in a murine model was measured by the mean linear intercept [Citation[19]], which was not increased in any group, but the specific lung volume was significantly greater in both smoking groups compared to the non-smoking groups. With this relatively short duration study of 8 weeks, we did not expect to demonstrate histologic differences of emphysema between different groups of mice, as it takes at least 6 months [Citation[19]] to induce these changes in a cigarette smoking murine model. We were interested in finding if the combination of cigarette smoking and Mp respiratory infection, in the short term, would induce potential changes antecedent to the development of histologic structural changes. Now that this model has been developed, longer-term studies can be initiated to determine if emphysema develops more rapidly and to a greater extent with the combination of cigarette smoke and infection.
Of interest, Choe and colleagues, recently reported on a rat model of emphysema induced by methylprednisolone [Citation[23]]. In this model, MMP-9 was the proteolytic enzyme that was felt to be responsible for the induction of emphysema. This model is similar to ours in regard to MMP-9 as an important proteolytic enzyme. Multiple other studies also support this link [Citation[24], Citation[25], Citation[26], Citation[27], Citation[28], Citation[29]].
Not all smokers develop COPD and it appears that only approximately 20% are affected [Citation[3]]. The question arises if other factors besides or in addition to genetic determinants are at the source of this finding. Childhood infection is considered a risk factor for the development of COPD in adult life [Citation[3]]. Thus, either the infecting agent is carried on into adulthood or airway anatomic changes occur which are amplified by cigarette smoke. Proteins expressed from adenovirus DNA persisted in the lungs following childhood infections and are capable of amplifying the airway inflammation produced by cigarette smoke [Citation[30], Citation[31]]. The atypical bacteria, Mp and Cp have been reported to be linked to chronic COPD [Citation[14], Citation[32], Citation[33], Citation[34], Citation[35]]. Our animal model suggests that at least Mp could interact with cigarette smoke to activate the cells and proteolytic enzymes that may lead to the development of chronic lung disease.
A model to study the interaction of cigarette smoke and atypical bacterial respiratory infection to induce inflammatory responses similar to those found in lung diseases is described in this report. Longer duration studies can now be undertaken to determine if this model more rapidly and/or to a greater extent, leads to structural changes of lung disease.
ACKNOWLEDGMENTS
The authors wish to thank Krista Campbell, Michael Goldsmith and Shanie McCarty for their technical assistance, and Gaynelle Ivandick and Barbara Ewing-Chow for manuscript formatting.
Supported by Colorado Tobacco Research Program.
REFERENCES
- Lopez A D, Murray C C. The global burden of disease, 1990–2020. Nat Med 1998; 4: 1241–1243, [PUBMED], [INFOTRIEVE], [CSA]
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003; 22(4)672–688, [PUBMED], [INFOTRIEVE], [CSA]
- The National Awareness Panel. Guidelines for the early detection and management of COPD. J Respir Dis 2000; 21(Suppl. 9)5–21, [CSA]
- Pietila M P, Thomas C F. Inflammation and infection in exacerbations of chronic obstructive pulmonary disease. Semin Respir Infect 2003; 18: 9–16, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Emre U, Roblin P M, Gelling M, Dumornay W, Rao M, Hammerschlag M R, Schacter J. The association of Chlamydia pneumoniae infection and reactive airway disease in children. Arch Pediatr Adolesc Med 1994; 148(7)727–732, [PUBMED], [INFOTRIEVE], [CSA]
- Hahn D L, Bukstein D, Luskin A, Zeitz H. Evidence for Chlamydia pneumoniae infection in steroid-dependent asthma. Ann Allergy Asthma Immunol 1998; 80(1)45–49, [PUBMED], [INFOTRIEVE], [CSA]
- Hahn D L, Dodge R W, Golubjatnikov R. Association of Chlamydia pneumoniae (strain TWAR) infection with wheezing, asthmatic bronchitis, and adult-onset asthma. JAMA 1991; 266(2)225–230, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Gil J C, Cedillo R L, Mayagoitia B G, Paz M D. Isolation of Mycoplasma pneumoniae from asthmatic patients. Ann Allergy 1993; 70(1)23–25, [PUBMED], [INFOTRIEVE], [CSA]
- Yano T, Ichikawa Y, Komatu S, Arai S, Oizumi K. Association of Mycoplasma pneumoniae antigen with initial onset of bronchial asthma. Am J Respir Crit Care Med 1994; 149(5)1348–1353, [PUBMED], [INFOTRIEVE], [CSA]
- Kraft M, Cassell G H, Henson J E, Watson H, Williamson J, Marmion B P, et al. Detection of Mycoplasma pneumoniae in the airways of adults with chronic asthma. Am J Respir Crit Care Med 1998; 158(3)998–1001, [PUBMED], [INFOTRIEVE], [CSA]
- Martin R J, Kraft M, Chu H W, Berns E A, Cassell G H. A link between chronic asthma and chronic infection. J Allergy Clin Immunol 2001; 107(4)595–601, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Kraft M, Cassell G H, Pak J, Martin R J. Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma: effect of clarithromycin. Chest 2002; 121(6)1782–1788, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Chu H W, Kraft M, Krause J E, Rex M D, Martin R J. Substance P and its receptor neurokinin 1 expression in asthmatic airways. J Allergy Clin Immunol 2000; 106(4)713–722, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Von Hertzen L, Alakarppa H, Koskinen R, Liippo K, Surcel H M, Leinonen M, et al. Chlamydia pneumoniae infection in patients with chronic obstructive pulmonary disease. Epidemiol Infect 1997; 118(2)155–164, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Martin R J, Chu H W, Honour J M, Harbeck R J. Airway inflammation and bronchial hyperresponsiveness after Mycoplasma pneumoniae infection in a murine model. Am J Respir Cell Mol Biol 2001; 24(5)577–582, [PUBMED], [INFOTRIEVE], [CSA]
- Chu H W, Honour J M, Rawlinson C A, Harbeck R J, Martin R J. Effects of respiratory Mycoplasma pneumoniae infection on allergen-induced bronchial hyperresponsiveness and lung inflammation in mice. Infect Immun 2003; 71(3)1520–1526, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Teague S V, Pinkerton K E, Goldsmith M, Gebremichael A, Chang S, Jenkins R A, et al. A sidestream cigarette smoking generator and exposure system for environmental tobacco smoke studies. J Inhal Toxicol 1994; 6: 79–93, [CSA]
- Smith K R, Uyeminami D L, Kodavanti U P, Crapo J D, Chang L Y, Pinkerton K E. Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidant. Free Radic Biol Med 2002; 33(8)1106–1114, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Takubo Y, Guerassimov A, Ghezzo H, Triantafillopoulos A, Bates J H, Hoidal J R, et al. Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J Respir Crit Care Med 2002; 166(12 Pt 1)1596–1603, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Elliot J G, Carroll N G, James A L, Robinson P J. Airway alveolar attachment points and exposure to cigarette smoke in utero. Am J Respir Crit Care Med 2003; 167(1)45–49, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Howard C V, Reed M G. Unbiased sterology: three-dimensional measurement in microscopy. Springer Verlag, New York 1998
- Wenzel S E, Trudeau J B, Barnes S, Zhou X, Cundall M, Westcott J Y, et al. TGF-beta and IL-13 synergistically increase eotaxin-1 production in human airway fibroblasts. J Immunol 2002; 169(8)4613–4619, [PUBMED], [INFOTRIEVE], [CSA]
- Choe K H, Taraseviciene-Stewart L, Scerbavicius R, Gera L, Tuder R M, Voelkel N F. Methylprednisolone causes matrix metalloproteinase-dependent emphysema in adult rats. Am J Respir Crit Care Med 2003; 167(11)1516–1521, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Culpitt S V, Maziak W, Loukidis S, Nightingale J A, Matthews J L, Barnes P J. Effect of high dose inhaled steroid on cells, cytokines, and proteases in induced sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 160(5 Pt 1)1635–1639, [PUBMED], [INFOTRIEVE], [CSA]
- Shapiro S D, Senior R M. Matrix metalloproteinases. Matrix degradation and more. Am J Respir Cell Mol Biol 1999; 20(6)1100–1102, [PUBMED], [INFOTRIEVE], [CSA]
- Finlay G A, O'Driscoll L R, Russell K J, D'Arcy E M, Masterson J B, Fitz Gerald M X, et al. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med 1997; 156(1)240–247, [PUBMED], [INFOTRIEVE], [CSA]
- Betsuyaku T, Nishimura M, Takeyabu K, Tanino M, Venge P, Xu S, et al. Neutrophil granule proteins in bronchoalveolar lavage fluid from subjects with subclinical emphysema. Am J Respir Crit Care Med 1999; 159(6)1985–1991, [PUBMED], [INFOTRIEVE], [CSA]
- Ohnishi K, Takagi M, Kurokawa Y, Satomi S, Konttinen Y T. Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysema. Lab Invest 1998; 78(9)1077–1087, [PUBMED], [INFOTRIEVE], [CSA]
- Russell R E, Culpitt S V, DeMatos C, Donnelly L, Smith M, Wiggins J, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2002; 26(5)602–609, [PUBMED], [INFOTRIEVE], [CSA]
- Elliott W M, Hayashi S, Hogg J C. Immunodetection of adenoviral E1A proteins in human lung tissue. Am J Respir Cell Mol Biol 1995; 12(6)642–648, [PUBMED], [INFOTRIEVE], [CSA]
- Matsuse T, Hayashi S, Kuwano K, Keunecke H, Jefferies W A, Hogg J C. Latent adenoviral infection in the pathogenesis of chronic airways obstruction. Am Rev Respir Dis 1992; 146(1)177–184, [PUBMED], [INFOTRIEVE], [CSA]
- Cherry J D, Taylor-Robinson D, Willers H, Stenhouse A C. A search for mycoplasma infections in patients with chronic bronchitis. Thorax 1971; 26(1)62–67, [PUBMED], [INFOTRIEVE], [CSA]
- Clementsen P, Farholt S, Permin H, Lange P, Stahl Skov P, Norn S. Chlamydia pneumoniae and chronic obstructive pulmonary disease: bronchial biopsies (PCR and culture) and specific serum antibodies in patients and controls. Inflamm Res 2000; 49(Suppl 1)S37–38, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Theegarten D, Mogileuski G, Anhenn O, Stamatis G, Jaeschock R, Morgenroth K. The role of chlamydia in the pathogenesis of pulmonary emphysema. Virchows Arch 2000; 437: 190–193, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Lambert H P. Antibody to Mycoplasma pneumoniae in normal subjects and in patients with chronic bronchitis. J Hyg Camb 1968; 66: 185–189, [PUBMED], [INFOTRIEVE], [CSA]