Abstract
A 26-year-old white male, lifelong non-smoker presented with a history of increased shortness of breath, for approximately 1 year. He had a history of welding aluminum parts. He had evidence of partially reversible reactive airways disease with a non obstructive component as well. VATS biopsy revealed evidence of airway and parenchymal inflammation consistent with aluminum pneumoconiosis. Approximately 5–10% of COPD is attributable to non-smoking causes including occupational exposures. There are studies to suggest that the persistence of aluminum particulate may cause ongoing inflammation despite removal from exposure. It is possible that the persistence of particulate matter from tobacco smoke remaining in the lung may contribute to the persistent inflammatory response found in former smokers. Further study is required to examine the importance of this potential inflammatory mechanism both in occupationally exposed and in cigarette smokers. Reduction of certain particulate components of cigarette smoke may have implications for prevention of disease or at least disease progression in some COPD patients.
HISTORY
Approximately 1 year prior, he developed diffuse body aches and pains particularly sub-costal pain that caused significant discomfort in trying to take deep-breaths. There was associated chest tightness and occasional wheezing. He had an associated dry cough but no fevers, chills or night sweats. He reported feeling short of breath even at rest or on trying to take any kind of deep breaths, coughing or yawning. He noted pain in the center of his chest, as well as in the sub-chondral region that is increased by any kind of exercise, emotions or cold air. He denied any significant upper airway congestion or post-nasal drip, and he denied any significant reflux symptoms. Aside from his symptoms listed, his review of systems was negative.
He also reported that work increased his symptoms, although he had not done peak flows away and at work. He has worked as a welder, welding aluminum parts for the last several years. This involved grinding and sanding of parts. No personal respiratory protection was ever used and there was no dedicated ventilation in a workplace with few windows or doors. He had been working in a supervisory position for approximately a year; however, in overseeing others' work, he reported that his exposures were only slightly less than when he was working as a welder himself. He reports no significant problems with water damage, and there were no pets or other significant exposures in his home environment or related to hobbies.
Several months prior to presentation to our clinic, he had an evaluation by a pulmonologist, including a pulmonary function test, a high-resolution CT scan and a bronchoscopy that was non-diagnostic, and he was told he had restrictive lung disease. Following this, he was empirically tried on prednisone for a month with some symptomatic improvement but no evidence of objective improvement on PFTs. When prednisone was retried 4 months later for 2 months, there was no substantial symptomatic or objective improvement.
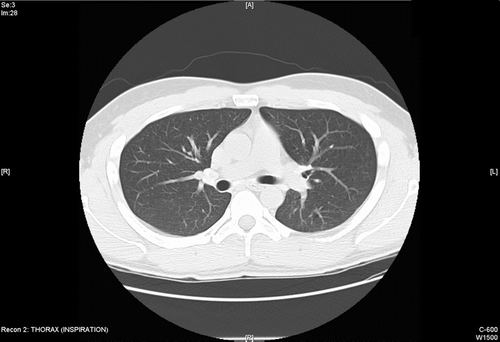
Figure 1 CT scan during baseline evaluation demonstrated mild-to-moderate hilar adenopathy with early calcification of lymph nodes. Mild air trapping with moderate bronchial wall thickening was noted. Linear band of opacity and ground galss opacities were noted in the right lower lobe. Minimal emphysema was reported in the right upper lobe.
He had no childhood history of asthma or allergies and no family history of asthma, allergies, COPD or other pulmonary or collagen vascular disease. He had no significant past medical history of other medical illnesses and he was on no medications. He was a lifelong non-smoker and denied use of alcohol or recreational drugs. His occupational history was essentially that of welding and he had no significant jobs prior to starting this trade.
PHYSICAL EXAM
Physical exam revealed a well-developed, well-nourished male appearing his stated age in no acute distress. Vital Signs: blood pressure 120/80, heart rate 70, respiratory rate 12. He demonstrated evidence for minimal nasal congestion. His chest exam revealed the trachea to be midline, no clubbing or cyanosis was present and breath sounds revealed occasional wheezes but no crackles or other sounds. The cardiovascular exam was normal as was the musculoskeletal exam and the remainder of the physical exam was unremarkable.
His previous pulmonary evaluation included a pulmonary embolus protocol CT scan that was negative for pulmonary emboli and showed some blunting in the costophrenic angle, but otherwise was negative. His Pulmonary Function Tests at that time included spirometry that was consistent with a mixed pattern FEV1 that was 43% predicted, FVC was 57% predicted, and the FEV1/FVC ratio was 71%. There was no significant bronchodilator response. His diffusing capacity was 63% predicted, but when accounting for alveolar volume it rose to 99% predicted. Hepatitis C, A and B antibodies were negative. Thyroid function tests were normal. ANA and ANCAs were normal. Myeloperoxidase was normal. Parvo virus was negative. His rheumatoid factor and HLA-B27 were negative.
TEST RESULTS
A high-resolution CT scan of his chest demonstrated moderate mediastinal bilateral hilar lymphadenopathy. Ground-glass and linear opacities were noted in the right lower lobe. There was minimal emphysema in the right upper lobe. The esophageal wall was presumed to be diffusely thickened. The examination was thought to be suspicious for Sarcoidosis ().
Pulmonary function tests indicated reduced total lung capacity that was only 67% of predicted, as was the TGV (65% predicted). However, he had a relatively increased residual volume at 99% predicted in a pattern that suggested a component of obstructive airways disease. His FEV1 was 2.84 L (53% predicted), his FVC was 3.9 L (61% predicted), and the ratio was 71%. He had no significant bronchodilator response time. His diffusion capacity was 28.7, which was 60% of predicted and improved to 97% of predicted when accounting for alveolar volume. His methacholine challenge revealed a PC20 FEV1 that was 0.5384 mg/ml. A pressure volume curve was within normal limits suggesting neither a predominantly restrictive or obstructive defect. Cardiopulmonary exercise testing showed that he indeed had borderline normal exercise tolerance being able to accomplish 86% of the predicted workload. He had a decrease in O2 saturation from 95 to 93%, but his Pa O2 dropped from 80 mmHg down to 70 mmHg. He had adequate tidal volume recruitment and respiratory rate recruitment.
Skin prick testing showed him to be atopic and his IgE level was 1,009. His CBC eosinophils were also borderline elevated at 6.9. Sputum eosinophils during a time off steroids for more than 2 months were 0. Steroid kinetics were performed and these showed that the patient had normal prednisone absorption and clearance and had lymphocytes that were sensitive to all steroids tested including prednisolone, dexamethasone, budesonide and fluticasone.
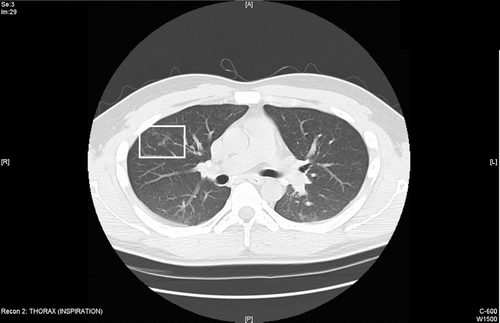
Figure 2 Follow up CT scan 3 months later while still exposed shows progressive bronchial wall thickening and development of new centriloblar nodules in the right upper lobe.
A bronchoscopy was done and showed evidence for ciliated columnar epithelium with marked chronic inflammation of the bronchial walls characterized by focally increased eosinophils. Some rare lymphoid aggregates with germinal centers were noted. There were also foci of organizing pneumonia. Small clusters of histiocytes were noted. No well-formed granulomas were noted. It was felt that the differential included hypersensitivity pneumonitis, collagen vascular diseases and infections, but not sarcoidosis. Peak flow studies away and at work showed a modest, yet not compelling, pattern of worsening during times at work. He was again tried on oral prednisone for approximately 1 month, and did not have a dramatic improvement. A follow up HRCT demonstrated new centrilobular nodules in the right upper lobe ().
Figure 3 H & E stain of VATS biopsy demonstrates airways with a patchy mild chronic inflammatory infiltrate and interstitial macules that are perivascular and peribronchiolar in distribution associated with mild collagen deposition. Finely granulated macrophages within the interstitium were reported as consistent with aluminum pneumoconiosis.
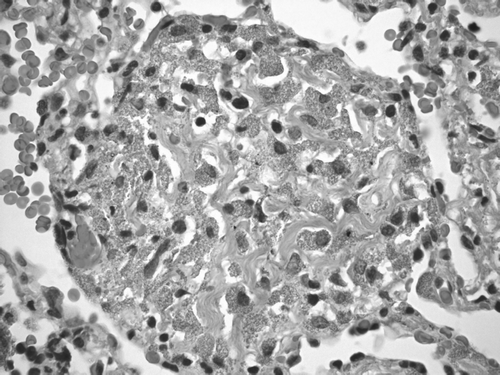
In light of these findings, the lack of clarity in the diagnosis and the failure to respond to prednisone, the patient underwent a video-assisted thoracoscopic lung biopsy. The specimens showed that he had finely granulated macrophages within the interstitium. There were interstitial macules that were perivascular and peribronchiolar in distribution and were associated with mild collagen deposition. Hemosiderin laden macrophages were also present. The airways and vessels show a patchy mild chronic inflammatory infiltrate (). The impression from this biopsy was that he had aluminum dust pneumoconiosis.
Given the patient's occupational history of being a welder of aluminum parts for the past several years, it was deduced that he did indeed have aluminum dust pneumoconiosis and possible asthma. He was removed from exposure for over 4 weeks and then was treated with prednisone 60 mg for 6 weeks without any subjective or objective improvement (). Currently the patient has been removed from ongoing exposure and has had no further deterioration in lung function or symptoms.
Table 1 Serial pulmonary function tests
DISCUSSION
This patient's final diagnosis was aluminum pneumoconiosis with both obstructive airway and parenchymal changes that were consistent with an interstitial pneumoconiosis process. While this case may not be typical of COPD related to non-smoking causes, it has several features that make it worthy of discussion. Between 5 and 10% of patients with COPD have no smoking history and their chronic airflow limitation is related to occupational, environmental or infectious etiologies [Citation[1], Citation[2]]. One could suggest that our patient had an asthmatic response (positive methacholine challenge) that may be related to environmental atopy or his exposure to aluminum. Despite the evidence for environmental atopy and bronchial hyperreactivity, he had no significant bronchodilator response and he had no improvement in pulmonary function on 3 separate oral corticosteroid trials during ongoing exposure and a 4th 6-week trial of prednisone while out of exposure. If we consider the definition of COPD to be airflow limitation that is only partially reversible, then this patient clearly fits the criteria. Further he had radiographic evidence of mild emphysema, bronchial wall thickening and centilobular nodules, which are commonly found in patients with COPD and have also been described in patients exposed to aluminum [Citation[3]]. Hilar adenopathy with aluminum deposits in the lymph nodes has also been described [Citation[4]].
There are previous reports of welders of aluminum parts developing asthma and/or pnuemoconiotic lesions similar to our patient [Citation[5], Citation[6], Citation[7], Citation[8], Citation[9], Citation[10], Citation[11]]. Aluminum has been recognized as capable of causing lung disease at least since the 1950s [Citation[12], Citation[13], Citation[14], Citation[15]]. There are several pathological lesions recognized as resulting from exposure to aluminum silicates including pnuemoconiosis [Citation[6], Citation[16]] and asthma (pot room asthma) [Citation[3], Citation[10], Citation[17], Citation[18], Citation[19], Citation[20], Citation[21]], but other entities such as small airways disease (bronchiolitis) [Citation[22]], eosinophilic pneumonia [Citation[23]] and desquamitive interstitial pneumonia [Citation[9]] have been reported. Sarcoid-like granulomatous reactions have also been described [Citation[5], Citation[6], Citation[8], Citation[24], Citation[25]]. The histopathology of the lungs response to aluminum is highly variable and likely is a function of both host (genetics, smoking, comorbidities) and environmental factors, (aluminum species, duration and co-exposures). In one report of a non-smoking drill polisher, a very similar pathological lesion was noted to our patient with bronchoalveolar lavage revealing high eosinophilia, and transbronchial biopsy specimens demonstrating interstitial pneumonia with giant cell infiltrates and peribronchiolar accumulation of macrophages laden with opaque dust. Mineralogic studies done from the tissue revealed a high concentration of exogenous particles that were identified as aluminum silicate [Citation[23]].
The pathogenesis of aluminum lung reactions may include antigenic Th-1 T cell responses such as are described in chronic beryllium disease leading to granulomatous reactions [Citation[5], Citation[6], Citation[8], Citation[24], Citation[25], Citation[26]] and possibly Th-2 antigenic/hapten responses leading to asthmatic changes in the airways (although this has not been confirmed) [Citation[8]]. Other studies have demonstrated increased expression of metalloproteinases [Citation[27]] and oxidative stress responses have also been described [Citation[8], Citation[28]]. Girod et al. [Citation[2]] hypothesized that the development of COPD may be the result of aluminum silicates and other particulates found in cigarettes inducing the airway and parenchymal changes, namely a respiratory bronchiolitis (See Journal club, this issue). They and others have made the observation that the inflammation in the airways persists and indeed may actually increase after cigarette smoking has ceased [Citation[2], Citation[29]]. The persistence of aluminum particles and fibres in lung tissue well after removal from occupational exposure has been documented [Citation[30]]. It is interesting to note that there is some indirect evidence that the inflammatory and clinical manifestations of chronic beryllium disease may be increased following smoking cessation [Citation[31]]. Perhaps in at least a subset of patients with COPD their airway and parenchymal changes are a result of pathogenic responses to mineral dusts found in tobacco that may perpetuate and drive and ongoing inflammatory response even after smoking cessation. Girod et al. [Citation[2], Citation[32]] point out that aluminum silicate dust (kaolinite) has been found in alveolar macrophages of smokers and has been reported as a component of tobacco products. There are several proposed pathogenic mechanisms for COPD pointing to the biological heterogeneity of this disorder and this patient's case supports the notion that COPD may be, at least in some subjects, driven by pathogenic responses to inhaled dusts such as aluminum silicates found in various occupational settings and tobacco smoke.
REFERENCES
- Global Initiative for Chronic Obstructive Lung Disease: Global Strategy for the Diagnosis. Management and Prevention of Chronic Obstructive Pulmonary Disease (GINA). National Institutes of Health, National Heart, Lung, and Blood Institute, Bethesda 2003
- Girod C E, King T E, Jr. COPD: a dust-induced disease?. Chest 2005; 128(4)3055–3064, [PUBMED], [INFOTRIEVE], [CSA]
- Kilburn K H, Warshaw R H. Irregular opacities in the lung, occupational asthma, and airways dysfunction in aluminum workers. Am J Ind Med 1992; 21(6)845–853, [PUBMED], [INFOTRIEVE], [CSA]
- Vahlensieck M, Overlack A, Muller K M. Computed tomographic high-attenuation mediastinal lymph nodes after aluminum exposition. Eur Radiol 2000; 10(12)1945–1946, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Fireman E, Goshen M, Ganon E, Spirer Z, Lerman Y. Induced sputum as an additional tool in the identification of metal-induced sarcoid-like reaction. Sarcoidosis Vasc Diffuse Lung Dis 2004; 21(2)152–156, [PUBMED], [INFOTRIEVE], [CSA]
- Hull M J, Abraham J L. Aluminum welding fume-induced pneumoconiosis. Hum Pathol 2002; 33(8)819–825, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Akira M. Uncommon pneumoconioses: CT and pathologic findings. Radiology 1995; 197(2)403–409, [PUBMED], [INFOTRIEVE], [CSA]
- Nemery B. Metal toxicity and the respiratory tract. Eur Respir J 1990; 3(2)202–219, [PUBMED], [INFOTRIEVE], [CSA]
- Herbert A, Sterling G, Abraham J, Corrin B. Desquamative interstitial pneumonia in an aluminum welder. Hum Pathol 1982; 13(8)694–699, [PUBMED], [INFOTRIEVE], [CSA]
- Vandenplas O, Delwiche J P, Vanbilsen M L, Joly J, Roosels D. Occupational asthma caused by aluminium welding. Eur Respir J 1998; 11(5)1182–1184, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Flodin U, Ziegler J, Jonnsen P, Axelson O. Bronchial asthma and air pollution at workplaces. Scand J Work Environ Health 1996; 22(6)451–456, [PUBMED], [INFOTRIEVE], [CSA]
- Corrin B. {Aluminium Pneumoconiosis. I in Vitro Comparison of Stamped Aluminium Powders Containing Different Lubricating Agents and a Granular Aluminium Powder. Br J Ind Med 1963; 20: 264–267, [PUBMED], [INFOTRIEVE], [CSA]
- Corrin B. Aluminium Pneumoconiosis. Ii Effect on the Rat Lung of Intratracheal Injections of Stamped Aluminium Powders Containing Different Lubricating Agents and of a Granular Aluminium Powder. Br J Ind Med 1963; 20: 268–276, [PUBMED], [INFOTRIEVE], [CSA]
- Edling N P. Aluminium pneumoconiosis A roentgendiagnostic study of five cases. Acta Radiol 1961; 56: 170–178, [PUBMED], [INFOTRIEVE], [CSA]
- Lynch K M, Mc I F. Pneumoconiosis from exposure to kaolin dust kaolinosis. Am J Pathol 1954; 30(6)1117–1127, [PUBMED], [INFOTRIEVE], [CSA]
- Sjogren B, Ljunggren K G, Almkvist O, Frech W, Basun H. Aluminosis and dementia. Lancet 1994; 344(8930)1154, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Burge P S, Scott J A, McCoach J. Occupational asthma caused by aluminum. Allergy 2000; 55(8)779–780, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Desjardins A, Bergeron J P, Ghezzo H, Cartier A, Malo J L. Aluminium potroom asthma confirmed by monitoring of forced expiratory volume in one second. Am J Respir Crit Care Med 1994; 150(6 Pt 1)1714–1717, [PUBMED], [INFOTRIEVE], [CSA]
- Kongerud J, Boe J, Soyseth V, Naalsund A, Magnus P. Aluminium potroom asthma: the Norwegian experience. Eur Respir J 1994; 7(1)165–172, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Lemiere C, Malo J L, Gautrin D. Nonsensitizing causes of occupational asthma. Med Clin North Am 1996; 80(4)749–774, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Soyseth V, Kongerud J, Kjuus H, Boe J. Bronchial responsiveness and decline in FEV1 in aluminium potroom workers. Eur Respir J 1994; 7(5)888–894, [PUBMED], [INFOTRIEVE], [CSA]
- Churg A, Wright J L. Small-airway lesions in patients exposed to nonasbestos mineral dusts. Hum Pathol 1983; 14(8)688–693, [PUBMED], [INFOTRIEVE], [CSA]
- Schwarz Y A, Kivity S, Fischbein A, Ribak Y, Fireman E, Struhar D, Topilsky M, Greif J. Eosinophilic lung reaction to aluminium and hard metal. Chest 1994; 105(4)1261–1263, [PUBMED], [INFOTRIEVE], [CSA]
- Drent M, Bomans P H, Van Suylen R J, Lamers R J, Bast A, Wouters E E. Association of man-made mineral fibre exposure and sarcoidlike granulomas. Respir Med 2000; 94(8)815–820, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Newman L S. Metals that cause sarcoidosis. Semin Respir Infect 1998; 13(3)212–220, [PUBMED], [INFOTRIEVE], [CSA]
- Brancaleone P, Weynand B, De Vuyst P, Stanescu D, Pieters T. Lung granulomatosis in a dental technician. Am J Ind Med 1998; 34(6)628–631, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Halatek T, Opalska B, Lao I, Stetkiewicz J, Rydzynski K. Pneumotoxicity of dust from aluminum foundry and pure alumina: a comparative study of morphology and biomarkers in rats. Int J Occup Med Environ Health 2005; 18(1)59–70, [PUBMED], [INFOTRIEVE], [CSA]
- Baser M E, Kennedy T P, Dodson R, Rao N V, Rawlings W, Jr., Hoidal J R. Hydroxyl radical generating activity of hydrous but not calcined kaolin is prevented by surface modification with dipalmitoyl lecithin. J Toxicol Environ Health 1990; 29(1)99–108, [PUBMED], [INFOTRIEVE], [CSA]
- Willemse B W, ten Hacken N H, Rutgers B, Lesman-Leegte I G, Postma D S, Timens W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J 2005; 26(5)835–845, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Voisin C, Fisekci F, Buclez B, Didier A, Couste B, Bastien F, Brochard P, Pairon J C. Mineralogical analysis of the respiratory tract in aluminium oxide-exposed workers. Eur Respir J 1996; 9(9)1874–1879, [PUBMED], [INFOTRIEVE], [CROSSREF], [CSA]
- Kreiss K, Mroz M M, Zhen B, Martyny J W, Newman L S. Epidemiology of beryllium sensitization and disease in nuclear workers. Am Rev Respir Dis 1993; 148(4 Pt 1)985–991, [PUBMED], [INFOTRIEVE], [CSA]
- Brody A R, Craighead J E. Cytoplasmic inclusions in pulmonary macrophages of cigarette smokers. Lab Invest 1975; 32(2)125–132, [PUBMED], [INFOTRIEVE], [CSA]