Abstract
Augmentation therapy with a plasma derived α1-Proteinase Inhibitor (α1-PI) has been demonstrated to be effective in restoring serum Alpha1-antitrypsin (AAT)* levels in individuals with AAT Deficiency (note: α1 PI and AAT are synonymous). The objective of this study was to demonstrate that the steady-state trough serum α1-PI levels, achieved by a new plasma derived α1-PI (Zemaira, study drug, ZLB Behring LLC, King of Prussia, Pennsylvania, USA), were bioequivalent to those achieved by the currently available α1-PI therapy, Prolastin (control drug, Bayer Corporation, Berkeley, California, USA), and maintained weekly trough serum antigenic α1-PI levels above the protective threshold of 11 μ M. This multi-center, controlled study randomized a total of 44 subjects to receive either study or control drug for a 10-week double-blind phase. The control group was then crossed over to receive the study drug for the remainder of the study (14 weeks). The difference in mean trough serum antigenic α1-PI level between the treatment groups was 1.45 μ M (90% CI-2.77, −0.13), signifying bioequivalence. The mean trough serum antigenic α1-PI level in the study drug group was greater than the therapeutic threshold of 11 μ M, achieving a level of 17.7 μ M during the steady-state period. Treatment-related adverse events (AEs) were seen in 7% and 21% of study and control drug treated subjects, respectively. No documented viral transmission occurred. These results demonstrate that the new plasma derived α1-PI (Zemaira) is bioequivalent to the currently available product Prolastin, is well tolerated, and safe with respect to the risk of viral transmission.
INTRODUCTION
Alpha1-antitrypsin Deficiency (hereafter referred to as α1-PI Deficiency) is one of the most common chronic, autosomally inherited disorders which may pre-dispose to life-threatening lung and liver disease. It is characterized by reduced serum levels of α1-PI, which is often associated with progressive and severe emphysema that becomes clinically apparent by the third to fourth decade of life. Less commonly, low blood levels of α1-PI are associated with liver disease and liver cirrhosis [Citation[1], Citation[2], Citation[3], Citation[4]]. It is of note that currently α1-PI Deficiency is significantly under-diagnosed [Citation[5]]. Approximately 100 genetic variants of α1-PI have been identified electrophoretically, of which only some are associated with clinical disease [Citation[4], Citation[6]]. Ninety-five percent of clinically affected α 1-PI deficient individuals are of the severe PiZZ genotype. Although the deficiency is found equally in males and females, there is a slight predominance of males with emphysema.
Approximately 45% of α 1-PI deficient patients also exhibit an asthmatic component to their lung disease, as evidenced by symptoms and/or signs of bronchial hyperreactivity [Citation[6]]. In addition, pulmonary infections, including pneumonia and acute bronchitis, are common and contribute significantly to the morbidity of the disease [Citation[7]]. α1-PI provides more than 90% of the lower respiratory tract's inhibition of neutrophil elastase (NE), a protease capable of destroying alveolar walls [Citation[8]]. Conditions that increase neutrophil accumulation and activation in the lung, such as respiratory infection and smoking, will in turn increase levels of NE. It is believed that this increase in uninhibited NE activity is the pathogenesis behind emphysema development in α1-PI deficient patients [Citation[8]]. Therefore, theoretically the most direct approach to therapy for α1-PI deficiency in patients with emphysema is to replace the missing protease inhibitor and thus re-establish anti-neutrophil elastase protection of the lower respiratory tract.
For almost 2 decades, the maintenance of blood levels of antigenic α1-PI above 11 μ M has been presumed to provide therapeutically relevant anti-neutrophil elastase protection [Citation[4]]. Previous research with the then only licensed α1-PI (Prolastin), has demonstrated that chronic α1-PI replacement therapy is safe and effective in raising serum α1-PI levels to the target levels while also significantly increasing α1-PI levels in lower lung epithelial lining fluid (ELF) [9, 10]. In addition, the National Heart Lung and Blood Institute registry study [Citation[11]] and two additional clinical studies [Citation[9], Citation[12]] have statistically suggested that long-term treatment with augmentation therapy in patients with α1-PI Deficiency with Prolastin can reduce the rate of decline of the forced expiratory volume in 1 second (FEV1) as compared to historical or concurrent controls. In these studies, evidence of improved survival was also demonstrated, especially in patients with FEV1 values between 35% and 49% of predicted.
Based upon this rationale and evidence, augmentation therapy was recommended by the American Thoracic Society for individuals with established airflow obstruction from α1-PI Deficiency [Citation[13]]. Unfortunately, the supply of existing α1-PI products has not been able to meet the demands of the patient population, requiring the development and registration of newer α1-PI products.
Zemaira is a highly purified, lyophilized intravenous α1-PI product derived from human plasma, which undergoes dual viral inactivation/removal processes. A recent pilot study has suggested that the different manufacturing processes for each commercially available α1-PI preparation may affect their purity, isoform composition and non-therapeutic protein content [Citation[14]].
The randomized and controlled study presented here is in line with the recommendations of the Blood Products Advisory Committee (BPAC) meeting of 19 June 1998. This served as an important forum to define the requirements for the registration of α1-PI products. The BPAC members voted to accept “the maintenance of a plasma level of 11 μ M α1-PI, in conjunction with the demonstration of an appropriately-defined increment in ELF α1-PI/neutrophil elastase-related analyte levels, as sufficient for demonstrating clinical evidence of efficacy of intravenously-administered alpha1-PI products in pivotal (phase III) studies.” The present study tested the capability of Zemaira to achieve the protective serum trough level of > 11 μ M, as well as to raise the concentration of α1-PI in the lower lung.
MATERIALS AND METHODS
Study drug
Zemaira is a highly purified, lyophilized intravenous α1-PI product derived from large pools of human plasma. Each donor unit of plasma has been tested and found non-reactive by serology for hepatitis B surface antigen (HBsAg); negative for antibodies to human immunodeficiency virus (HIV-1/2) and to hepatitis C virus (HCV); and to have an alanine aminotransferase (ALT) level of no more than twice the upper limit of normal. The plasma used in the manufacture of this product has also been tested and found negative for HBV, HCV, and HIV-1 using polymerase chain reaction (PCR) technology.
To provide additional safeguards in an effort to reduce potential exposure to viral agents that may be present in human plasma, two viral reduction steps, pasteurization (60°C for 10 hours in an aqueous solution with stabilizers) and dual ultrafiltration, are included in the manufacturing process. The specific activity is ≥ 0.7 mg of functional α1-PI per milligram of total protein with functional activity determined by capacity to neutralize human NE. The purity is ≥90% α1-PI. The study drug and control drug were analyzed to establish antigenic and functional α1-PI, total protein (mg/vial) and specific activity.
Study design
This was a multi-center, randomized and controlled study. All sites received institutional review board approval to conduct the study. Following informed consent, subjects were randomized in a 2:1 ratio to receive either Zemaira (study drug) or Prolastin (control drug) for a 10-week double-blind phase. The control group was then crossed over to an open study phase where all subjects received Zemaira for the remainder of the study (14 weeks). α1-PI treatment with study drug or control drug was administered by weekly intravenous infusion at a dose of 60 mg/kg functionally active α1-PI. All subjects were followed for a total of 24 weeks according to the assessment schedule in . In a subset of 15 subjects with FEV1 ≥50% (10 in the study drug group and 5 in the control drug group) bronchoalveolar lavage (BAL) was performed to evaluate levels of α 1-PI in the epithelial lining fluid (ELF). This was carried out in 2 centers and performed on each of these 15 subjects at baseline and 7 ± 1 days after the last blinded infusion.
Table 1 Assessment schedule
As the control drug has an infusion volume and required infusion time which is twice that of the study drug, along with a slightly different visual appearance, blinding was achieved through the following methods. The α1-PI infusion was divided into 2 opaque intravenous bags in the hospital pharmacy and normal saline was added to the second bag to equalize the infusion volumes if the subject was randomized to the study drug. In the home setting, the α1-PI infusion was in plain packaging and administered by an individually assigned experienced infusion nurse with no prior experience of α 1-PI product infusion.
The primary objectives of this study were to demonstrate that the steady-state trough serum α1-PI levels achieved by weekly infusion of the study drug were bioequivalent (i.e., not inferior) to those achieved by weekly infusion of the control drug. In addition, weekly trough serum antigenic α 1-PI levels were to be maintained above the protective threshold of 11 μ M. The secondary objectives were to compare the safety and tolerability of both drugs and to demonstrate increases of the antigenic α1-PI levels in the ELF of the lower lung. In addition the following exploratory analyses were to be performed: the weekly trough serum functionally active α1-PI levels during steady-state period (Week 7–Week 24) to demonstrate no significant downward trend and the serum functional α1-PI levels expressed as a percentage of antigenic levels.
Patient population
Subject inclusion criteria included: age 18 to 70 years; willingness to sign informed consent and participate in the 6-month study; males, post-menopausal females, and non-pregnant, non-lactating females who were using reliable contraceptive methods and had a documented negative pregnancy test; diagnosis of α1-PI Deficiency (serum α1-PI level < 11 μ M) and the following genotypes: PiZZ, PiZ null, or Pi null null; clinical evidence of emphysema as determined by thin section computerized tomography scan; plus at least one of the following: chest X-ray evidence of lung destruction, abnormal lung function studies (FEV1 ≤80% of predicted), FEV1 decline of ≥35 mL/year or FEV1 ≥18% of predicted; no treatment with an α1-PI concentrate or plasma-derived product for at least 4 weeks prior to the screening visit, with the exception of subjects who had participated in a previous clinical trial with the study drug.
Study exclusion criteria included: significant concomitant disease; current evidence of alcohol or illicit drug use; a history of allergy to human plasma derived products or mannitol; selective IgA deficiency; current smoker; administration of any other experimental new drug or participation in a study of a marketed product within 3 months and evidence of prior viral infection with HIV, HBV and HCV. Evidence of prior hepatitis A virus (HAV) infection by reactive serology was not to be considered a basis for exclusion.
Analytical methods
Assays of serum α1-PI levels and ELF analytes were performed by Mark Brantly (Alpha1-Antitrypsin Genetics Laboratory, University of Florida, Gainesville, FL). Serum antigenic α1-PI levels were measured by nephelometry (Behring Nephelometer Analyzer, Behring Diagnostics, Westwood, MA) using a true laboratory standard (in-house standard serum calibrated against the M1(V213) standard). Serum functional α1-PI levels were measured by an anti-neutrophil elastase capacity assay utilizing the same standard.
Statistical methods
For the primary biochemical efficacy analysis the mean steady-state antigenic trough α1-PI serum level from Week 7 to Week 11 was calculated for each subject. The bioequivalence hypothesis H01 (difference of mean steady state α1-PI serum levels between study drug and comparator drug ≤ −3 μM) was tested using a 2-group, 1-sided t-test at a level of α = 5%. A 2-sided 90% confidence interval was provided for the difference of mean steady-state trough antigenic α1-PI serum levels between treatment groups. If the lower limit of the confidence interval was > −3 μM, hypothesis H01 was rejected and bioequivalence was concluded. The second hypothesis H02 (mean α1-PI steady state trough serum levels in study drug group ≤11 μM) was tested using a 1-group, 1-sided t-test at a level of α = 5%. A 2-sided 90% confidence interval was provided for the mean steady-state trough antigenic α 1-PI serum level in the study drug group. If the lower limit of the confidence interval was > 11 μ M, hypothesis H02 was rejected [Citation[15], Citation[16]].
With respect to secondary endpoints, safety data analysis, in the form of descriptive analyses only, was based on all subjects who had received at least 1 dose of study medication. This included the evaluation of all adverse events (AEs), especially serious and severe AEs, judged by the investigators as either related or unrelated to treatment with the study medications. Serious adverse events are those that are fatal, life threatening, permanently disabling, or result in, or prolong, hospitalization. Otherwise, adverse events were rated as mild, moderate or severe in intensity. In the subset of 15 subjects who underwent bronchoscopy, ELF obtained by BAL at Week 11 was compared to baseline. Descriptive statistics of BAL results were presented for each time point by treatment group. Statistical analyses were performed for antigenic levels of α1-PI. The difference between Week 11 and baseline was evaluated and summarized by means of a 2-sided 90% confidence interval for the mean change based on a t-distribution. For the between group comparison, a 2-sided 90% confidence interval was provided for the difference of mean changes in the ELF between treatment groups.
To evaluate the possibility of a significant downward trend in the weekly trough antigenic α1-PI serum levels, the slope of a linear regression line on the weekly trough serum levels in steady-state (Week 7 to Week 24) was estimated for each subject and a 90% confidence interval was calculated for the mean slope. If the lower limit of the confidence interval was > −0.1 μ M/week and the upper limit was > 0.0 μ M/week, it was concluded that there was no significant downward trend in the steady-state serum levels.
RESULTS
A total of 44 subjects were enrolled in the study; 30 subjects were randomized to the Zemaira (study drug) treatment group and 14 subjects were randomized to the Prolastin (control drug) treatment group. Nine subjects had participated in a previous clinical trial with the study drug. Forty-three subjects completed the blind phase; 1 subject died in the final week of the blind phase of the control drug treatment arm. Forty-two subjects completed the open phase; 1 subject voluntarily withdrew from the study and 41 subjects completed the viral safety follow-up phase. A subset of 15 subjects (10 in the study drug group and 5 in the control drug group) underwent bronchoscopy and BAL. The groups were similar in characteristics () with the exception of the study drug group being comprised of more males (70% vs. 50%, not statistically significant), and were younger (50 vs. 56 years, p = 0.0095).
Table 2 Demographic and baseline characteristics
Efficacy results
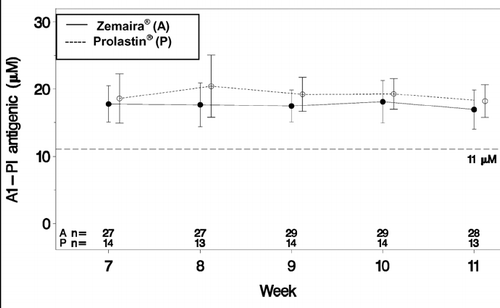
The primary objectives were met in this study. The mean trough serum antigenic α1-PI levels of the two groups were within 3 μM (actual value 1.45 μM) with the lower limit of the 90% confidence interval being −2.77 μ M (). These results enable rejection of the null hypothesis and support the demonstration of bioequivalence of the two drugs studied. The difference between the two groups was statistically significant (p = 0.0722) at a 2-sided α level of 10% (the upper limit of the 90% CI was −0.13, i.e., less than 0). The mean trough serum antigenic α1-PI level in the study drug group from Week 7 to Week 11 was 17.7 μ M (90% CI: 16.9, 18.5), greater than the therapeutic threshold of 11 μ M.
Figure 1 Mean ± SD of trough serum antigenic α1-PI during weeks 7–11.
Within each treatment group, ELF levels of antigenic α1-PI demonstrated a statistically significant increase from baseline to Week 11; however, no significant difference was seen between the two products ().
Table 3 Summary of ELF antigenic α1-PI results at baseline and week 11
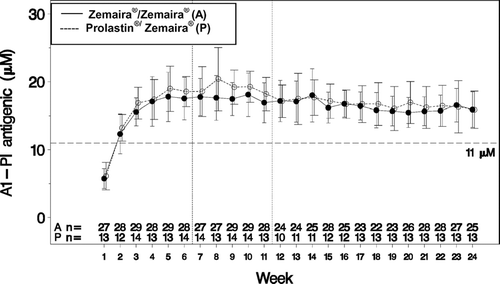
Examination of the steady-state trough serum antigenic α1-PI level between Week 7 to Week 24 revealed a downward trend as determined by the mean slope of −0.13 μM/week; 90% CI −0.18 to −0.09 (). Serum levels at steady state of functionally active α1-PI remained stable during the entire treatment period (slope = 0.00, 90% CI −0.03 to 0.03).
Figure 2 Mean ± SD of trough serum antigenic α1-PI over entire study.
Safety results
No serious or severe AEs were judged by the investigators to be related to either the study drug or control drug. In the blinded phase of the study, 90% of subjects in the study drug group and 100% of subjects in the control drug group reported at least 1 treatment-emergent AE. The most frequently reported AEs were headache, sinusitis, upper respiratory infection, increased cough, sore throat, fatigue, fever, bronchitis, bronchospasm, and flushing. No relevant differences were detected between the treatment groups. The investigators judged treatment-emergent non-serious AEs as related to the medication in 2 (7%) subjects in the study drug group and in 3 (21%) subjects in the control drug group (). The related AEs in the study drug group included injection site pain and paresthesia (cold arms), lasting 1 minute and two hours, respectively. Both were rated as being of mild severity as judged by the investigator. The 3 related AEs reported in the control drug group were: an asymptomatic transient seroconversion of hepatitis B core antibody coded by the investigator as viral hepatitis B, vasodilatation/facial flushing, and a parvovirus B19 infection. The asymptomatic transient seroconversion for hepatitis B consisted of IgG core antibody positivity at Week 11, but reverted to negative at the 6-month follow-up. Hepatitis core IgM antibody as well as liver transaminases remained negative during the entire time of the subject's participation in the study. The subject had also received hepatitis B vaccine in the past. This AE was thought to represent a passive transfer of antibodies against hepatitis B core antigen or a laboratory artifact. The parvovirus B19 related AE was seen in a subject who had no antibodies against parvovirus B19 prior to start of treatment, but became positive for IgG and IgM after 10 weeks of treatment with the control drug. The investigator noted that the AE could be due to a recent current community infection and it was judged to be of mild severity.
Table 4 Summary of adverse events during 10-week blinded study period. Rates per infusion presented in parentheses
There was a statistically non-significant lower incidence of serious AEs and of the overall AE rate in the study drug group compared to the control group during the blinded treatment phase (). One death occurred during the blinded phase of the study in a 72-year-old female with a medical history of chronic obstructive pulmonary disease (COPD) and osteoporosis, assigned to the control drug treatment group. The cause of death was listed as respiratory arrest due to COPD, which the investigator considered to be unrelated to study medication. No other subject discontinued the study due to an AE.
In the open phase, 1 additional AE was considered by the investigator to be related to the study drug. One subject experienced mild fatigue 1 day after the second open-label treatment, which continued for 7 days. In all subjects there were no clinically relevant changes in clinical laboratory values, pulmonary function tests, vital signs, physical examinations, EKGs, and chest X-rays which were judged by the investigator to be related to the study drug or control drug. No subjects developed detectable antibodies to α1-PI.
Viral seroconversion
Subjects were monitored for the presence of antibodies to HIV and markers for viral hepatitis. Subjects who had been negative for HBsAg at screening were vaccinated against HBV. Subjects treated with the study drug were tested 6 months after the end of treatment for HAV, HBV, HCV, HIV, and parvovirus B19 and no evidence of viral transmission was observed.
Study and control drug analysis
Analysis via an antigenic assay indicated a significantly higher level of antigenic α1-PI per unit of functionally active α1-PI in the control drug preparation, demonstrating a significant amount of inactive α1-PI protein (). The potency of the products, as determined by functional assay was comparable and correlated well with the labeled amount. The mean specific activity of the study drug was 1.07, higher than that of the control drug (0.77).
Table 5 Study drug analysis
DISCUSSION
This was a randomized, controlled, multi-center study, which compared the biochemical efficacy, safety, and tolerability of Zemaira, a newly developed plasma derived α1-PI, with that of Prolastin, in subjects with α1-PI Deficiency and emphysema. The primary endpoint was a comparison of the mean steady-state trough serum antigenic α1-PI levels between the treatment groups, with the aim to demonstrate bioequivalence. The design of this study was comparable to another bioequivalence study comparing a solvent-detergent treated plasma derived α1-PI from another manufacturer, with that of Prolastin [Citation[17]].
Based on previous clinical studies and consistent with the terminal half-life of approximately 5 days, all subjects were expected to have achieved steady-state trough serum α1-PI levels after 6 doses of study treatment (i.e., 42 days after the first dose). The comparison was therefore conducted upon α1-PI levels achieved between Week 7 and 11. In addition the study design addressed recommendations made to the FDA by its Blood Products Advisory Committee. Therefore the study aimed to demonstrate the maintenance of a plasma level of 11 μ M α1-PI, in conjunction with the demonstration of an appropriately defined increment in ELF α1-PI analyte levels.
All study subjects were diagnosed with α1-PI Deficiency of PiZZ genotype, and all showed evidence of emphysema on a CT scan performed prior to randomization. The two treatment groups were well matched in terms of demographics, disease characteristics and medical history.
The study results demonstrate the bioequivalence of Zemaira (study drug) and Prolastin (control drug), as the mean trough serum antigenic α1-PI levels of the two groups, from Week 7 to Week 11, were within 3 μ M. Of greater clinical relevance was the mean level of serum antigenic α1-PI achieved during this period, which was significantly greater than the accepted therapeutic threshold of 11 μ M. These results also demonstrate that the treatment protocol of weekly infusions of 60 mg/kg of functionally active α1-PI delivers the therapeutically relevant anti-neutrophil elastase protection. The implications of the observed negative slope of trough serum levels are uncertain.
Although the trough serum antigenic α1-PI levels in the study drug group were not inferior to trough levels achieved after treatment with the control drug, thus fulfilling the primary objective, the difference between the two groups was nevertheless statistically significant. In order to investigate this difference, the study and control drugs were analyzed for antigenic and functionally active α 1-PI levels. This is relevant, as the dose of study and control medication had been based on the labeled potency, which is determined by a functional assay and was accurate for both products. However, the primary endpoint of this study was the measurement of the serum antigenic level of α1-PI achieved. Despite both treatment arms having received the same amount of functionally active α 1-PI, analysis revealed a significantly higher level of antigenic α1-PI per unit of functionally active α1-PI in the control drug compared with the study drug. The antigenic assay measures immunoreactive α1-PI, and is not necessarily an indicator of potency of α 1-PI as determined by anti-neutrophil elastase capacity. Due to the identified product differences and the dosing regimen, which was based on the functional α1-PI assay, the subjects had received relatively larger amounts of antigenic α1-PI in the control drug group, which may have contributed to the higher antigenic α1-PI levels observed in the serum. Within each group, ELF levels of antigenic α1-PI increased from baseline to Week 11, providing direct evidence that intravenous administration of α1-PI delivers α1-PI to the target site in the lung, and supporting the hypothesis that intravenous administration of α1-PI confers anti-neutrophil elastase protection to the lung. The results seen were also in line with the historical data reported by Wewers et al. [Citation[10]].
The study drug was well tolerated. No statistically significant differences in AE rates were seen between groups in the blinded phase; however, there were a lower number of both total reported and treatment-related AEs in the study drug group compared with the control drug group. One AE of particular interest and judged to be possibly treatment related, was the parvovirus B19 seroconversion in a patient who had received the control drug. In this subject no clinical symptoms of a parvovirus infection were reported and the source of infection was most likely community acquired, as documented by the investigator. Parvovirus B19 is a non-enveloped virus, which in turn makes it either resistant or partially resistant to some viral inactivation techniques. Pasteurization has documented efficacy against parvovirus B19, supporting the community-acquired theory of transmission in this subject [Citation[18], Citation[19]].
In summary, the primary and secondary objectives in this study were achieved demonstrating bioequivalence and comparability of Zemaira to Prolastin in treating patients with α1-PI Deficiency and emphysema. This means that Zemaira is a viable treatment option in α 1-PI Deficient patients. This is especially important when considering the current ongoing screening efforts and the percentage of under-diagnosed patients in the population.
This research was fully funded by ZLB Behring LLC.
REFERENCES
- Eriksson S. Pulmonary emphysema and alpha1-antitrypsin deficiency. Acta Med Scand 1964; 175(2)197–205, [PUBMED], [INFOTRIEVE], [CSA]
- Eriksson S. Studies in alpha1-antitrypsin deficiency. Acta Med Scand (Suppl) 1965; 432: 1–85, [CSA]
- Morse J O. Alpha1-antitrypsin deficiency. NEJM 1978; 299: 1045–8, 1099-1105[PUBMED], [INFOTRIEVE], [CSA]
- Crystal R G. α 1-antitrypsin deficiency, emphysema, and liver disease. J Clin Invest 1990; 85: 1343–1352, [PUBMED], [INFOTRIEVE], [CSA]
- Stoller J K. Clinical features and natural history of severe α1-antitrypsin deficiency. Chest 1997; 111: 123S–128S, [PUBMED], [INFOTRIEVE], [CSA]
- World Health Organization. Alpha-1-antitrypsin deficiency. Report of WHO Meeting, World Health Organization, GenevaSwitzerland, March, 18–201996
- Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest 2000; 118(5)1480–1485, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Gadek J E, Fells G A, Zimmerman R L, Rennard S I, Crystal R G. Antielastases of the human alveolar structures; implications for the protease-antiprotease theory of emphysema. J Clin Invest 1981; 68: 889–898, [PUBMED], [INFOTRIEVE], [CSA]
- Wencker M, Banik N, Buhl R, Seidel R, Konietzko N. Long-term treatment of alpha-1-antitrypsin deficiency-related pulmonary emphysema with human alpha-1-antitrypsin. Eur Resp J 1998; 11: 428–433, [CROSSREF], [CSA]
- Wewers M D, Casolaro M A, Sellers S E, Swayze S C, McPhaul K M, Wittes J T, Crystal R G. Replacement therapy for alpha1-antitrypsin deficiency associated with emphysema. NEJM 1987; 316: 1055–1062, [PUBMED], [INFOTRIEVE], [CSA]
- Alpha1-Antitrypsin Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Resp Crit Care Med 1998; 158: 49–59, [CSA]
- Seersholm N. Does alpha-1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha-1-antitrypsin deficiency?. Eur Resp J 1997; 10(10)2260–2263, [CROSSREF], [CSA]
- American Thoracic Society/European Respiratory Society Statement. Standards for the Diagnosis and Management of Individuals with Alpha-1 Antitrypsin Deficiency. Am J Resp Crit Care Med 2003; 168: 818–900, [CROSSREF], [CSA]
- Cowden D I, Fisher G E, Weeks R L. A pilot study comparing the purity, functionality and isoform composition of alpha-1-proteinase inhibitor (human) products. Curr Med Res Opin 2005; 21(6)877–883, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Morikawa T, Yoshida M A. Useful testing strategy in phase iii trials: combined test of superiority and test of equivalence. J Biopharm Stat 1995; 5(3)297–306, [PUBMED], [INFOTRIEVE], [CSA]
- Dunnet C W, Gent M. An alternative to the use of two-sided tests in clinical trials. Stat Med 1996; 15: 1729–1738, [CROSSREF], [CSA]
- Stoller J K, Rouhani F, Brantly M, Shahi S, Dweik R A, Stocks J M, Clausen J, Campbell E, Norton F. Biochemical efficacy and safety of a new pooled human plasma α 1-antitrypsin, respitin. Chest 2002; 122: 66–74, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Bernhardt D, Schäfer W, Mehdi S, Nowak T, Weimer T, Gröner A. Advances in Transfusion Safety. Paul-Ehrlich-Institute, LangenGermany June, 2001, 7–8
- Gröner A, Nowak T, Römisch J. Purity, activity and virus safety of a pasteurized antithrombin concentrate. Semin Thromb Hemost 2002; 28(Suppl. 1)79–85, [CSA]