Abstract
Acute exacerbations are significant events in the course of COPD. The pathogenesis of exacerbations was poorly understood, specifically, the role of bacteria was highly controversial. Recent observations have demonstrated that bacterial infection is involved in about half of the exacerbations. The predominant mechanism of bacterial exacerbation in COPD appears to be acquisition of new strains of bacterial pathogens from the environment that are able to establish infection in the tracheobronchial tree in COPD because of compromised innate lung defenses. These pathogens interact with airway cells, elicit an inflammatory response, which underlies the pathophysiology and symptoms characteristic of exacerbation. An immune response that can be mucosal, systemic or both develops to the infecting bacterial strain. This immune response contains the infectious process, could eradicate the infecting pathogen and prevent re-infection with the same strain. However, because of antigenic diversity among bacterial strains, this immunity tends to be strain-specific rather than widely protective. Other mechanisms, including increase in bacterial load and interaction with other etiologies such as viruses, also could contribute to bacterial exacerbations. Improved understanding of the host-pathogen interaction in the airways in COPD will lead to novel approaches to prevention and treatment of exacerbations.
Exacerbations are an integral manifestation of chronic obstructive pulmonary disease (COPD). However, until recently they were assigned a nuisance value and thought to be of little importance in the pathogenesis and clinical course of COPD. Accumulating evidence from several sources has now clearly established that exacerbations are a major determinant of health-related quality-of-life in COPD, a significant contributor to health care expenditures for this disease and a dominant cause of mortality among these patients (Citation[1], Citation[2], Citation[3]). Even the long-held belief that exacerbations do not contribute to the progressive airflow obstruction characteristic of COPD has been refuted by carefully conducted studies in well-characterized patient populations (Citation[4], Citation[5], Citation[6]). With this rekindled interest in exacerbations, understanding their etiology and pathogenesis has received increased attention. Application of new research techniques to this question has been very productive with several new insights (Citation[7], Citation[8]).
The clinical course of exacerbations, with their acute to sub-acute onset, followed by slow resolution over days and weeks, is strongly suggestive of an infectious cause (Citation[9]). Among the possible infectious agents of exacerbation, the role of bacteria has been the most controversial (Citation[10], Citation[11], Citation[12], Citation[13]). Several innovative investigations have now clearly established that a substantial proportion of exacerbations are indeed bacterial in origin (Citation[7], Citation[8]). These recent findings also suggest new mechanisms for exacerbations, in contrast to the long-held belief that an increase in bacterial load in the airways is the primary mechanism for exacerbations.
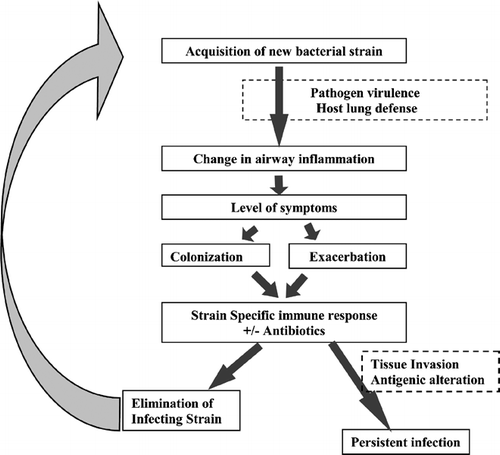
COPD is now thought to be inflammatory disease. In a current model of pathogenesis of exacerbations, an increase from baseline airway inflammation is regarded as the mechanism that leads to the clinical manifestations of an exacerbation (Citation[7], Citation[8]). Several etiologies can contribute to this increase in airway inflammation. The likely pathway by which bacteria cause this increase in airway inflammation is depicted in . This potential mechanism of recurrent bacterial exacerbations in COPD will be explored in this paper, and supporting evidence for this pathway will be presented. Other potential mechanisms undoubtedly contribute to the pathogenesis of bacterial exacerbations and will also be discussed.
Figure 1. Proposed model of bacterial infection in COPD.
Acquisition of new strains of bacteria and exacerbation
Abnormal presence in the lower airway of bacterial pathogens in stable COPD was recognized more than four decades ago in an elegant bronchoscopic sampling study by Laurenzi et al. (Citation[14]). This presence was termed ‘colonization’, though it was never established that specific immune response to these pathogens or detrimental effects to the host were absent, which are required to define ‘colonization’. Following this, several longitudinal cohort investigations with repeated sputum cultures for isolation of bacteria were conducted to elucidate the role of bacteria in exacerbations of COPD (Citation[15], Citation[16]). The expected result of these studies was a higher incidence of bacterial isolation during exacerbations than during stable COPD, thereby explaining the pathogenesis of bacterial exacerbations. This expectation was not met and from the negative results of these studies, two opposing concepts emerged that subsequently dominated our thinking about bacterial exacerbations. One concept was that exacerbations are caused by other etiologies rather than bacteria, who are innocent bystanders in this disease, and their isolation from sputum during exacerbation is purely coincidental secondary to chronic colonization (Citation[10], Citation[12]). The other school of thought was that exacerbations were related to bacterial load, with increased concentrations of bacteria in the airways causing symptoms and lower concentrations being compatible with periods of quiescence (Citation[17]).
Substantial progress has been made in recent years in molecular characterization of bacterial pathogens to readily distinguish among strains of a species. This has led to better appreciation of the level of diversity among strains of a pathogenic species (Citation[18]). In fact, antigenic diversity is a major mechanism of recurrent infection, as antigenically diverse strains can subvert immune responses developed to other strains of the same species (Citation[19]). These developments have made it clear that simply culturing and enumerating colony counts of pathogens from bodily fluids, as was done in earlier studies of bacterial infection in COPD, is inadequate in understanding this infectious process (Citation[15], Citation[16]).
A recent study has addressed the limitations of previous studies. This study, in common with previous investigations, enrolled a COPD cohort with longitudinal collection of clinical information and sputum for culture (Citation[20]). However, in contrast to previous studies, strains of potential respiratory pathogens isolated from sputum were characterized by molecular techniques, to identify when new strains were acquired by a patient and when those strains were cleared from the respiratory tract. Using this approach, acquisition of new strains of certain bacterial species has been shown to be clearly associated with a greater than two-fold increased risk of exacerbation of COPD (Citation[20]) (, ). The time frame of increased risk appears to be up to 4–8 weeks after acquisition of a new strain. Specifically by pathogen, the increased risk of exacerbations with new strain acquisition was seen for Nontypeable Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pneumoniae, but not for Pseudomonas aeruginosa (). Strain characterization for Haemophilus parainfluenzae, Staphylococcus aureus and Enterobacteriaceae was not performed in this study.
Figure 2. Time lines and molecular typing for patients with COPD. The horizontal line is a time line, with each number indicating a clinic visit. The arrows indicate exacerbations. Isolates of H. influenzae and M. catarrhalis were assigned types based on sodium dodecyl sulfate-polyacrylamide gel and pulsed-field gel electrophoresis respectively. The types are indicated by letters A to E. Molecular mass standards are noted on the left of the gels. Reproduced with permission from Sethi et al. (Citation[20]).
![Figure 2. Time lines and molecular typing for patients with COPD. The horizontal line is a time line, with each number indicating a clinic visit. The arrows indicate exacerbations. Isolates of H. influenzae and M. catarrhalis were assigned types based on sodium dodecyl sulfate-polyacrylamide gel and pulsed-field gel electrophoresis respectively. The types are indicated by letters A to E. Molecular mass standards are noted on the left of the gels. Reproduced with permission from Sethi et al. (Citation[20]).](/cms/asset/17f4eb10-81de-4887-8968-79b9be74f50f/icop_a_165108_uf0002_b.gif)
Table 1. Isolation of a new strain of a bacterial pathogen and increases the risk of exacerbation of COPD
Another approach to better characterize airway bacterial flora during exacerbations has been by bronchoscopic sampling of the tracheobronchial tree with either protected specimen brushings and/or bronchoalveolar lavage. In combination with quantitative cultures, such sampling provides an accurate picture of the bacteriology at the site of infection during exacerbations. Rosell et al. recently reported a pooled analysis of clinical studies published between 1993 and 2002 that performed quantitative cultures of protected specimen brushing in stable COPD, during exacerbations and in healthy controls (Citation[21]). Bacterial pathogens were rarely isolated from healthy individuals and when isolated were at < 102 colony forming units/ml (cfu/ml). Bacterial concentrations of 102 to 103 cfu/ml were defined as low and ≥ 104 cfu/ml was considered high bacterial load in the airways. Low microbial load was found in 25% of stable patients and 50% of exacerbations. High microbial load was found in slightly less than 10% of stable COPD and 20% of exacerbations. When adjustment for the degree of airway obstruction (forced expiratory volume in 1 second percent predicted, FEV1%) was made, a statistically significant dose relationship was seen with high microbial load and exacerbations for H. influenzae but not for P. aeruginosa.
These findings would suggest that bacterial load is indeed important in the pathogenesis of exacerbations. However, the design of the bronchoscopic studies does not allow such a conclusion. Because of practical reasons, bronchoscopic studies are cross-sectional rather than longitudinal. Therefore, whether the bacterial strains isolated at bronchoscopy are new acquisitions or not is not known. One could speculate that when new strains are acquired, because of the lack of an effective host immune response, there is uninhibited growth of these strains in the airways resulting in higher concentrations. Once an immune response develops there is reduction in the airway concentrations of bacteria, and therefore, lower concentrations are seen in stable disease. Therefore, the occurrence of exacerbation and increased concentration of bacteria in the airways, both related to new strain acquisition, could appear to be related to each other.
The bronchoscopic studies unequivocally demonstrate the presence in the tracheobronchial tree of concentrations of bacteria associated with tissue infection. The strain analysis data provides a clear mechanistic explanation of the mechanism of recurrent exacerbation based on antigenic diversity. These innovative studies provide solid support to the concept that a significant proportion of exacerbations of COPD are bacterial in origin. Unfortunately, quantitative cultures, bronchoscopic sampling and strain characterization cannot be applied easily in the clinical setting to determine the etiology of an exacerbation in an individual patient.
Pathogen virulence as a determinant of exacerbation
Not every new strain acquisition of bacterial pathogens is associated with exacerbation (Citation[20]). A complex host-pathogen interaction in the airways likely determines the outcome of each new bacterial strain acquisition in COPD. The balance between host defense and pathogen virulence determines the level of airway inflammation, which in turn determines the level of symptoms in the patient (). Of course, patient perception and physician interpretation of symptoms are additional determinants of exacerbation.
Though H. influenzae, M. catarrhalis and S. pneumoniae are well recognized respiratory pathogens, there is little known regarding their virulence determinants in airway mucosal infection. Putative factors of pathogen virulence include adhesion to respiratory epithelial cells, invasion of respiratory epithelial cells, inactivation of host defense mechanisms and elicitation of inflammatory mediators from airway cells. Chin et al. compared in vitro and in vivo virulence of H. influenzae strains isolated during COPD exacerbations with colonizing strains from the cohort study discussed above (Citation[22]). Strains isolated during exacerbations caused more airway neutrophil recruitment in the mouse pulmonary clearance model, adhered to primary airway human airway epithelial cells in culture in significantly higher numbers, and elicited more interleukin-8 (IL-8) when compared to colonizing strains from epithelial cells (). Strains of H. influenzae associated with symptomatic infection are more likely to produce IgA protease than strains that colonize the nasopharynx (Citation[23]). Thus, inactivation of host defenses is an important determinant of disease expression among bacterial strains. These observations support pathogen virulence as an important determinant of clinical manifestations of new bacterial strain acquisition in COPD. Additional observations of pathogen virulence with relevance to COPD are needed.
Figure 3. Comparison of H. influenzae exacerbation (Exac) and colonization (Col) strains. *denotes p < 0.05. Reproduced with permission from Chin et al. (Citation[22]). (A) Neutophils in bronchoalveolar lavage (BAL) in response to mouse airway infection with bacterial strains. (B) Adherence of bacterial strains to tracheobronchial epithelial cells in culture. (C) Production of interleukin-8 (IL-8) by tracheobronchial epithelial cells in culture on exposure to bacterial strains.
![Figure 3. Comparison of H. influenzae exacerbation (Exac) and colonization (Col) strains. *denotes p < 0.05. Reproduced with permission from Chin et al. (Citation[22]). (A) Neutophils in bronchoalveolar lavage (BAL) in response to mouse airway infection with bacterial strains. (B) Adherence of bacterial strains to tracheobronchial epithelial cells in culture. (C) Production of interleukin-8 (IL-8) by tracheobronchial epithelial cells in culture on exposure to bacterial strains.](/cms/asset/240c9541-4b07-4508-bd1c-62fef733fb62/icop_a_165108_uf0003_b.gif)
Host defense as a determinant of exacerbation
The healthy lung has several innate defense mechanisms that keep it sterile, in spite of the daily assault of large numbers of inhaled and aspirated pathogens. Tobacco smoking and the development of COPD are associated with significant disruption of these innate immune mechanisms, allowing the colonization of the lower respiratory tract with a variety of microbial pathogens (Citation[24]).
On the acquisition of a bacterial pathogen in to the lower airway, adaptive immunity attempts to control and eradicate the infection. When immune responses to new strains of M. catarrhalis associated with COPD exacerbation and colonization were compared, a mucosal IgA response to the infecting strain was more common and vigorous with colonization, while a systemic IgG immune response was more common and vigorous with exacerbations (Citation[25]) (). This raises the interesting possibility that the host immune response could dictate the clinical expression of a bacterial strain acquisition in COPD, with a vigorous mucosal immune response possibly excluding the bacteria from interaction with the epithelial mucosa, resulting in less airway inflammation and therefore favoring colonization. An alternative explanation is that colonization results in more of a mucosal than systemic antigen burden. The consequent immune response therefore differs with the clinical scenario, rather than determining it.
Figure 4. Comparison of serum IgG and sputum IgA antibody response to homologous strains of M. catarrhalis following colonization or exacerbation. Based on data from Murphy et al. (Citation[25]). (A) Percent of episodes of colonization or exacerbation that were associated with an antibody response. (B) Intensity of antibody response as a percent change from baseline level of antibody. Median values are shown.
![Figure 4. Comparison of serum IgG and sputum IgA antibody response to homologous strains of M. catarrhalis following colonization or exacerbation. Based on data from Murphy et al. (Citation[25]). (A) Percent of episodes of colonization or exacerbation that were associated with an antibody response. (B) Intensity of antibody response as a percent change from baseline level of antibody. Median values are shown.](/cms/asset/a72eb78d-7d8b-4fcb-8c03-3110568e9f41/icop_a_165108_uf0004_b.gif)
Another example of host defense determining exacerbation occurrence in COPD was found in peripheral blood mononuclear cell proliferation on exposure to a conserved H. influenzae antigen, outer membrane protein P6 (Citation[26]). Patients with COPD who had suffered from one or more exacerbation with H. influenzae in the preceding 12 months demonstrated diminished response to P6, while the response of those who had not experienced such exacerbations was comparable to healthy controls. One can speculate that a vigorous cellular response to H. influenzae antigens suppresses or even eradicates newly acquired strains of H. influenzae and therefore exacerbations are less likely.
Much more needs to be learned about the determinants of host immune response that determine the outcome of a bacterial acquisition by a patient with COPD. This knowledge could lead to development of new and unique approaches to prevent and treat exacerbations.
Airway inflammation as a determinant of exacerbation
As inflammation appears to underlie the symptoms of chronic phase of COPD, a reasonable supposition is that the increased symptoms of exacerbation are related to increases in airway inflammation (). Studies comparing airway inflammation in stable COPD with exacerbation are few, but consistently show increased airway inflammation with exacerbations (Citation[27], Citation[28]). This increased inflammation could induce airway mucosal edema, bronchospasm and airway mucus hypersecretion, which in turn can be related to the classical symptoms of exacerbation of dyspnea, wheezing, cough and sputum production.
Though exacerbations of COPD appear to be initiated by inflammation, this inflammatory process is not uniform and is related to the etiology of the exacerbation (Citation[29], Citation[30]). Exacerbations associated with bacterial pathogens exhibit significantly more neutrophilic inflammation than non-bacterial episodes (Citation[29], Citation[30]). Specifically, exacerbations associated with H. influenzae demonstrated significantly higher levels of IL-8, tumor necrosis factor −α (TNFα) and neutrophil elastase (NE) whereas exacerbations associated with M. catarrhalis had significantly higher TNFα and NE when compared to non-bacterial exacerbations. Airway inflammation seen with H. parainfluenzae was heterogeneous but as a group had an inflammatory profile similar to non-bacterial exacerbations (Citation[30]). Higher concentrations of bacteria have also been associated with greater amounts of inflammation, suggesting a stimulus-response relationship (Citation[29], Citation[30]). Furthermore, a significant correlation between the clinical severity of an exacerbation and the level of sputum neutrophil elastase has been demonstrated (Citation[30]).
Sputum purulence by gross examination, when carefully assessed by trained observers with standardized methodology, is a good marker for the presence of bacterial pathogens in exacerbations (Citation[31]). Furthermore, with resolution of symptoms of purulent exacerbations, there is a consistent decrease in neutrophilic airway inflammation, as reflected in decreases in sputum levels of IL-8, Leukotriene B4 (LTB4), NE and myeloperoxidase (MPO) (Citation[28]). When such clinical resolution is accompanied by bacteriological eradication of the offending pathogen, there is a more marked reduction in airway inflammation, compared to those instances where bacterial pathogens persist in the airway in spite of clinical resolution (Citation[28]).
The above diverse findings clearly support the role of bacteria inducing airway inflammation that manifests clinically as exacerbations. Bacterial colonization has also been associated with airway inflammation in COPD (Citation[32]). Our model would suggest that airway inflammation with colonization is less than with exacerbations, therefore is not accompanied by symptoms greater than baseline. Empiric evidence for this concept is lacking at present.
Strain-specific immune response
Adaptive immunity, especially the production of antibodies, plays a large role in defense against bacterial infections (). Several older studies examined the humoral immune response to bacterial pathogens in COPD. In these studies, sera from COPD patients were compared with sera obtained from healthy controls with a single strain or a panel of a few bacterial strains as an antigen (Citation[11]). Results of these studies were contradictory and confusing, which is not surprising considering our current understanding of the antigenic diversity among bacterial strains. In order to obtain reliable results in the study of immune response to bacterial infection, several considerations are important. Homologous or infecting strains should be used as the antigen to account for antigenic diversity among strains. Samples (serum or mucosal secretions) obtained after infection should be compared with pre-infection samples to clearly distinguish antibodies that develop following infection from baseline antibodies. Immunoassays used should be specific for antibodies that bind to surface antigens of the bacterial pathogen, in order to avoid muddying the results with cross-reactive antibodies that often bind to non-surface-exposed epitopes (Citation[11]).
When these criteria are met, development of antibodies that bind to the bacterial cell surface has been demonstrated following exacerbations associated with H. influenzae, M. catarrhalis and S. pneumoniae as well as following colonization with M. catarrhalis (Citation[25], Citation[33], Citation[34]). Furthermore, for H. influenzae, these antibodies have been demonstrated to be bactericidal for the strain and to have a strong degree of strain specificity (Citation[33]). Though strain-specificity of the antibodies directed against M. catarrhalis and S. pneumoniae has not been directly demonstrated, it is likely, based on the observation that once a strain of these pathogens is eliminated from the airway in COPD, re-acquisition of the same strain is a rare event (Citation[25]).
Development of adaptive immune responses following exacerbation and colonization with these respiratory pathogens supports the pathogenic role of these organisms in the lower airway. Furthermore, the strain-specificity of these immune responses explains why they are not protective against antigenically different strains of the same species, accounting for the recurrent exacerbations seen in COPD.
Exceptions to the model of recurrent exacerbation
It is quite likely that the model shown in does not explain all bacterial exacerbations. Specifically, Pseudomonas aeruginosa, a significant airway pathogen in bronchiectasis and cystic fibrosis, has been isolated from sputum and bronchoscopic samples in exacerbations, usually in severe COPD. However, an association between exacerbations and new strain isolation was not identified for P. aeruginosa in COPD as well as in cystic fibrosis (Citation[20], Citation[35]). This suggests alternative mechanisms, including increased bacterial load or re-infection from an endogenous site. P. aeruginosa forms complex communities enclosed in matrix of extracellular polymeric substances, known as biofilms, in the airways of patients with cystic fibrosis. A change from this biofilm state to a free floating planktonic state has been associated with exacerbations of cystic fibrosis and a similar mechanism may exist among patients with COPD (Citation[36]).
Other gram-negative enteric bacteria and S. aureus are often isolated from sputum, and in severe COPD, from bronchoscopy samples obtained during exacerbations. However, no information is available regarding strain changes and immune and inflammatory responses, to determine the mechanism of exacerbations with these pathogens.
In order to elicit the exact role of bacterial load in the causation of exacerbation, it would be essential to demonstrate that an increase in concentration of a colonizing strain in the airways is sufficient in its own to cause an exacerbation. Such a mechanism could exist, especially with intercedent viral infection or other insults to the airway, which could alter the equilibrium between a colonizing strain and the host defense mechanisms. This could allow the strain to proliferate and contribute to increased airway inflammation and the manifestations of the exacerbation.
Viral and bacterial interaction in exacerbations of COPD
It is commonly believed that antecedent viral infection is essential for the development of bacterial exacerbations of COPD, though little data exists to support this notion. In one small study, exacerbations in which viral infection was determined by culture and serology and H. influenzae was isolated from sputum were studied further (Citation[37]). Development of serum IgG antibodies to the H. influenzae strain isolated was regarded as evidence for bacterial causation. In this study, exacerbations could be attributed to virus alone, to both viral and H. influenzae infection and to bacterial infection alone. Therefore, though antecedent or simultaneous viral infection does occur, it is likely that several bacterial exacerbations occur de novo. Whether co-infection by these two classes of pathogens alters the nature of the exacerbation is not known.
Specific viruses may predispose to specific bacteria, for example, the association between influenza virus and S. pneumoniae and S. aureus in pneumonia may extend to exacerbations of COPD (Citation[38]). M. catarrhalis exacerbations demonstrate a seasonal pattern reminiscent of rhinovirus infections, suggesting an association between these two pathogens (Citation[25]). In one study of severe exacerbations, co-infection with C. pneumoniae and bacterial infection was described (Citation[39]).
CONCLUSIONS
Substantial progress has been made in the understanding of the mechanism of bacterial exacerbations in COPD. Availability of animal models of smoking-induced airway disease that could be infected with the respiratory pathogens that cause exacerbations in COPD will substantially accelerate research in this area. Further examination of cellular and molecular mechanisms in human subjects will add to our knowledge regarding bacterial exacerbations. Understanding the virulence determinants of pathogens in the airway and their interaction with airway epithelial cells and macrophages would be invaluable. Exacerbations can be related to more than one inciting factor simultaneously. Limited knowledge of the cooperation of these inciting factors is available at present and is a fertile area of research. Insight in to the mechanisms and pathophysiology of exacerbations should eventually lead to novel methods of treatment and prevention.
REFERENCES
- Andersson F, Borg S, Jansson S A, Jonsson A C, Ericsson A, Prutz C, Ronmark E, Lundback B. The costs of exacerbations in chronic obstructive pulmonary disease (COPD). Respir Med 2002; 96: 700–708, [INFOTRIEVE], [CSA]
- Seemungal T AR, Donaldson G C, Paul E A, Bestall J C, Jeffries D J, Wedzicha J A. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157: 1418–1422, [INFOTRIEVE], [CSA]
- Celli B R, Cote C G, Marin J M, Casanova C, Montes de Oca M, Mendez R A, Pinto Plata V, Cabral H J. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 1005–1012, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Fletcher F, Peto R. The natural history of chronic airflow obstruction. Br Med J 1977; 1: 1645–1648, [INFOTRIEVE], [CSA]
- Kanner R, Anthonisen N R, Connett J E. The Lung Health Study Research Group. Lower respiratory illnesses promote FEV(1) decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am J Respir Crit Care Med 2001; 164: 358–364, [INFOTRIEVE], [CSA]
- Donaldson G C, Seemungal T A, Bhowmik A, Wedzicha J A. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57: 847–852, [INFOTRIEVE], [CSA], [CROSSREF]
- Sethi S. New developments in the pathogenesis of acute exacerbations of chronic obstructive pulmonary disease. Curr Opin Infect Dis 2004; 17: 113–119, [INFOTRIEVE], [CSA], [CROSSREF]
- White A J, Gompertz S, Stockley R A. Chronic obstructive pulmonary disease 6: The aetiology of exacerbations of chronic obstructive pulmonary disease. Thorax 2003; 58: 73–80, [INFOTRIEVE], [CSA], [CROSSREF]
- Leidy N K, Rennard S I, Schmier J, Jones M K, Goldman M. The breathlessness, cough, and sputum scale: the development of empirically based guidelines for interpretation. Chest 2003; 124: 2182–2191, [INFOTRIEVE], [CSA], [CROSSREF]
- Tager I, Speizer F E. Role of infection in chronic bronchitis. N Engl J Med 1975; 292: 563–571, [INFOTRIEVE], [CSA]
- Murphy T F, Sethi S. Bacterial infection in chronic obstructive pulmonary disease. Am Rev Respir Dis 1992; 146: 1067–1083, [INFOTRIEVE], [CSA]
- Hirschmann J V. Do bacteria cause exacerbations of COPD?. Chest 2000; 118: 193–203, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Murphy T F, Sethi S, Niederman M S. The role of bacteria in exacerbations of COPD. A constructive view. Chest 2000; 118: 204–209, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Laurenzi G A, Potter R T, Kass E H. Bacteriologic flora of the lower respiratory tract. N Engl J Med 1961; 265: 1273–1278, [INFOTRIEVE], [CSA]
- Gump D W, Phillips C A, Forsyth B R, McIntosh F K, Lamborn K R, Stouch W H. Role of infection in chronic bronchitis. Am Rev Respir Dis 1976; 113: 465–473, [INFOTRIEVE], [CSA]
- McHardy V U, Inglis J M, Calder M A, Crofton J W. A study of infective and other factors in exacerbations of chronic bronchitis. Br J Dis Chest 1980; 74: 228–238, [INFOTRIEVE], [CSA], [CROSSREF]
- Miravitlles M. Exacerbations of chronic obstructive pulmonary disease: when are bacteria important?. Eur Respir J 2002; 36: 9s–19s, [CSA], [CROSSREF]
- Maslow J N, Mulligan M E, Arbeit R D. Molecular epidemiology: application of contemporary techniques to the typing of microorganisms. Clin Infect Dis 1993; 17: 153–164, [PUBMED], [INFOTRIEVE], [CSA]
- Brunham R C, Plummer F A, Stephens R S. Bacterial antigenic variation, host immune response and pathogen-host coevolution. Infect Immun 1993; 61: 2273–2276, [INFOTRIEVE], [CSA]
- Sethi S, Evans N, Grant B JB, Murphy T F. Acquisition of a new bacterial strain and occurrence of exacerbations of chronic obstructive pulmonary disease. N Engl J Med 2002; 347: 465–471, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Rosell A, Monso E, Soler N, Torres F, Angrill J, Riise G, Zalacain R, Morera J, Torres A. Microbiologic determinants of exacerbation in chronic obstructive pulmonary disease. Arch Intern Med 2005; 165: 891–897, [INFOTRIEVE], [CSA], [CROSSREF]
- Chin C L, Manzel L J, Lehman E E, Humlicek A L, Shi L, Starner T D, et al. Haemophilus influenzae from patients with chronic obstructive pulmonary disease exacerbation induce more inflammation than colonizers. Am J Respir Crit Care Med 2005; 172: 85–91, [INFOTRIEVE], [CSA], [CROSSREF]
- Vitovski S, Dunkin K T, Howard A J, Sayers J R. Nontypeable Haemophilus influenzae in carriage and disease: a difference in IgA1 protease activity levels. JAMA 2002; 287: 1699–1705, [INFOTRIEVE], [CSA], [CROSSREF]
- Bals R, Hiemstra P S. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J 2004; 23: 327–333, [INFOTRIEVE], [CSA], [CROSSREF]
- Murphy T F, Brauer A L, Grant B J, Sethi S. Moraxella catarrhalis in chronic obstructive pulmonary disease: burden of disease and immune response. Am J Respir Crit Care Med 2005; 172: 195–199, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Abe Y, Murphy T F, Sethi S, Faden H S, Dmochowski J, Harabuchi Y, et al. Lymphocyte proliferative response to P6 of Haemophilus influenzae is associated with relative protection from exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 165: 967–971, [INFOTRIEVE], [CSA]
- Aaron S D, Angel J B, Lunau M, Wright K, Fex C, Le Saux N, et al. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163: 349–355, [PUBMED], [INFOTRIEVE], [CSA]
- White A J, Gompertz S, Bayley D L, Hill S L, O'Brien C, Unsal I, et al. Resolution of bronchial inflammation is related to bacterial eradication following treatment of exacerbations of chronic bronchitis. Thorax 2003; 58: 680–685, [INFOTRIEVE], [CSA], [CROSSREF]
- Gompertz S, O'Brien C, Bayley D L, Hill S L, Stockley R A. Changes in bronchial inflammation during acute exacerbations of chronic bronchitis. Eur Respir J 2001; 17: 1112–1119, [INFOTRIEVE], [CSA], [CROSSREF]
- Sethi S, Muscarella K, Evans N, Klingman K L, Grant B JB, Murphy T F. Airway inflammation and etiology of acute exacerbations of chronic bronchitis. Chest 2000; 118: 1557–1565, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Stockley R A, O'Brien C, Pye A, Hill S L. Relationship of sputum color to nature and outpatient management of acute exacerbations of COPD. Chest 2000; 117: 1638–1645, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Banerjee D, Khair O A, Honeybourne D. Impact of sputum bacteria on airway inflammation and health status in clinical stable COPD. Eur Respir J 2004; 23: 685–691, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Sethi S, Wrona C, Grant B, Murphy T. Strain specific immune response to Haemophilus influenzae in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004; 169: 448–453, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Bogaert D, van der Valk P, Ramdin R, Sluijter M, Monninkhof E, Hendrix R, de Groot R, Hermans P W. Host-pathogen interaction during pneumococcal infection in patients with chronic obstructive pulmonary disease. Infect Immun 2004; 72: 818–823, [INFOTRIEVE], [CSA], [CROSSREF]
- Aaron S D, Ramotar K, Ferris W, Vandemheen K, Saginur R, Tullis E, Haase D, Kottachchi D, St. Deni M, Chan F. Adult cystic fibrosis exacerbations and new strains of Pseudomonas aeruginosa. Am J Respir Crit Care Med 2004; 169: 811–815, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Gibson R L, Burns J L, Ramsey B W. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003; 168: 918–951, [INFOTRIEVE], [CSA], [CROSSREF]
- Bandi V, Jakubowycz M, Kinyon C, Mason E O, Atmar R L, Greenberg S B, Murphy T F. Infectious exacerbations of chronic obstructive pulmonary disease associated with respiratory viruses and non-typeable Haemophilus influenzae. FEMS Immunol Med Microbiol 2003; 37: 69–75, [INFOTRIEVE], [CSA]
- Beadling C, Slifka M K. How do viral infections predispose patients to bacterial infections?. Curr Opin Infect Dis 2004; 17: 185–191, [INFOTRIEVE], [CSA], [CROSSREF]
- Soler N, Torres A, Ewig S, Gonzalez J, Celis R, El-Ebiary M, Hernandez C, Rodriguez-Roisin R. Bronchial microbial patterns in severe exacerbations of chronic obstructive pulmonary disease (COPD) requiring mechanical ventilation. Am J Respir Crit Care Med 1998; 157: 1498–1505, [INFOTRIEVE], [CSA]