Abstract
Chronic obstructive pulmonary disease (COPD) is a highly prevalent airway disease characterized by an abnormal inflammatory response of the lungs to noxious particles and gases. Cigarette smoking remains a major risk factor in COPD development. Accumulating evidence suggests that apoptosis, a regulated form of cell death, may play an important role in COPD pathogenesis. Increased numbers of apoptotic cells can be detected in lung tissue and airwaysof human subjects with COPD, relative to normal lungs or those from smokers without COPD. Alveolar wall destruction associated with emphysema development, may involve increased apoptosis of alveolar structural cells. Several intervention-induced apoptotic models (e.g., cigarette smoke, vascular-endothelial growth factor inhibition, and interferon-γ) cause emphysematous changes in vitro and in vivo. Increased apoptosis in COPD can also imply defects in the normal physiological clearance of apoptotic cells. Additional factors that relate to perpetuation of the pathogenesis of COPD, including protease/antiprotease imbalance, inflammation and oxidative stress, may mutually promote apoptosis or contribute to impaired clearance of apoptotic cells. Given that cigarette smoking is the most common cause of COPD, identification of the pathways of cigarette smoke-induced apoptosis may further the understanding of COPD pathogenesis. However, apoptosis rate is not diminished after cessation of cigarette smoking, indicating that other mechanisms perpetuate apoptosis in COPD. Therefore, understanding functional relationships between apoptosis and protease/antiprotease imbalance, inflammation, oxidative stress and other factors potentially involved in COPD pathogenesis may uncover crucial therapeutic targets.
Key words: :
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) has emerged as a major global health problem, predicted to become the third most common cause of death and the fifth most common cause of disability in the world by 2020 (Citation[1]). Despite enormous global importance, relatively little research has addressed the pathogenesis and management of COPD as compared to asthma.
The Global Initiative on Obstructive Lung Disease (GOLD) defined COPD as a disease state characterized by progressive airflow limitation that is not fully reversible, associated with an abnormal inflammatory response of the lungs to noxious particles and gases. This definition implies that COPD is a chronic inflammatory disease. Over the past 30 years, the prevailing concept to explain the pathogenesis of COPD has implicated lung inflammation caused by cigarette smoke, environmental pollutants, or bacterial products (Citation[2]). Lung inflammation caused by noxious particles (e.g., cigarette smoke) is generally associated with an influx of inflammatory cells (macrophages, neutrophils, CD8+ T lymphocytes) into the airways and lungs. Furthermore, a disruption of the balance between protease and antiprotease activities in COPD patients, resulting in the development of emphysema, is believed to represent a major component of COPD pathogenesis (Citation[3]) ().
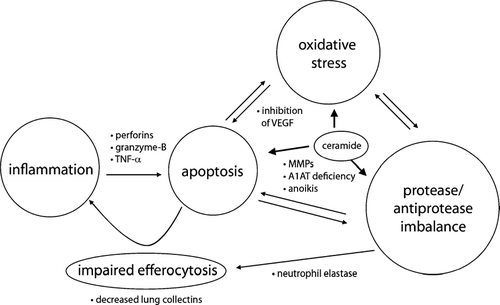
Figure 1 Interaction of apoptosis, inflammation, proteolysis and oxidative stress. Increased inflammatory cells (neutrophils, macrophages, cytotoxic CD8+ T-cells) cause apoptosis of alveolar epithelial cells through the release of perforins, granzyme-B, and TNFα. Anoikis, apoptosis by inadequate or inappropriate cell–matrix contacts, and direct proteolysis of death-inducing signals by MMP-7 mediate apoptosis. Alpha-1 antitrypsin (A1AT) inhibits active caspase-3, suggesting A1AT deficiency involves apoptotic mechanisms relevant to disease processes characterized by excessive structural cell apoptosis as emphysema. Oxidative stress could lead to a reduction of vascular endothelial growth factor (VEGF) levels, resulting in apoptosis of alveolar endothelial cells. Neutrophil elastase (NE) causes impaired clearance of apoptotic cells and sustained inflammation by acting on the phosphatidylserine receptor of macrophages. Up-regulation of ceramide production mediates alveolar cell apoptosis, oxidative stress, matrix proteolysis, and alveolar destruction in the emphysema model of VEGF receptor blockade.
This protease/antiprotease imbalance may arise from the activity of inflammatory cells recruited by cigarette smoke, or may evolve from genetically-related conditions such as alpha-1 antitrypsin deficiency. While the latter represents a typical example of protease/antiprotease imbalance, it accounts for the development of very few cases of COPD. Oxidative stress, which can amplify inflammation, may represent another important mechanism involved in the pathogenesis of COPD. Oxidative stress occurs when reactive oxygen species (ROS) are produced in excess of endogenous antioxidant defense mechanisms, resulting in cumulative damage to lipids, proteins and deoxyribonucleic acid (DNA) (Citation[4]).
Cigarette smoke itself contains a high concentration of ROS (Citation[5]), and may trigger additional ROS generation by inflammatory cells such as activated macrophages and neutrophils. More recently, many investigators have proposed that apoptosis subsequent to efferocytosis, which may be defective, plays an important role in the pathogenesis of emphysema (Citation[6]). This review will discuss recent data on the functional significance of apoptosis in COPD, with an emphasis on emphysema.
Identification of apoptosis in human lungs
The identification of alveolar cell apoptosis was initially described in emphysematous human lung tissue sections using in situ end labeling of fragmented DNA. Among the cells that showed positive signals, most were endothelial cells from capillaries and arterioles. Although less frequently, prominent intranuclear staining was also described in alveolar epithelial cells, interstitial cells, and inflammatory cells, while control lungs were negative (Citation[7]). Yokohori and colleagues showed that both apoptosis and proliferation of alveolar wall cells were significantly increased in patients with emphysema than in asymptomatic smokers and nonsmokers, using terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) and proliferation cell nuclear antigen (PCNA) staining (Citation[8]).
Similarly, lung cell (alveolar epithelial, endothelial, and mesenchymal) apoptosis and cell proliferation were increased in emphysematous lung tissue. Moreover, the activated subunits of caspase-3 and increased expression of pro-apoptotic proteins (ie., Bax and Bad) were detected in emphysema lungs, while the antiapoptotic protein Bcl-2 was not detected in either normal or emphysematous lung tissue (Citation[9]). Significant contacts exist among alveolar type I and type II epithelial, endothelial, and fibroblastic cells such that severe damage to one could reasonably lead to disruption of the other, and presumably of the matrix and interstitium as well (Citation[10]). Alveolar destruction requires that type II, type I, endothelial, and myofibroblastic cells are destroyed concordantly (Citation[11]).
In this regard, these structural contacts provide a potential mechanism for the pathogenic dysfunction of all alveolar septal cells caused by cigarette smoke exposure. Kasahara and colleagues demonstrated an increase in apoptotic epithelial and endothelial alveolar septal cells in emphysematous lungs compared to the lungs from nonsmokers, healthy smokers, and patients with primary pulmonary hypertension (Citation[12]). In addition, they described no significant difference in apoptosis indicators between healthy nonsmokers and smokers without emphysema (Citation[12]).
In agreement with this finding, recent data has demonstrated that apoptosis persists despite smoking cessation, suggesting that cigarette smoke itself is not the sole agent causing apoptosis once COPD is established, although COPD individuals are more likely susceptible to smoke-induced cell damage and apoptosis (Citation[13]). This finding indicates that factors including protease/antiprotease imbalance, inflammation and oxidative stress that relate to the pathogenesis of emphysema may also contribute to increased apoptosis (Citation[13]). Furthermore, genetic alterations in cellular responses to apoptotic stimuli may also play contributory roles.
INTERVENTION-INDUCED APOPTOSIS IN EMPHYSEMA
Cigarette smoke
Cigarette smoke, a complex mixture of more than 4,800 chemical compounds, contains high concentrations of free radicals and other oxidants (Citation[14]). Free radicals in cigarette smoke are derived from both the gas and the tar phases. The gas-phase cigarette smoke contains approximately 1015 radicals per puff, primarily of the alkyl and peroxyl types. In addition, nitric oxide is present in cigarette smoke in concentrations of 500–1,000 ppm (Citation[15]). The radicals in the tar phase of cigarette are more stable and are predominantly organic, such as the semiquinone radical. Superoxide (O2· −), hydroxyl radical (· OH) and hydrogen peroxide (H2O2), have also been detected in the tar phase, and arise during the autoxidation of catechol and hydroquinone components of tar, possibly involving interactions with metal ions (Citation[15], Citation[16]).
Cigarette smoke tar contains more than 1018 free radicals per gram (Citation[16]). Whereas short-lived radicals in the gas phase of cigarette smoke may be quenched immediately in the epithelial lining fluid (ELF), redox reactions in cigarette smoke condensate, which forms in the epithelial lining fluid, may produce reactive oxygen species (ROS) for a considerable time (Citation[17]). Although the mechanisms by which cigarette smoke causes deleterious effects on cells are not well understood, free radicals and ROS may play a major role in cigarette smoke-induced apoptosis.
In support of this hypothesis, both oxidative stress and apoptosis exhibited a dose-response relationship with cigarette smoke extract (CSE) concentrations in human lung fibroblasts (Citation[18]). Cigarette smoke-induced apoptosis, now thought to be one of the pathogenic mechanisms in emphysema, was observed in a variety of human cells including monocytic cell line U-937, umbilical vein endothelial cells, alveolar macrophages, bronchial epithelial cells, and the HFL-1 fibroblast strain (Citation[18], Citation[19], Citation[20], Citation[21], Citation[22]). In in vivo studies, exposure of rats to mainstream cigarette smoke for either 18 or 100 consecutive days produced a significant and time-dependent increase in the proportion of apoptotic cells in the bronchial and bronchiolar epithelium (Citation[23]).
Smoke treatment produced an increase in the level of proapoptotic proteins in the terminal bronchiole areas of the lung tissue from rats (Citation[24]). In vitro studies using alveolar macrophages showed that cell death by CSE occurred in a dose- and time-dependent manner, with 93% of cells showing apoptosis after 24 hr exposure to 10% CSE solution (Citation[21]). CSE induced apoptosis at lower concentrations (10–25%) and necrosis at higher concentrations (50–100%) in human lung fibroblasts (Citation[25]). CSE activated the caspases-3 and -8, the p38 mitogen activated protein kinase (p38 MAPK), and the stress activated protein kinase/c-Jun N-terminal protein kinase (SAPK/JNK) in human aortic endothelial cells (HAEC) (Citation[26]). On the other hand, some contradiction has arisen in terms of how cigarette smoking affects this apoptotic process, because both stimulatory and inhibitory potential of smoking on apoptosis have been described.
Cigarette smoke stimulated caspase 3 precursors as well as intact poly (ADP-ribose) polymerase (PARP) production, but did not activate caspase 3 or cleave PARP in human bronchial epithelial cells (Citation[27]). Furthermore CSE-induced DNA damage was reversible in human lung fibroblasts, and cells proliferated when CSE was removed after 24 h exposure (Citation[28]). Cell death in response to cigarette smoke by necrosis may be responsible for the loss of alveolar walls and inflammation observed in emphysema (Citation[29]).
However, some limitations in CSE experiments may occur in such studies, which can complicate interpretation. First, variations in the method of 100% CSE preparation can occur between laboratories, for example, prepared at a concentration of 1 cigarette/25 ml in double-distilled water (Citation[27]), RMPI (Citation[28]) or 1 cigarette/ml in calcium magnesium-free phosphate-buffered saline (Citation[29]). Because the effect on cells is dependent on the duration of smoke exposure and the concentration of smoke, such variations can lead to differential results even in the same cell lines with regard to cell death. Secondly, several kinds of detection techniques for apoptosis have been developed and used, each with its advantages and limitations. For example, cells with positive TUNEL staining or single-stranded-DNA breaks may or may not proceed to apoptotic death. Furthermore, the annexin-V affinity assay is not suitable for attached cells because the enzymatic detaching of the cells may cause false-positive results.
Considering these limitations, there is no doubt that cigarette smoking causes apoptosis in a variety of cells. It was reported that the level of proapoptotic proteins (caspase-8, caspase-3, Bax, t-Bid, and cytochrome c) was increased in the lungs of cigarette smoke exposed rats (Citation[30]) with immunochemical staining, indicating cigarette smoke sensitizes the lung structural cells toward receptor-dependent as well as mitochondrial-dependent apoptotic pathways. In addition, the level of cytokines (e.g., TNF-α, TGF-β 1), and Fas was increased in the peripheral blood of COPD patients (Citation[31]).
These findings suggest that the Fas-mediated apoptotic pathway may participate in the pathogenesis of COPD (). Smoking-induced mitochondrial dysfunction has also been suggested as a prerequisite step toward cell death through the intrinsic apoptotic pathway (Citation[32]). Our laboratory demonstrated that CSE caused loss of cellular ATP and rapid depolarization of mitochondrial membrane potential. Furthermore, we have recently described the functional compartmentalization of heme oxygenase-1 (HO-1) in the mitochondria of lung epithelial cells, and its potential role in defense against mitochondria-mediated cell death during cigarette smoke extract exposure (Citation[33]).
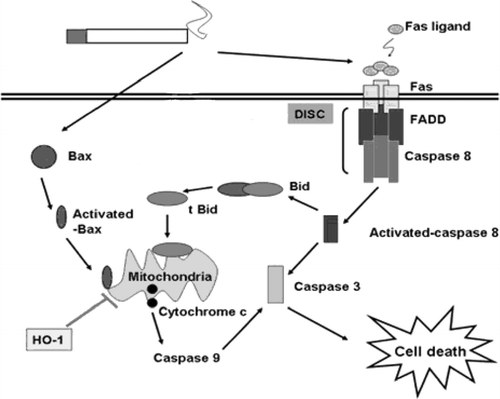
Figure 2 Pathways of cigarette smoke-induced cell death. The diagram depicts the pathways by which cigarette smoke triggers cell death. Cigarette smoke triggers both death receptor-dependent (extrinsic) apoptotic and mitochondrial (intrinsic) pathways. In the extrinsic apoptotic pathway, cigarette smoke may trigger a Fas ligand independent pathway. Cigarette smoke induces the assimilation of the death inducing signaling complex (DISC) in the plasma membrane. HO-1 inhibits mitochondria-mediated cell death during cigarette smoke extract exposure.
Inhibition of vascular endothelial growth factor (VEGF)
VEGF is a family of ligands (A–D), which binds two related receptor tyrosine kinases (RTK), VEGF receptor-1 (VEGFR-1) and VEGFR-2. VEGF-A, a key regulator of blood vessel growth, stimulates endothelial cell and type II cell growth and survival by binding VEGFR-2 (Citation[34]). Importantly, increasing evidence suggests that epithelial and endothelial alveolar septal death due to a decrease of endothelial cell maintenance factors may contribute to the pathogenesis of emphysema, as demonstrated by VEGF-R blockade (Citation[35]), VEGF gene reduction (Citation[36]), or anti-endothelial cell antibodies (Citation[37]), which cause increased alveolar cell apoptosis and airspace enlargement in animal studies. VEGF and VEGFR-2 protein and mRNA expression are decreased in human emphysematous lungs (Citation[12]).
A mimetic of superoxide dismutase prevented alveolar enlargement, alveolar septal cell apoptosis, and expression of markers of oxidative stress in an animal model treated with the VEGF receptor blocker (Citation[38]), which supports a relationship between oxidative stress and apoptosis in the pathogenesis of COPD. Recently, it was reported that cigarette smoke exposure reduced neuropilin (NRP)-1 and glypican (GYP)-1, which form the VEGFR-2 tertiary complex, as well as VEGF and VEGFR-2 levels in rat lungs. Furthermore, VEGF, VEGFR-2, GYP-1, and NRP-1 expression were decreased in the lungs of both smokers and COPD patients compared with nonsmokers (Citation[39]). Collectively, apoptosis plays an important role of alveolar destruction in a variety of study models involving VEGF inhibition.
Interferon gamma
Interferon-γ (IFN-γ), originally defined based on its antiviral activity, is now recognized as the prototypic Th1/Tc1 cytokine and exerts key roles in immune regulation, cellular proliferation, microbicidal effector activation, and leukocyte trafficking (Citation[40]). Wang and colleagues first reported that IFN-γ, a prominent product of CD8+ cells, causes emphysema with alveolar enlargement, enhanced lung volumes, enhanced pulmonary compliance, and macrophage- and neutrophil-rich inflammation in a transgenic adult murine lung (Citation[41]). They also demonstrated that, in transgenic mice lung, IFN-γ caused epithelial cell DNA damage and apoptosis, which were associated with death receptor and mitochondrial apoptosis pathway activation. In this model, cathepsin S plays an important role in IFN-γ -induced DNA damage, apoptosis, emphysema, inflammation, and protease accumulation (Citation[42]).
INTERACTION OF APOPTOSIS, INFLAMMATION, PROTEOLYSIS AND OXIDATIVE STRESS
Efferocytosis, inflammation and apoptosis
Cells undergoing programmed cell death can be removed rapidly and efficiently in situ by not only the mononuclear phagocyte system but also by other cell types, including fibroblasts and endothelial, epithelial, smooth muscle, and stromal cells. Removal of apoptotic bodies precludes damage to the tissue by pro-inflammatory processes. This uptake process is highly conserved, uses unique receptors and signaling pathways and is referred to as efferocytosis (Citation[43], Citation[44]).
Recently, it has been shown that apoptotic cell recognition can also induce production of tissue cell growth factors or other stimuli that may contribute to replenishment of the damaged cells under normal circumstances (Citation[45]). There is compelling evidence that alveolar macrophages from patients with COPD are less effective in the phagocytosis of apoptotic airway epithelial cells compared to controls (Citation[6]). There are several potential explanations for defective phagocytosis.
Increased neutrophil elastase production in COPD patients cleaves the phosphatidylserine receptor on macrophages, resulting in impaired clearance of apoptotic cells and sustained inflammation (Citation[44], Citation[46]). This effect could link the protease balance hypothesis for COPD with the efficiency of apoptotic cell uptake. Another factor, the lung collectins, surfactant proteins A and D (SP-A and SP-D), which were decreased in the lungs of smokers, have both been shown to promote the uptake of apoptotic cells. SP-D-/- mice were shown to exhibit a lung clearance defect in vivo (Citation[47]).
Although it is demonstrated that apoptosis of alveolar wall or endothelial cells can cause pulmonary emphysema without the accumulation of inflammatory cells in some animal models, there is no doubt there is an impressive influx of inflammatory cells, and corresponding interactions between inflammatory and apoptotic mechanisms likely take place in the lungs of COPD patients. This might be mediated by the presence of activated neutrophils resistant to apoptosis in COPD (Citation[50], Citation[51]). In addition to neutrophils and macrophages, CD8+ T-lymphocytes, which could cause apoptosis of alveolar epithelial cells through the release of perforins, granzyme-B and TNF-α, are increased in the lungs of COPD patients (Citation[48], Citation[49]).
In conclusion, impaired apoptotic cell clearance, as it may aggravate inflammation, may have many consequences on the pathogenesis of COPD, and also may represent a potential target for therapy.
Protease/anti-protease imbalance and apoptosis
For a long time, protease/antiprotease imbalance has been a main stem theory in COPD. However, some evidence suggests potential interactions between apoptosis and matrix proteolysis. Apoptosis may be affected by direct proteolysis of death-inducing signaling components. The Fas ligand was identified as a matrix metalloproteinase (MMP) substrate although the activity of the solubilized Fas ligand is disputed (Citation[52]). MMP-7 activated soluble Fas ligand, which appears to be important for apoptosis during prostate involution (Citation[53]). Neutrophil elastase cleaves the phosphatidylserine receptor on macrophages, causing the impaired clearance of apoptotic cells in cystic fibrosis and bronchiectasis (Citation[46]). Cigarette smoke stimulates the production of the sphingolipid, ceramide, which has been shown to mediate alveolar cell apoptosis, oxidative stress, matrix proteolysis, and alveolar destruction in the emphysema model of VEGF receptor blockade (Citation[54]).
Anoikis, the subset of apoptosis triggered by inadequate or inappropriate cell–matrix contacts, maintains the correct cell number in high-turnover epithelial tissues. The structural interaction among alveolar type I and type II, endothelial, and fibroblastic cells in the alveolar structure is uniquely close such that it is conceivable that they coordinate their response to injury or stresses, and that coordinated cell death and extracellular matrix degradation can cause emphysematous lung (Citation[10]).
Aoshiba et al. suggested that cell-extracellular matrix interactions modulate apoptosis in bronchial epithelium (Citation[55]). However, the cross-talk among alveolar septal cells remains unclear at present. On the other hand, the well known protease inhibitor, alpha-1 antitrypsin (A1AT) inhibits staurosporine-induced caspase-3 activation, suggesting that direct inhibition of active caspase-3 by A1AT may represent a novel anti-apoptotic mechanism relevant to disease processes characterized by excessive structural cell apoptosis, such as emphysema (Citation[56]).
Oxidative stress and apoptosis
It is well known that oxidative stress plays a major role in COPD (Citation[57]), with increased expression of markers of oxidative stress systemically and locally in patients with COPD (Citation[58], Citation[59]). The mechanisms by which increased endogenous ROS production triggers apoptosis are incompletely understood, but appear to involve mitochondrial dysfunction, and release of apoptogenic factors (Citation[60]). Modulations of major MAPK activities, as well as the generation of lipid mediators such as 4-hydoxynonenal and ceramide may be involved (Citation[60]). Furthermore ROS may induce apoptosis by direct DNA damage and activate polyadenosine diphosphate ribose polymerase.
Activation of the NF-κ B pathway, which regulates multiple pro-inflammatory genes, may lead to amplification of the inflammatory response. It was demonstrated that antioxidant defenses determine susceptibility to emphysema in mice deficient in the master transcription factor NF-E2 related factor-2 (Nrf2), which regulates multiple critical antioxidant enzymes (Citation[61]). The interaction between oxidative stress and apoptosis was shown in a rat model of experimental emphysema induced by VEGFR blockade. In this study, apoptosis predominated in areas of oxidative stress, and apoptosis blockade by a broad spectrum caspase-inhibitor markedly reduced the expression of markers of oxidative stress. Because blockade of apoptosis prevented oxidative stress and emphysema in this model, it was proposed that oxidative stress and apoptosis mutually interact in the process of alveolar cell destruction (Citation[39]).
SUMMARY
Ample studies have shown increased apoptosis and expression of apoptogenic factors in COPD, both from human subjects and animal models. As many studies show increasing evidence of interactions of apoptosis with other important factors; such as inflammation, protease-antiprotease imbalance and oxidative stress, the functional role of apoptosis in COPD pathogenesis has been unveiled (). Recent studies have also related these mechanisms with defects in the clearance of apoptotic cells. Although cigarette smoking is not the sole agent causing apoptosis in COPD once it is established, a better understanding of the apoptotic signaling pathways triggered by cigarette smoke, may provide novel therapies for COPD.
REFERENCES
- Lopez A D, Murray C C. The global burden of disease, 1990−2020. Nature Med 1998; 4: 1241–1243
- Shapiro S D. The pathogenesis of emphysema: the elastase:antielastase hypothesis 30 years later. Proc Assoc Am Physicians 1995; 107: 346–352
- Demedts I K, Brusselle G G, Bracke K R, Vermaelen K Y, Pauwels R A. Matrix metalloproteinases in asthma and COPD. Curr Opin Pharmacol 2005; 5: 257–263
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 2003; 22: 672–688
- Pryor W A, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann NY Acad Sci 1993; 686: 12–27
- Hodge S, Hodge G, Scicchitano R, Reynolds P N, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol 2003; 81: 289–296
- Segura-Valdez L, Pardo A, Gaxiola M, Uhal B D, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000; 117: 684–694
- Yokohori N, Aoshiba K, Nagai A. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 2004; 125: 626–632
- Imai K, Mercer B A, Schulman L L, Sonett J R, D'Armiento J M. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J 2005; 25: 250–258
- Sirianni F E, Chu F SF, Walker D C. Human alveolar wall fibroblasts directly link epithelial type 2 cells to capillary endothelium. Am J Respir Crit Care Med 2003; 168: 1532–1537
- Tuder R M, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. cellular and molecular mechanisms of alveolar destruction in emphysema. Proc Am Thorac Soc 2006; 3: 503–510
- Kasahara Y, Tuder R M, Cool C D, Lynch D A, Flores S C, Voelkel N F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 2001; 163: 737–744
- Hodge S, Hodge G, Holmes M, Reynolds P N. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J 2005; 25: 447–454
- Church D F, Pryor W A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect 1985; 64: 111–126
- Pryor W A, Stone K. Oxidants in cigarette smoke: radicals, hydrogenperoxides, peroxynitrate, and peroxynitrite. Ann N Y Acad Sci 1993; 686: 12–28
- Zang L Y, Stone K, Pryor W A. Detection of free radicals in aqueous extracts of cigarette tar by electron spin resonance. Free Radic Biol Med 1995; 19: 161–167
- MacNee W, Rahman I. Oxidants and antioxidants as therapeutic targets in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 160: S58–S65
- Carnevali S, Petruzzelli S, Longoni B, Vanacore R, Barale R, Cipollini M, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2003; 284: L955–L963
- Sugano N, Ito K. Nicotine switches the form of H2O2 induced cell death from apoptosis to necrosis in U937 cells. Immunol Lett 2000; 70: 163–166
- Wang J, Wilcken D E, Wang X L. Cigarette smoke activates caspase-3 to induce apoptosis of human umbilical venous endothelial cells. Mol Genet Metab 2001; 72: 82–88
- Aoshiba K, Tamaoki J, Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2001; 281: L1392–L1401
- Hoshino Y, Mio T, Nagai S, Miki H, Ito I, Izumi T. Cytotoxic effects of cigarette smoke extract on an alveolar type II cell-derived cell line. Am J Physiol Lung Cell Mol Physiol 2001; 281: L509–L516
- D'Agostini F, Balansky R M, Izzotti A, Lubet R A, Kelloff G J, De Flora S. Modulation of apoptosis by cigarette smoke and cancer chemopreventive agents in the respiratory tract of rats. Carcinogenesis 2001; 22: 375–380
- Kuo W H, Chen J H, Lin H H, Chen B C, Hsu J D, Wang C J. Induction of apoptosis in the lung tissue from rats exposed to cigarette smoke involves p38/JNK MAPK pathway. Chem Biol Interact 2005; 155: 31–42
- Ishii T, Matsuse T, Igarashi H, Masuda M, Teramoto S, Ouchi Y. Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am J Physiol Lung Cell Mol Physiol 2001; 280: L1189–L1195
- Raveendran M, Wang J, Senthil D, Wang J, Utama B, Shen Y, et al. Endogenous nitric oxide activation protects against cigarette smoking induced apoptosis in endothelial cells. FEBS Lett 2005; 579: 733–740
- Liu X, Conner H, Kobayashi T, Kim H, Wen F, Abe S, et al. Cigarette smoke extract induces DNA damage but not apoptosis in human bronchial epithelial cells. Am J Respir Cell Mol Biol 2005; 33: 121–129
- Kim H, Liu X, Kobayashi T, Conner H, Kohyama T, Wen F Q, Fang, et al. Reversible cigarette smoke extract–induced DNA damage in human lung fibroblasts. Am J Respir Cell Mol Biol 2004; 31: 483–490
- Wickenden J A, Clarke M C, Rossi A G, Rahman I, Faux S P, Donaldson K, et al. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am J Respir Cell Mol Biol 2003; 29: 562–570
- Wu C H, Lin H H, Yan F P, Wu C H, Wang C J. Immunohistochemical detection of apoptotic proteins, p53/Bax and JNK/FasL cascade, in the lung of rats exposed to cigarette smoke. Arch Toxicol 2005; 9: 1–9
- Hodge S J, Hodge G L, Reynolds P N, Scicchitano R, Holmes M. Increased production of TGF-beta and apoptosis of T lymphocytes isolated from peripheral blood in COPD. Am J Physiol Lung Cell Mol Physiol 2003; 285: L492–L499
- Banzet N, Francois D, Polla B S. Tobacco smoke induces mitochondrial depolarization along with cell death effects of antioxidants. Redox Rep 1999; 4: 229–236
- Slebos D J, Ryter S W, van der Toorn M, Liu F, Guo F, Baty C J, et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol 2006, (in press)
- Ferrara N, Gerber H P, Le Couter J. The biology of VEGF and its receptors. Nat Med 2003; 9: 669–676
- Kasahara Y, Tuder R M, Taraseviciene-Stewart L, Le Cras T D, Abman S H, Hirth P, et al. Inhibition of vascular endothelial growth factor receptors causes lung cell apoptosis and emphysema. J Clin Invest 2000; 106: 1311–1319
- Tang K, Rossiter H B, Wagner P D, Breen E C. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J Appl Physiol 2004; 97: 1559–1566
- Taraseviciene-Stewart L, Scerbavicius R, Choe K H, Moore M, Sullivan A, Nicolls M R, et al. An animal model of autoimmune emphysema. Am J Respir Crit Care Med 2005; 171: 734–742
- Tuder R M, Zhen L, Cho C Y, Taraseviciene-Stewart L, Kasahara Y, Salvemini D, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol 2003; 29: 88–97
- Marwick J A, Stevenson C S, Giddings J, MacNee W, Butler K, Rahman I, Kirkham P A. Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol 2006; 290: L897–L908
- Schroder K, Hertzog P J, Ravasi T, Hume D A. Interferon-γ: an overview of signals, mechanisms and functions. J Leukocyte Biol 2004; 75: 163–189
- Wang Z, Zheng T, Zhu Z, Homer R J, Riese R J, Chapman H A, et al. Interferon-γ induction of pulmonary emphysema in the adult murine lung. J Exp Med 2000; 192: 1587–1600
- Zheng T, Kang M J, Crothers K, Zhu Z, Liu W, Lee C G, et al. Role of Cathepsin S-dependent epithelial cell poptosis in IFN-γ -Induced alveolar remodeling and pulmonary emphysema. J Immunol 2005; 174: 8106–8115
- deCathelineau A M, Henson P M. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem 2003; 39: 105–117
- Gardai S J, Bratton D L, Ogden C A, Henson P M. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol 2006; 79: 896–903
- Golpon H A, Fadok V A, Taraseviciene-Stewart L, Scerbavicius R, Sauer C, Welte T, et al. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J 2004; 18: 1716–1718
- Vandivier R W, Fadok V A, Hoffmann P R, Bratton D L, Penvari C, Brown K K, et al. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest 2002; 109: 661–670
- Vandivier R W, Ogden C A, Fadok V A, Hoffmann P R, Brown K K, Botto M, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol 2002; 169: 3978–3986
- Keatings V M, Collins P D, Scott D M, Barnes P J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153: 530–534
- Lacoste J Y, Bousquet J, Chanez P, Van Vyve T, Simony-Lafontaine J, Lequeu N, et al. Eosinophilic and neutrophilic inflammation in asthma, chronic bronchitis, and chronic obstructive pulmonary disease. J Allergy Clin Immunol 1993; 92: 537–548
- Saetta M, Di Stefano A, Turato G, Facchini F M, Corbino L, Mapp C E, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157: 822–826
- Liu A N, Mohammed A Z, Rice W R, Fiedeldey D T, Liebermann J S, Whitsett J A, et al. Perforin-independent CD8(+) T-cell-mediated cytotoxicity of alveolar epithelial cells is preferentially mediated by tumor necrosis factor-alpha: relative insensitivity to Fas ligand. Am J Respir Cell Mol Biol. 1999; 20: 849–858
- Kayagaki N, Kawasaki A, Ebata T, Ohmoto H, Ikeda S, Inoue S, Yoshino K, Okumura K, Yagita H. Metalloproteinase-mediated release of human Fas ligand. J Exp Med 1995; 182: 1777–1783
- Powell W C, Fingleton B, Wilson C L, Boothby M, Matrisian L M. The metalloproteinase matrilysin (MMP-7) proteolytically generates active soluble Fas ligand and potentiates epithelial cell apoptosis. Curr Biol 1999; 9: 1441–1447
- Petrache I, Natarajan V, Zhen L, Medler T R, Richter A T, Cho C, Hubbard W C, Berdyshev E V, Tuder R M. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 2005; 11: 491–498
- Aoshiba K, Rennard S I, Spurzem J R. Cell-matrix and cell-cell interactions modulate apoptosis of bronchial epithelial cells. Am J Physiol 1997; 272: L28–L37
- Petrache I, Fijalkowska I, Medler T R, Skirball J, Cruz P, Zhen L, Petrache H I, Flotte T R, Tuder R M. α -1 Antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol 2006; 169: 1155–1166
- MacNee W. Oxidants/antioxidants and chronic obstructive pulmonary disease: pathogenesis to therapy. Novartis Found Symp 2001; 234: 169–185
- Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996; 154: 1055–1060
- Rahman I, van Schadewijk A AM, Crowther A JL, Hiemstra P S, Stolk J, MacNee W, De Boer W I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166: 490–495
- Ryter S, Kim H P, Hoetzel A, Park J W, Wang X, Choi A M. Mechanisms of cell death in oxidative stress. Antioxidants Redox Signali 2007; 9: 49–89
- Rangasamy T, Cho C Y, Thimmulappa R K, Zhen L, Srisuma S S, Kensler T W, Yamamoto M, Petrache I, Tuder R M, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 2004; 114: 1248–1259