Abstract
The current clinical classification of smoking-related lung disease fails to take into account the heterogeneity of chronic obstructive pulmonary disease (COPD). With an increased understanding of pathophysiologic variation, COPD now clearly represents a spectrum of overlapping diseases with important extrapulmonary consequences. A “phenotype” describes the outward physical manifestations of a particular disease, and compromises anything that is part of the observable structure, function or behavior of an individual. Such phenotypic distinctions in COPD include: frequent exacerbator, pulmonary cachectic, rapid decliner, airways hyperresponsiveness, impaired exercise tolerance, and emphysema versus airways disease. These variable manifestations, each with unique prognostic, clinical and physiologic ramifications, represent distinct phenotypes within COPD. While all of these phenotypes have smoking as a common risk factor, the other risk factors that determine these phenotypes remain poorly understood. An individual smoker has variable expression of each phenotype and there is mounting evidence that COPD phenotypes have different clinical outcomes. These phenotypes can be broadly classified into one of three groups: clinical, physiologic and radiographic. This review presents the evidence for the spectrum of COPD phenotypes with a focused discussion on the pathophysiologic, epidemiologic and clinical significance of each subtype.
INTRODUCTION
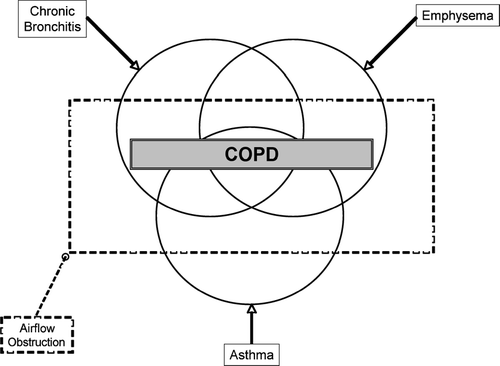
Clinicians have been long aware that neither the traditional distinctions of “emphysema” versus “chronic bronchitis” nor the traditional clinical phenotypes of “blue bloater” and “pink puffer” are sufficient to categorize patents that suffer form smoking-related lung disease. More recently, the Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop has used quantitative measures (FEV1 and FEV1/FVC ratio) to define COPD, despite the fact that the neither emphysema nor chronic bronchitis have airflow limitation in their definition (Citation[1], Citation[2]). Furthermore, the historical Venn diagram illustrating the overlapping disease entities included in the definition of chronic airflow obstruction () fails to take into account the full heterogeneity of chronic obstruction pulmonary disease (COPD) (Citation[3]). While all of these phenotypes have smoking as a common risk factor, the other risk factors that determine these phenotypes remain poorly understood. Furthermore, an individual smoker has variable expression of each phenotype and there is mounting evidence that COPD phenotypes have different clinical outcomes.
Figure 1 The classic Venn diagram used to describe the overlapping disease entities included in the definition of COPD (Source: (3), , p. 578).
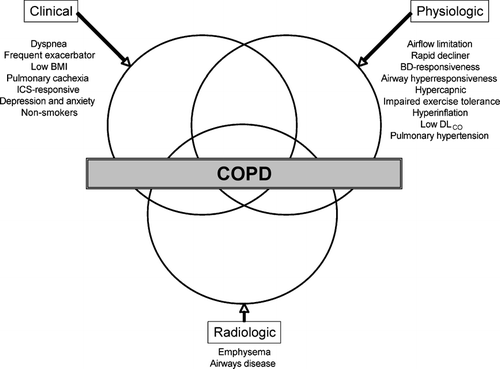
Over the past several decades, with an increased understanding of the heterogeneity and pathophysiologic variation, COPD now clearly represents a spectrum of overlapping diseases. These variable manifestations, each with unique prognostic, clinical and physiologic ramifications, represent distinct phenotypes within COPD. These phenotypes can be broadly classified into one of three groups: clinical, physiologic and radiographic (). The aim of this review is to present the evidence for the spectrum of COPD phenotypes with a focused discussion on the pathophysiologic, epidemiologic and clinical significance of each subtype.
Figure 2 Revised Venn diagram taking into account the expanded heterogeneity of COPD (BMI, body-mass index; ICS, inhaled corticosteroid; BD, bronchodilator).
CLINICAL PHENOTYPES OF COPD
There has been an increased understanding over the past several years that certain clinical features, when present, can predict worsening functional status, and thereby are associated with increased morbidity and mortality. Historically, these prognostic factors were age, resting hypoxemia and the post-bronchodilator FEV1 (Citation[2], Citation[4], Citation[5]). However, there exists considerable variation in outcome, performance status and symptomatology, especially among elderly patients with severe COPD (Citation[6]).
Dyspnea
Although the FEV1 correlates with mortality and morbidity, dyspnea varies considerably for the same degree of airflow limitation (Citation[7]). The degree of breathlessness is one of the major symptoms that impacts patients' quality of daily life (Citation[8], Citation[9]). From both a clinical and research perspective, patients with COPD can be categorized into subgroups based upon their level of dyspnea. Although there may be, in general, overlap between degree of dyspnea and airflow limitation, an assessment of breathlessness is independent of a given FEV1(Citation[7]).
The most commonly used tool to assess severity of breathlessness is the modified Medical Research Council (MRC) dyspnea scale (Citation[9], Citation[10]). The use of this scale is in contrast to disease-specific or generic measures of health related quality of life (HRQOL), such as the St. George's respiratory questionnaire (SGRQ) or the Medical Outcomes Study Short Form 36-item questionnaire (SF-36), respectively. These, and other, quality of life assessments can be used in conjunction with subjective measures of dyspnea and objective determinants of disease severity (such as the FEV1) to better categorize patients with COPD. The simplest example of this approach is to categorize patients into 3 levels: no or little dyspnea, dyspnea on exertion, and dyspnea on any exertion or at rest (Citation[9], Citation[11]).
Hajiro et al. used a more comprehensive approach in 194 male patients with stable, mild-to-severe COPD, and was able to show that, by using the SGRQ and SF-36 in conjunction with the MRC dyspnea scale, the HRQOL of patients was more clearly separated by the level of dyspnea than by the ATS disease staging (Citation[9]). A more recent 5-year prospective Japanese study of 183 patients using the same methodology showed the level of dyspnea was significantly correlated with survival (p < 0.001), while the effect of disease staging (as evaluated by the percentage of predicted FEV1) on the 5-year survival rate was not significant (p = 0.08) (Citation[12]). The increased realization that measurements of breathlessness, in conjunction with degree of airflow limitation and other objective surrogates of disease, are important to fully assess the complex systemic nature of COPD have led to the development of composite indices of severity and prognosis. The best-validated example of this is the BODE index (body-mass index, degree of airflow obstruction, dyspnea, and exercise capacity), which has been prospectively shown to be a better predictor of mortality than the FEV1 (Citation[13]) ().
Figure 3 Kaplan–Meier survival curves for the 4 quartiles on the BODE index (Panel A) and the 3 stages of severity of COPD as defined by the ATS (Panel B). Source: (Citation[13]), , p. 1010. In Panel A, quartile 1 is a score of 0 to 2, quartile 2 is a score of 3 to 4, quartile 3 a score of 5 to 6, and quartile 4 a score of 7 to 10. Survival differed significantly among the 4 groups (p < 0.0001). In Panel B, stage I is defined by an FEV1 > 50% predicted, stage II by an FEV1 36–50% predicted, and stage III by an FEV1 ≤ 35% predicted. Survival differed significantly among the 3 groups (p < 0.001).
![Figure 3 Kaplan–Meier survival curves for the 4 quartiles on the BODE index (Panel A) and the 3 stages of severity of COPD as defined by the ATS (Panel B). Source: (Citation[13]), Fig. 1, p. 1010. In Panel A, quartile 1 is a score of 0 to 2, quartile 2 is a score of 3 to 4, quartile 3 a score of 5 to 6, and quartile 4 a score of 7 to 10. Survival differed significantly among the 4 groups (p < 0.0001). In Panel B, stage I is defined by an FEV1 > 50% predicted, stage II by an FEV1 36–50% predicted, and stage III by an FEV1 ≤ 35% predicted. Survival differed significantly among the 3 groups (p < 0.001).](/cms/asset/3274b8e1-afc9-4f00-bf98-02cc7d0451ec/icop_a_262814_uf0003_b.gif)
Frequent exacerbator
When assessing prognosis, there has also been a shift to look at the effects of “hard” intermediate clinical endpoints, such as hospitalization for an acute exacerbation of COPD (AECOPD). A prior exacerbation has been shown in multiple studies to be an independent risk factor for future hospitalization from COPD (Citation[14],Citation[15],Citation[16],Citation[17],Citation[18]). Exacerbations not only lead to increased morbidity and mortality from COPD, but are a primary contributor to the cost of illness in both the hospital and outpatient setting (Citation[19]). The direct and indirect costs of COPD are estimated to be at around $32 billion yearly; in 2002, the costs of hospitalization for an exacerbation constituted 40.6% ($7.3 billion) of direct COPD costs (Citation[19]). On a national scale, an estimated 25% of cases presenting to the emergency room for dyspnea have been attributed to COPD-related exacerbations (Citation[19], Citation[20]). It can be safely concluded that any ongoing and future attempts to reduce the economic and health burden from COPD needs to focus on further defining and identifying the frequent exacerbator phenotype.
Hospitalization has both an immediate- and long-term risk for mortality. In an observational study of more than 72,000 patients admitted for AECOPD, there was a 2.5% in-hospital mortality rate, and those that died were older, had more comorbidities, and a longer length of hospitalization (Citation[21]). In an uncontrolled prospective study of 171 patients with severe COPD (mean FEV1 34.6%) presenting with an acute exacerbation, there was a 23% observed mortality rate in the year after the index hospitalization (Citation[22]). The sustained effects of hospitalization for AECOPD have been found to extend beyond one year. The APACHE II score on admission to a general medicine ward correlates with 3-year mortality, with 50% mortality at 3 years for an admission APACHE II score greater than 20 (Citation[23]).
Hospitalization in the year prior to a subsequent admission for AECOPD has also been found to be a strong predictor for early mortality (Citation[24]). This observation helps define an important subgroup of COPD patients with advanced disease that may represent a “frequent exacerbator” phenotype. Additionally, this is the group that benefits most from inhaled corticosteroids (Citation[25], Citation[26]), with a pooled analysis suggesting a 25% reduction in all-cause mortality over 1 year as compared to placebo (Citation[27]). Identification and close follow-up of these patients, with an emphasis placed on early recognition and treatment of acute exacerbations, will likely lead to decreased morbidity, improved healthcare utilization, and, hopefully, improved mortality.
Low BMI
Unexplained weight loss in patients with COPD is an independent predictor of increased mortality (Citation[28],Citation[29],Citation[30]) and impacts functional capacity and health-related quality of life (Citation[31]). Body-composition categories are associated with GOLD stage of disease severity (Citation[29]) (). In a French national database cohort study, 4,088 patients with chronic bronchitis or emphysema requiring long-term oxygen therapy were identified; at baseline, 23% of men and 30% of women were malnourished (BMI < 20), and BMI was significantly correlated with degree of airflow limitation (Citation[32]). At a mean follow-up of 7.5 years, BMI correlated with mortality, and the effect of BMI on survival was independent of age, FEV1, PaO2, and sex ().
Figure 4 Body-composition categories are linked to GOLD disease severity. Source: (Citation[29]), , p. 56. Cachexia (N = 117; men, BMI < 21 and FFMI < 16; women, BMI ≥ 21 and FFMI < 5). Semistarvation (N = 23; men BMI < 21 and FFMI ≥ 16; women, BMI < 21 and FFMI ≥ 15). Muscle atrophy (N = 40; men, BMI ≥ 21 and FFMI < 16; women, BMI ≥ 21 and FFMI < 15). No impairment (N = 232; men, BMI ≥ 21 and FFMI ≥ 16; women, BMI ≥ 21 and FFMI ≥ 15). Cachexia was significantly (p = 0.001) more prevalent in GOLD stage IV (N = 207) than in GOLD stage II (N = 71) or III (N = 134). BMI, body mass index (kg/m2); FFMI, fat-free mass index (kg/m2).
![Figure 4 Body-composition categories are linked to GOLD disease severity. Source: (Citation[29]), Fig. 1, p. 56. Cachexia (N = 117; men, BMI < 21 and FFMI < 16; women, BMI ≥ 21 and FFMI < 5). Semistarvation (N = 23; men BMI < 21 and FFMI ≥ 16; women, BMI < 21 and FFMI ≥ 15). Muscle atrophy (N = 40; men, BMI ≥ 21 and FFMI < 16; women, BMI ≥ 21 and FFMI < 15). No impairment (N = 232; men, BMI ≥ 21 and FFMI ≥ 16; women, BMI ≥ 21 and FFMI ≥ 15). Cachexia was significantly (p = 0.001) more prevalent in GOLD stage IV (N = 207) than in GOLD stage II (N = 71) or III (N = 134). BMI, body mass index (kg/m2); FFMI, fat-free mass index (kg/m2).](/cms/asset/f851f366-09e8-44af-aff6-c0109c93ce93/icop_a_262814_uf0004_b.gif)
Figure 5 Prognostic influence of BMI on survival in patients with COPD receiving long-term oxygen therapy. Source: (32), , p. 1463. The initial numbers in each group were 990 (BMI < 20), 1,731 (BMI, 20 to 24), 1,003 (BMI, 25 to 29), and 364 (BMI > 30) [p < 0.001].
![Figure 5 Prognostic influence of BMI on survival in patients with COPD receiving long-term oxygen therapy. Source: (32), Fig. 3, p. 1463. The initial numbers in each group were 990 (BMI < 20), 1,731 (BMI, 20 to 24), 1,003 (BMI, 25 to 29), and 364 (BMI > 30) [p < 0.001].](/cms/asset/60a152a2-7909-4d80-89d7-27100a5f5f27/icop_a_262814_uf0005_b.gif)
Thus, the low BMI phenotype likely includes up to a quarter of patients with severe COPD. Impressively, BMI was the most powerful predictor of duration and rate of hospitalization, independent of blood gas levels and respiratory function (Citation[32]). In the Copenhagen City Heart Study, the proportion of subjects who lost > 1 unit BMI (∼ 3.8 kg) at 5-year follow-up was significantly associated with the severity of airflow limitation, reaching 30% in subjects with severe COPD (Citation[30]). In these subjects, there was a significant risk ratio interaction between the effect of baseline BMI and weight change; in the normal-to-underweight (BMI < 25), best survival was seen in those who were able to gain weight, whereas for the overweight and obese (BMI ≥ 25), best survival was seen in stable weight (Citation[30]).
Various studies have described the presence of nutritional abnormalities in patients with COPD, including alterations in caloric intake, basal metabolic rate, intermediate metabolism and body composition (Citation[33],Citation[34],Citation[35],Citation[36]). Unexplained weight loss occurs in almost 50% of patients with severe COPD, but is also seen in 10–15% of patients with mild-to-moderate disease (Citation[34]). The incidence of weight loss in COPD has been shown to correlate with severity of disease, with a body mass index (BMI) value of 25 kg/m2 or below associated with increased mortality (Citation[28]). These studies highlight that weight loss is not necessarily due to inadequate nutrition, and that both the rate of weight loss and the resultant BMI need to be taken into account when assessing patient with COPD who are at risk for “pulmonary cachexia” (see next section).
The actual prevalence and magnitude of muscle wasting are probably underestimated when extrapolated from body weight measurements, since lean body mass (an index of muscle mass) may be reduced despite preservation of body weight (Citation[34], Citation[37]). Recent published data from the Copenhagen City Heart Study supports that the free-fat mass index (fat-free mass/height2, which can be measured using bioelectrical impendence analysis) provides information in addition to BMI in patients with COPD, and is associated with both overall and disease-specific mortality independent of FEV1 (Citation[38]).
Pulmonary cachexia
There is accumulating evidence that the inflammatory effects of COPD extend beyond the lungs () (Citation[39]). The best understood systemic effect is the muscular dysfunction and wasting seen in many patients with COPD, and defines a subgroup with important associated morbidities and impairments in functional capacity (Citation[40]). The term “cachexia” highlights the preferential loss of muscle over fat, with evidence of increased protein degradation (Citation[41], Citation[42]).
Table 1 Systemic effects of COPD
Musculoskeletal derangements that can be seen in COPD include structural changes in muscle fiber type and function, such as a shift in muscle fiber type from I (“slow twitch”) to II (“fast twitch”) with selective atrophy, fibrosis and fat replacement of IIx (“very fast twitch”) fibers (Citation[43]). Type I (slow) fibers are resistant to fatigue, whereas type II (fast) fibers are easily fatigable. A reduced proportion of type I fibers in patients with COPD has been observed as compared to age-matched controls (Citation[44], Citation[45]) and diameter of muscle fibers is reduced in proportion to the severity of COPD or reduction in FEV1 (Citation[46]). The prevalence of these histological muscle abnormalities in all COPD patients is unknown, since only 15 weight-stable patients were studied.
Historically, the peripheral muscle dysfunction seen in COPD patients was explained by their sedentary lifestyle; indeed, there have been a series of studies that show physical training improves peripheral muscle function, exercise tolerance, and quality of life (Citation[47],Citation[48],Citation[49]). More recent data argues strongly in favor of an acquired myopathy, implying at least one or more pharmacologic and/or pathophysiologic mechanisms responsible for the peripheral muscle dysfunction seen in COPD (Citation[50]). There is also evidence that skeletal muscle apoptosis is increased in patients with COPD and a low BMI as compared to COPD patients with a normal BMI, age-matched healthy volunteers and sedentary subjects without COPD (Citation[51]) (). The degree of skeletal muscle apoptosis is also associated with a lower exercise tolerance despite a similar degree of lung function impairment, supporting that pulmonary cachexia is a phenotypic distinction independent of FEV1. Additionally, a recent study demonstrated that quadriceps maximal voluntary contraction force, a simple functional measurement of strength, provides more prognostic information than age, BMI and FEV1 in patients with COPD (Citation[52]).
Figure 6 Skeletal muscle apoptosis in COPD. Source: (Citation[51]), , p. 487. Top panel: representative example of a muscle biopsy obtained in a control subject (a) and in a patient with COPD and low BMI (b). Nuclei are stained blue in the former (non-apoptotic), whereas most of them are stained brown (apoptotic) in the latter. Bottom panel: Western blot showing the relative intensities of the two poly(ADP-ribose) polymerase (PARP, a marker of cellular apoptosis) bands (89- and 117-kD fragments) in a representative subject of each of the 4 groups studied.
![Figure 6 Skeletal muscle apoptosis in COPD. Source: (Citation[51]), Fig. 2, p. 487. Top panel: representative example of a muscle biopsy obtained in a control subject (a) and in a patient with COPD and low BMI (b). Nuclei are stained blue in the former (non-apoptotic), whereas most of them are stained brown (apoptotic) in the latter. Bottom panel: Western blot showing the relative intensities of the two poly(ADP-ribose) polymerase (PARP, a marker of cellular apoptosis) bands (89- and 117-kD fragments) in a representative subject of each of the 4 groups studied.](/cms/asset/51415b3a-d074-40aa-a591-81e0b70819ef/icop_a_262814_uf0006_b.gif)
The pathogenesis of cachexia in COPD, although not fully understood, is a complex interplay of increased muscle protein breakdown, inflammation, negative energy balance, hypoxemia, corticosteroid therapy, insulin resistance, and decreased testosterone levels (Citation[37], Citation[39], Citation[40], Citation[42]) (). Inflammatory cytokines, such as TNF-α and IL-1, have been shown to play a central role in the enhanced protein catabolism seen in COPD patients (Citation[40], Citation[53]). Although the precise mechanisms for these effects remain unknown, there is a suggestion that COPD patients with cachexia may represent an “inflammatory phenotype.” This hypothesis is supported by a study showing that non-response to nutritional therapy in COPD is associated with an elevated systemic inflammatory response (Citation[54]). Thus, it is hopeful that pharmacologic interventions targeting systemic inflammation and/or hormonal imbalances may reverse cachexia in these patients, and possibly prevent further morbidity.
Figure 7 Possible mechanisms of cachexia in COPD. Source: (Citation[37]), , p. 1715. Growth factors such as insulin-like growth factor-I (IGF-I) and MyoD have their anabolic effects by activating myofibrils synthesis. Myofibrillar proteins are marked for degradation by ubiquitin (Ub) and are then processed through the 26 S proteasome. This protein degradation pathway can be activated by muscle inactivity. Proinflammatory cytokines such as TNF (shown here), IL-1, and IL-6, for instance, exert their catabolic action by activating NF-B, which can enter the nucleus after dissociating from its inhibitor (IB). One catabolic action of NF-B is to repress the gene expression of MyoD. Proinflammatory cytokines may also activate the ubiquitin-proteasome pathway. A reasonable hypothesis is that systemic (TNF, growth factors) as well as local factors (inactivity, acidosis) interact in the development of cachexia. Ub = ubiquitin; NF-B = nuclear factor B; IB = NF-B natural inhibitor; MHC = myosin heavy chain; IGF = insulin-like growth factor; IGFR = insulin-like growth factor receptor; TNF = tumor necrosis factor; TNFR I and II = tumor necrosis factor receptor I and II.
![Figure 7 Possible mechanisms of cachexia in COPD. Source: (Citation[37]), Fig. 1, p. 1715. Growth factors such as insulin-like growth factor-I (IGF-I) and MyoD have their anabolic effects by activating myofibrils synthesis. Myofibrillar proteins are marked for degradation by ubiquitin (Ub) and are then processed through the 26 S proteasome. This protein degradation pathway can be activated by muscle inactivity. Proinflammatory cytokines such as TNF (shown here), IL-1, and IL-6, for instance, exert their catabolic action by activating NF-B, which can enter the nucleus after dissociating from its inhibitor (IB). One catabolic action of NF-B is to repress the gene expression of MyoD. Proinflammatory cytokines may also activate the ubiquitin-proteasome pathway. A reasonable hypothesis is that systemic (TNF, growth factors) as well as local factors (inactivity, acidosis) interact in the development of cachexia. Ub = ubiquitin; NF-B = nuclear factor B; IB = NF-B natural inhibitor; MHC = myosin heavy chain; IGF = insulin-like growth factor; IGFR = insulin-like growth factor receptor; TNF = tumor necrosis factor; TNFR I and II = tumor necrosis factor receptor I and II.](/cms/asset/294bf8be-b8de-44d5-af4c-a0f1efdbede3/icop_a_262814_uf0007_b.gif)
There is also evidence that increased muscle oxidant production and/or reduced muscle antioxidant capacity may contribute to the myopathy seen in many patients with COPD (Citation[50]). Activation of the nuclear transcription factor NF-κβ and upregulation of the inducible form of nitric oxide synthetase has been shown to occur in the skeletal muscle of COPD patients with low body weight as compared to those with a normal BMI (Citation[55]). A recent study demonstrated impaired mitochondrial respiratory function in COPD patients with low BMI compared to both patients with normal BMI and healthy controls, suggesting that altered oxidative phosphorylation in low BMI patients indicates an abnormal mitochondrial adaptation to long-term repeated episodes of cell hypoxia (Citation[56]). Since plasma and muscle antioxidants are reduced at rest in COPD (Citation[57], Citation[58]), some authors argue that physical training in combination with antioxidant supplementation needs to be further explored (Citation[50], Citation[59]). However, pulmonary rehabilitation alone has not been shown to improve BMI in patients with COPD.
Other strategies to improve BMI include the institution of non-invasive positive pressure ventilation (NIPPV). In COPD patients with severe chronic respiratory failure (mean FEV1 80% and mean PaCO2 56 mmHg), NIPPV led to significant increases in body weight at 6 and 12 months in malnourished patients (BMI < 20 kg/m2); there were no significant changes in the overall population (Citation[60]).
Inhaled corticosteroid-responsive
The routine use of inhaled corticosteroids (ICS) as adjuvant therapy to long-acting bronchodilators remains, arguably, the most controversial area in the management of patients with COPD. With that, it remains unclear if an ICS-responsive COPD phenotype can be defined. Clinically, there appears to be a sub-group of patients with severe airflow limitation that demonstrate spirometric improvement in response to a trial of ICS (Citation[2]). Much of the controversy arises from debate surrounding what may dictate a meaningful clinical response, such as an improvement in FEV1 or a slowing in the rate of lung function decline in comparison to a reduction in exacerbation rate or an improved mortality (Citation[61]).
Earlier studies of ICS in COPD conducted in the 1990s had mixed results, with no study showing a slowing in the rate of FEV1 decline with the addition of an ICS (Citation[62],Citation[63],Citation[64],Citation[65],Citation[66],Citation[67]). However, more recent randomized clinical trials have shown that ICS are associated with improved baseline pulmonary function, reduced exacerbation frequency and severity, and improved quality of life (Citation[68],Citation[69],Citation[70],Citation[71],Citation[72]). A meta-analysis suggested that ICS may modify the natural history of COPD, with treatment for ≥ 2 years slowing the rate of lung function decline (Citation[72]). Pooled analysis of individual patient data from 7 randomized trials and over 5,000 subjects showed that ICS reduced all-cause mortality in COPD (Citation[27]).
The earliest large prospective randomized clinical trial evaluating the efficacy of combined ICS and a long acting β2-agonist (LABA) versus either alone and placebo over one year showed rapid effects of both on peak flows and breathlessness (Citation[73]). However, combination therapy was not superior to either agent alone, and there was no significant benefit of the ICS over the LABA. This was despite earlier trials that showed combination ICS and LABA therapy provide a significant improvement in trough, peak and mean FEV1 compared with monotherapy (Citation[68], Citation[69], Citation[71]). Thus, from this variable and sometimes conflicting data, it can be postulated that there may exist an “ICS-responsive” phenotype, possibly characterized by increased small-airways inflammation.
Towards a Revolution in COPD Health (TORCH), published in early 2007, sought to definitively determine whether ICS, alone or in combination with a LABA, imparted a survival benefit in patients with moderate-to-severe COPD (Citation[74]). Although the reduction in all-cause mortality in the combination-therapy (ICS and LABA) did not reach the predetermined level of statistical significance, there were significant benefits in regard to exacerbation rate and quality-of-life indices. Thus, TORCH added little to what was previously known about the role of ICS as a potential disease modifier in COPD. Consequently, since TORCH lacked precise characterization of study subjects, the results cannot be used to support or argue against the existence of an ICS-responsive phenotype.
The ISEEC study, published shortly after TORCH, is a pooled analysis of almost 4,000 participants from 7 long-term randomized controlled trials of ICS versus placebo (Citation[75]). In the first 6 months after randomization, ICS use was associated with a significant mean relative increase in FEV1 of 2.42% compared with placebo (Citation[75]). Beyond 6 months of therapy, there was no significant difference between placebo and ICS in terms of FEV1 decline. Interestingly, the initial treatment effect observed was dependent on smoking status and gender, with more of an effect in ex-smokers (compared to current smokers) and women.
This suggests that the pathologic correlate of increased small airways inflammation in an ICS-responsive population may relate to underlying smoking status and gender. Extending this further, it can be postulated that the minority of former smokers who develop COPD have persistent pulmonary inflammation leading to chronic airflow obstruction and, subsequently, are more ICS-responsive than current smokers whose airflow limitation is primarily a result of parenchymal destruction. However, this argument is purely speculative, and further research into defining which patients have a meaningful response to ICS and the underlying mechanisms for these differences is needed.
In one arm of the Lung Health Study, 1,116 patients with mild-to-moderate COPD were randomized to inhaled triamcinolone versus placebo (Citation[67]). The addition of an ICS did not slow rate of decline in lung function, but did improve airway hyperresponsiveness and respiratory symptoms, as well as decreased the use of health care services. The reduction in airway hyperresponsiveness may indicate that those patients with methacholine reactivity would gain the most from an ICS, but this has not been shown prospectively. It could be argued that COPD patients with airway reactivity have an “asthmatic phenotype,” and should gain more benefit from the addition of an ICS. It is also possible that these patients simply have both asthma and COPD. Some experts have hypothesized that alveolar macrophages in patients with COPD have “acquired” corticosteroid resistance from impaired histone deacetylase 2 activity, which is necessary to unwind DNA for anti-inflammatory gene transcription (Citation[76], Citation[77]). Thus, novel therapeutic approaches that unlock this corticosteroid resistance may lead to more effective anti-inflammatory treatment for COPD.
The clinical challenge of the appropriate use of ICS underscores the need to fully characterize all patients with COPD, for both prognostication and potential response to therapy. This approach will allow better clinical decision making when weighing the potential benefit of an ICS in an individual patient against the possibility of long-term systemic side effects (e.g., infectious complications, weight gain, glucose intolerance, cataracts, osteoporosis). Hopefully, future studies investigating ICS (and other agents) for COPD will prospectively take phenotypic distinction into consideration.
Depression and anxiety
Psychological distress is common in patients with COPD, which underscores the need to understand the disease as a multi-systemic disorder. Both depression and anxiety, and their interaction, have been shown to significantly impact quality of life, adherence to therapy, medical burden and functional capacity (Citation[78],Citation[79],Citation[80]). Clinically, we understand that mood and anxiety disorders greatly compound the emotional and physical suffering from this disease. The prevalence of depression among patients with COPD, based upon a systematic review, is estimated to be between 6% and 42%; 5 studies reported the prevalence to be 12% or higher (Citation[81]). Most studies support that up to 35–50% of the COPD population show signs of depression, which is higher than that of the general population (Citation[81]). In the community, approximately 15–25% of people have a depressive disorder. Clearly, the data is somewhat inconclusive as to the exact prevalence of depression in COPD. Nevertheless, there is agreement that people with COPD are at higher risk for depression than the general population.
The estimated prevalence of anxiety disorders in COPD ranges up to 96% (Citation[82]). Anxiety disorders include panic, anxiety or a predominance of these symptoms. While 96% may be erroneously high, a systematic review of 14 studies concluded that the prevalence of anxiety in COPD ranged between 10% and 94% (Citation[83]). An earlier study to determine the predictors of quality of life in patients with mild hypoxemia and COPD showed that patients with COPD (N = 985) scored significantly higher than healthy controls (N = 25) on a tension-anxiety scale; however, the degree and prevalence of this effect was not determined (Citation[84]). The best estimates suggest that the prevalence of generalized anxiety disorder among patients with COPD ranges from 10% to 15.8% when using standard diagnostic procedures, compared with lifetime rates of 3.6% to 5.1% in the general population (Citation[83]). Rates of anxiety symptoms are even higher, ranging from 13% to 51%, and are higher than in patients with heart failure, cancer, and other medical conditions (Citation[83], Citation[85]). Unfortunately, many of the epidemiological studies have been limited by small sample sizes, lacks of diversity and/or varied screening methods. Consequently, agreement on the exact prevalence of mood and anxiety disorders in COPD is difficult to surmise.
In one of the largest prospective studies conducted to determine the prevalence of depression and anxiety in patients with chronic pulmonary disease, Kunik et al. helped clarify the picture. He followed 1,334 patients who were screened using the Primary Care Evaluation of Mental Disorders (PRIME-MD) (Citation[86]). In their study, 80% of patients had evidence of anxiety, depression, or both. Depression and anxiety appear to be prevalent across all stages of the disease, but is particularly prevalent in its severe forms and in women (Citation[87], Citation[88]). One study showed an 5.4% absolute increased risk of depression in patients with severe COPD (FEV1 < 50% predicted) as compared to patients with mild or moderate disease, and a 7.5% increased risk as compared to healthy controls (Citation[87]).
The emotional aspect of COPD is an important predictor of functional capacity, quality of life and health status. Kim et al. examined depression and anxiety's contribution to overall variance in functional health status as measured by the SF-36 (Citation[80]). Depression and anxiety significantly contributed to impairment above and beyond that of disease severity (FEV1) and medical burden. Similarly, Felker et al. found that 11% to 18% of the change in physical function on both general- and disease-specific HRQOL measures was attributed to depressive symptoms (Citation[89]). Treatment failures are also highly related to depression and anxiety.
Dahlen et al. showed a significant association between hospital admissions and relapse due to COPD exacerbations and the presence of depressive and anxious symptoms according to the Hospital Anxiety and Depression scale (Citation[79]). Since the majority of these studies are cross-sectional, causation has not yet been determined. In addition, as with the prevalence studies, most of the studied populations lacked diversity, making generalizability difficult. Interestingly, a recent prospective study examined mood and respiratory symptoms in 515 adults over a 9-year period and found a causal relationship between anxiety and dyspnea (Citation[90]). However, this study was not limited to patients with COPD, and investigations like this one will need to be replicated to discover if causality exists.
While direct causation continues to elude investigators, the mechanisms behind the strong connection between mood symptoms may be described as a complex interplay between behavior and physiology. Depression in COPD has previously been understood as unprocessed grief from the losses in physical functioning (Citation[91]). There is also strong reasoning to suggest tobacco dependence as a mediating variable (Citation[92]). Early and lifelong depression often precedes adolescent smoking. While both the above suggestions are part of the problem, recent advances in imaging describe physiological changes as well. Borson et al. suggest a connection between neuropsychological changes due to chronic hypoxemia and long-term exposure to tobacco (Citation[93]).
Anxiety, given its relationship to dysregulation of normal breathing patterns, dynamic hyperinflation and dyspnea, can be closer linked to physiologic changes (Citation[94]). Chemoreceptors in the brainstem respond to increasing levels of CO2. The consequence is a “false suffocation alarm” and a subjective experience of anxiety and panic. Late-stage patients become increasingly vigilant of their shortness of breath and actually learn the panic response. Brenes provides an exceptional illustration of this cognitive-physiologic association (Citation[83]):
According to psychological theories of anxiety, individuals panic when they misinterpret bodily sensations as more dangerous than they truly are and as a signal of impending danger (Citation[95]). When a patient with COPD experiences dyspnea, he or she presumably makes incorrect assumptions (e.g., “I am going to die”) in response to the feeling of breathlessness. This in turn leads to a heightened state of physiological arousal accompanied by additional sensations and misinterpretations. While breathlessness may represent actual danger in patients with COPD, COPD patients with panic disorder have more negative cognitions than COPD patients without panic disorder, but they do not differ with respect to pulmonary functioning (Citation[96]). This suggests that anxiety in COPD patients with panic disorder reflects inaccurate cognitions rather than greater disease severity.
Thus, there is a subset of patients with COPD whose dyspnea may be driven more by anxiety and depression than degree of airflow limitation. It can be postulated that these patients may have more hyperinflation at baseline (prior to the development of a clinically apparent emotional disorder) and thus are more prone to panic-induced breathlessness. Identifying an understanding the biopsychosocial mechanisms behind co-morbid mood and anxiety disorders is crucial to the treatment of the disease and is the next frontier of psychological research in COPD.
Non-smokers with COPD
COPD in non-smokers may be more common than has been previously realized. In 7,526 non-smokers (defined as < 5 pack years) who underwent spirometry screening in the 3rd National Health and Nutrition Examination Survey (NHANES-III), mild COPD was present in 5.3% and moderate-to-severe COPD was present in 1.2% (Citation[97]). In this general population survey, non-smokers accounted for 25% of COPD cases. Similar rates have been reported in the United Kingdom and Spain (Citation[98], Citation[99]). Only 12% of nonsmokers with COPD had ever received a physician diagnosis of COPD.
Demographic differences in non-smokers with COPD suggest that it might be a distinct disease, especially for the majority (81%) of non-smokers with COPD who have mild disease. Several studies have reported that 80–90% of non-smokers with COPD are women (Citation[97], Citation[98]); however, there are no gender differences in those with moderate to severe COPD (Citation[97]). Age is also a risk factor for mild COPD in the non-smoker. The risk of moderate-to-severe COPD in non-smokers is markedly increased in middle-aged males and inversely associated with non-White ethnicity.
Both environmental/occupational exposures and autoimmune disorders have been postulated to cause COPD in non-smokers. In a study of 395 non-smoking women living in Turkey, those exposed to indoor pollutants produced by inefficient biomass stoves are 2.5 times more likely to have chronic bronchitis (Citation[100]). An Australian study of 608 non-smokers exposed to occupational dusts, COPD was 2–3 times more prevalent in those with exposure to biologic dusts or gases and fumes (Citation[101]). In the United States, non-smoking women living in areas with high levels of particles, nitrogen oxides and sulphates (e.g., Long Beach, Ca.) have higher rates of decline in FEV1 (Citation[102]). A small study (N = 22) of non-smokers with COPD found that they had an increased incidence of autoimmune (particularly thyroid) disease and sputum neutrophilia (Citation[98]). However, these associations were not risk factors in NHANES III (Citation[97]).
Misdiagnosis, particularly asthma, may also contribute to the reported higher than expected prevalence of COPD in non-smokers. In the Birring et al. study (Citation[98]), there were a large number of subjects with sputum eosinophilia. In the NHANES III population, only age and a history of asthma were associated with mild-to-moderate COPD in younger patients (Citation[97]). In a longitudinal cohort from Tucson, subjects with active asthma had a 17-times higher risk of receiving a diagnosis of emphysema and a 12.5-times higher risk of having COPD even after adjusting for smoking (Citation[103]).
Airway hyperresponsiveness may play a role in progression to fixed airflow obstruction since it is associated with a higher rate of decline in FEV1 (Citation[104]). α1-antitrypsin deficiency has been associated with a rapid rate of decline in FEV1 in non-smokers (Citation[105]). Since the majority of the larger population studies did not fully phenotype subjects, it is possible that non-COPD obstructive lung diseases accounted for these numbers. For instance, COPD has been noted in constrictive bronchiolitis (Citation[106]), rheumatoid arthritis (Citation[107]), panbronchiolitis (Citation[108]), and diffuse idiopathic neuroendocrine cell hyperplasia (Citation[109]).
PHYSIOLOGIC PHENOTYPES IN COPD
Physiologic phenotypes in COPD have overlap with most of the clinical phenotypes previously discussed. This is not only biologically predicted, but underscores the need to full characterize patients not just by clinical symptoms and functional capacity, but also by standardized assessments of pulmonary function and gas exchange. Despite the overlap, an expanded discussion of distinct physiologic phenotypes observed is warranted to guide therapy and continue to unravel the molecular mechanisms of the disease processes.
Degree of airflow limitation
Categorizing COPD by severity of airflow limitation, as assessed by the percentage of the predicted post-bronchodilator FEV1, remains the most common method to stage patients (Citation[2], Citation[110]). Indeed, the FEV1 is highly correlated with morbidity and mortality (Citation[2], Citation[111], Citation[112]). However, there is an increased awareness that FEV1 alone is not the best predictor of mortality, and several other factors are now recognized to be important prognostic variables (). Additionally, the severity of airflow limitation often under-or over-estimates an individual patient's symptoms or functional capacity. For example, a patient with severe diffuse emphysema may have a relatively preserved FEV1 as compared to another patient with marked small airways inflammation and minimal emphysema, but have significantly more dyspnea and exercise limitation as a result of hyperinflation.
Table 2 Prognostic factors in COPD
Conversely, a patient with long-standing severe airways disease and associated very low FEV1 may be much more responsive to inhaled agents and, thus, have minimal exertional dyspnea or functional limitation. This increased understanding that patients with COPD have systemic manifestations that are not reflected by the FEV1 have led to multidimensional grading systems to better categorize and predict outcomes (Citation[13]). The use of these composite indices, which include, but are not limited to, the degree airflow limitation, are becoming the primary way in which the FEV1 is used to assess the severity of COPD.
Rapid decliner
Airflow limitation, as defined by a reduced FEV1/FVC (< 70%) and further quantified by the FEV1, remains the primary component in defining COPD (Citation[110]) (). Lung function normally increases to a maximal value at adulthood, remains stable for 10–15 years, and then declines (Citation[113]). Thus, COPD can develop because of a reduced maximal lung function, an earlier age of onset of decline, or an accelerated rate of decline, likely in the setting of chronic exposure to noxious inhalants (i.e., cigarette smoke). Although the rate of FEV1 loss accelerates with age, it has been shown that non-smokers lose lung function slowly and almost never develop clinically significant airflow obstruction (Citation[114]).
Table 3 Spirometric classification of COPD severity based on post-bronchodilator FEV1
Extrapolating from data generated by Fletcher and Peto in the 1960s, “rapid decliners” can be arbitrarily defined as those patients who lose lung function (FEV1) at a rate greater than 60 mL per year. A complementary approach to defining rapid decline in lung function, as described by Mannino et al., is to follow patients prospectively and determine the quartiles of change in FEV1 from baseline and classify those in the most negative category as “rapid decliners” and the other three categories as the referent group (Citation[115], Citation[116]). Using this methodology, the rapid decliners had a mean annual loss of 110 mL, which was 5.4% (range 3.5–22.7%) of their baseline prebronchodilator FEV1 per year [the referent groups had a mean change in FEV1 of −59 to 41 mL per year (−2.7–3.6%)]. However, this approach skews the classification of rapid decliners towards more severe stages of COPD (GOLD 3 or 4) at baseline. There is evidence that rapid lung function decline is independently associated with an increased risk of COPD-related hospitalizations and death in both elderly (Citation[115]) and adult (Citation[116]) populations.
The Lung Health Study, a multicenter clinical trial of smoking intervention and inhaled bronchodilator in almost 6,000 middle-aged smokers with mild-to-moderate COPD, provided the best understanding of the relationships between loss of lung function and various other risk factors beyond tobacco use (Citation[117]). As predicted, smoking cessation significantly reduced decline in lung function, with continuing smokers losing 300 mL of FEV1 over 5 years, whereas sustained quitters lost less than 50 mL.
Methacholine reactivity was tested at entry into the Lung Health Study, and found to be a powerful predictor of rate of loss of FEV1 (second only to smoking habit) (Citation[118]). This lends some weight to the “Dutch hypothesis” that decline in lung function is related to preexisting airway reactivity. Airway hyperresponsiveness had relatively little effect in people who stopped smoking but was important in those who did not, suggesting that airway reactivity is not the only factor responsible for the development of chronic irreversible airflow limitation in smokers. Baseline FEV1 did not influence subsequent decline once smoking status, age, sex, and reactivity had been considered (Citation[118]).
The role of repeated respiratory infections or bacterial colonization of the respiratory tract on the decline in lung function was also investigated in the Lung Health Study. Participants were questioned regarding physician visits for bronchitis, pneumonia, influenza, or chest colds during the preceding year at their annual visits, and positive responses were related to lung function data. Respiratory infections were common, averaging 0.2 episodes per participant per year (Citation[119]). In continuing smokers, one respiratory infection per year was associated with an excess loss of FEV1 of 7 mL, whereas there was no significant loss of lung function in ex-smokers (Citation[120]).
The authors comment that although the effect of infections seemed small, it would not have been trivial in patients who had as many as two such episodes per year over many years. This finding contraindicated earlier work by Fletcher and colleagues (Citation[121]), but has support in a prior more recent study (Citation[122]). In the Copenhagen City Heart Study, the presence of mucus hypersecretion was associated with a significantly greater decline in FEV1 in asthmatics as compared to non-asthmatics (Citation[123]). Thus, the precise role of respiratory infections on the decline in lung function in patients with COPD remains unknown, but there is evidence suggesting that recurrent infections in smokers impart an increased rate of decline.
Concomitant respiratory disease, such as asthma, may contribute to rapid decline in FEV1 in patients with COPD. There are several large, prospective observational studies suggesting that asthmatics have steeper FEV1 declines than non-asthmatics (Citation[123], Citation[124]). However, neither of these studies adjusted for comorbid respiratory diseases. A more recent epidemiologic study investigated this further and attempted to define the excess decline in FEV1 in asthma and COPD independent of comorbid respiratory disease (Citation[125]). Of the 2,926 participants, 7.3% had asthma, 11.1% had COPD and 4.3% had both, all of which were “physician-confirmed” diagnoses. Although subjects with both asthma and COPD had the lowest absolute levels of FEV1, they did not have a rate of decline that was increased as compared to non-asthmatics. Only those with COPD in the absence of asthma had both a low initial FEV1 and a significantly steeper rate of decline. These observations are contradictory to data from the Lung Health Study that suggested airway reactivity was a strong predictor of decline in lung function, and interpretation of this study is limited by the imprecise manner in which COPD and asthma were defined, selection bias and the analytic models used.
There are clear genetic risk factors for the rapid decline in FEV1, most notably homozygosity for the α1-antitrypsin Z allele (). The Lung Health Study further investigated the hypothesis that accelerated decline in lung function is related to genetic susceptibility to cigarette smoke beyond reduced antiproteolysis seen in PiZZ α 1-antitrypsin deficiency. At 5-year follow-up, from the 3,216 participants that continued to smoke, 283 were selected with the fastest rate of decline of FEV1 and 308 with no decline (Citation[126]). These subjects were genotyped for polymorphisms in the α1-antitrypsin, microsomal epoxide hydrolase, vitamin D-binding protein, and tumor necrosis factor (TNF) genes. Rapid decline was significantly associated with the MZ genotype of the α 1-antitrypsin gene. Haplotype frequencies of the microsomal epoxide hydrolase gene, which is expressed in bronchial epithelial cells and involved in the metabolism of highly reactive oxygen species in cigarette smoke, were significantly different between rapid decliners and non-decliners.
Table 4 Potential susceptibility genes for rapid decline in lung function
Vitamin D-binding protein isoforms and TNF polymorphisms were not associated with rate of decline of lung function. Similar studies looking at polymorphisms in the matrix metalloproteinase (MMP) gene family, which play an essential role in tissue remodeling and may contribute to the development of emphysema, suggest some MMP genes may contribute to decline in lung function (Citation[127]). Polymorphisms in the transforming growth factor-β 1 gene contribute to airway hyperresponsiveness but not to rapid decline in lung function (Citation[128]).
Interestingly, heterozygosity at position 27 (Gln27 → Glu) of the β2-adrenergic receptor has been shown to be protective against an accelerated rate of decline in lung function in smokers; polymorphisms at position 16 (Agr16 → Gly) did not contribute to the rate of decline in lung function, measures of bronchial hyperresponsiveness or bronchodilator responsiveness (Citation[127]). These investigations suggest that variations in α1-antitrypsin expression, response to reactive oxygen species, and/or imbalances in the proteinase-antiproteinase system may contribute to the “rapid decliner” phenotype, and further shed light onto possible pathophysiologic mechanisms responsible for the variation seen in COPD. Since only those participants who continued to smoke were studied, these results need to be interpreted with caution when trying to define effects independent of continued tobacco exposure.
Although the definition of rapid decliners is not fully characterized, these patients are more likely to have airways hyperresponsiveness by a positive methacholine challenge. Recurrent respiratory infections may also impart increased risk for loss of lung function, but it remains unclear if this is a cause or effect of bacterial colonization and mucus hypersecretion. It is possible that this group of patients have an attenuated inflammatory response that persists well after smoking cessation, and likely contributes to the subgroup with frequent exacerbations.
Bronchodilator-responsiveness
Although COPD is classically defined by irreversible airflow obstruction, there is a significant subgroup of patients who exhibit bronchodilator responsiveness (improved FEV1 or FVC to a short acting β-agonist) during pulmonary function testing based upon ATS and/or ERS guidelines. Whether these patients also have asthma or, more likely, represent an “asthmatic phenotype” of COPD, remains controversial and largely a matter of expert opinion. Although there exists important overlap between bronchodilator responsiveness (BDR) and airway hyperresponsiveness (AHR), these physiologic definitions should be viewed as distinct entities when defining separate phenotypes that have variable risks and outcomes.
Bronchodilators, especially the long acting β2-agonists (LABA), remain the mainstay for maintenance treatment of COPD due to their effective symptomatic response (Citation[129],Citation[130],Citation[131],Citation[132]). Although there is some limited evidence that LABAs show improvement in health status (Citation[133], Citation[134]) and exercise capacity (Citation[135]), whether they affect disease progression remains unclear. From the limited number of studies comparing the long-term effects of LABAs to short-acting agents, ICS and/or placebo on lung function decline, frequency of exacerbations and mortality, it is impossible to determine if there is a “bronchodilator-responsive” phenotype (Citation[25], Citation[69], Citation[70], Citation[134], Citation[136], Citation[137]).
In the Lung Health Study, changes in FEV1 in response to isoproterenol were measured in 4,194 participants annually for 5 years, and again 11 years after study entry (Citation[138]). At study entry, approximately 20% of participants demonstrated an initial response that was > 200 mL, but responses of > 15% of the pre-bronchodilator value or 12% of the predicted normal value were uncommon (< 3%). BDR did not predict the rate of lung function decline. More notably, intra-individual responses were quite variable and changed unpredictably over time; of participants who had an absolute response of < 200 mL at the first annual visit, about 20% had a larger response at subsequent testing. Conversely, of those with responses ≥ 200 mL at the first annual visit, just under half had responses < 200 mL at subsequent visits.
The observed tremendous variability in BDR was supported by results form the ISOLDE study, which clearly showed that BDR is a continuous variable over time, and classifying patients as “responders” and “non-responders” can be misleading and does not predict disease progression (Citation[139]) (). This study also showed that smoking status, atopy, and withdrawing ICS were unrelated to BDR, as was the rate of decline in FEV1, decline in health status, and exacerbation rate. Thus, there is compelling evidence that BDR in COPD is not a distinct phenotype and provides little clinical or prognostic information.
Figure 8 Changes in bronchodilator responder classification and corresponding subgroup mean FEV1 at each visit. Source: (Citation[139]), , p. 662. Numbers in circles refer to the total classified as non-responders at that visit and those in squares are the positive responders on the same occasion. Note that some patients in the ERS criteria group exhibited a “response” after both drugs at the first visit despite being classified as non-responsive to salbutamol alone.
![Figure 8 Changes in bronchodilator responder classification and corresponding subgroup mean FEV1 at each visit. Source: (Citation[139]), Fig. 5, p. 662. Numbers in circles refer to the total classified as non-responders at that visit and those in squares are the positive responders on the same occasion. Note that some patients in the ERS criteria group exhibited a “response” after both drugs at the first visit despite being classified as non-responsive to salbutamol alone.](/cms/asset/374bda75-bfad-4dd3-a27b-5ffd467faee0/icop_a_262814_uf0008_b.gif)
There is an important role of BDR in association with other physiologic and molecular phenotypes seen in COPD in further defining genetic risk factors for the development and progression of the disease. In a large cohort of severe, early-onset COPD patients, Celedon et al. has found greater BDR among current smokers or ex-smokers who were first-degree relatives of these early-onset probands than currently or ex-smoking controls, suggesting that these individuals may have enhanced susceptibility to the detrimental effects of cigarette smoking (Citation[140]). In the same cohort, there is evidence for significant linkage to airflow obstruction susceptibility loci on chromosome 2q and 8p, providing further evidence that post-bronchodilator spirometry are optimal phenotypes for COPD genetic studies (Citation[141]). These studies have identified several genomic regions that could contain loci regulating BDR in early-onset COPD families. Although there is evidence that BDR is not a stable phenotype over time, these studies suggest that there may be a group of patients with COPD who do exhibit persistent reversible airflow limitation, and that there is possibly a genetic basis for this observation.
With the recent availability and increased use of tiotropium bromide, there is evolving evidence that there may exist a “long-acting anticholinergic phenotype” with specific effects on improved lung function and exercise capacity. Long-term studies over one year comparing tiotropium to placebo (Citation[142], Citation[143]) and ipratropium have shown clinical benefits on pulmonary function, breathlessness, use of rescue short acting β-agonists, health-related quality of life, COPD exacerbations and hospitalizations. There is also evolving evidence that long-term use of tiotropium has sustained effects on exercise tolerance, primarily by reduction of hyperinflation and increased inspiratory capacity both at rest and with exercise (Citation[144], Citation[145]). Expectedly, there is tremendous overlap between this potential tiotropium-responsive phenotype and those patients with resting dyspnea form hyperinflation and/or limited exercise tolerance from a reduced ability to recruit inspiratory capacity and tidal volume. It is hopeful that, with longer prospective trials being completed, tiotropium may be a true disease-modifying agent in COPD.
Airway hyperresponsiveness
The observed role of BDR is in stark contrast to the presence of airway hyperresponsiveness (AHR) in COPD, which has been shown not only to alter the natural history of the disease, but may be an independent risk factor for the development of smoking-induced irreversible airflow obstruction. AHR, as defined by a ≥ 20% fall in FEV1 with inhalation of ≤ 25 mg/mL methacholine, was assessed in the Lung Health Study at enrollment and at 5-year follow-up. Initially, 63% of men and 87% of women had AHR by this criteria, with 46% of men and 74% of women showing AHR at methacholine concentrations ≤ 10 mg/ mL (Citation[146]). During 5 years of follow-up, participants with greater degrees of AHR at study entry showed a greater longitudinal decline in FEV1 (Citation[118]). Changes in AHR over this 5-year period were primarily related to change in the FEV1, with greater a decline in FEV1 reflecting a greater increase in AHR (Citation[147]). Interestingly, smoking cessation had a small additional benefit in AHR beyond its favorable effects on FEV1 changes, supporting the hypothesis that COPD is, in part, the result of a complex gene-environment interaction.
These results have been supported by a population-based cohort Swiss study on Air Pollution and Lung Disease in Adults (SAPALDIA), which sought to determine if AHR contributed to the development of COPD and asthma (Citation[148]). Respiratory symptoms and AHR were assessed in 7,216 participants at enrollment. Eleven years later, 5,825 of these participants were re-evaluated. This study found that AHR was associated with nearly a 3-fold increase risk of wheezing at follow-up among asymptomatic participants. Participants who went on to develop COPD (defined by FEV1/FVC < 70% at follow-up) had a 4.5-fold higher odds of having AHR at enrollment. In addition, AHR was associated with a significantly accelerated decline in FEV1 in current, former and ex-smokers, supporting what was seen in the Lung Health Study.
Thus, the presence of AHR not only represents a distinct COPD phenotype, but clearly impacts the natural history of the disease, and may suggest a more pathophysiologic relevance of AHR in COPD (Citation[149]). However, the presence of AHR in COPD does not play as central a role as it does in asthma. These observations highlight that similar genetic mechanisms are likely present in both diseases, but result in different phenotypic expression of disease (Citation[150]).
Hypercapnic
The development of chronic CO2 retention in some patients with COPD, and its relationship to an abnormal breathing pattern, is one of the oldest described physiologic derangements seen with severe airflow obstruction. This heralded the description of the “blue bloater” with an attenuated respiratory drive and being less dyspnic than their “pink puffer” counterpart (Citation[151]). However, there is no evidence that a depressed respiratory drive in patients with COPD is associated with less dyspnea or greater exercise tolerance when patients are matched for degree of airflow limitation (Citation[151]). A study of patients with and without CO2 retention during exercise across a wide range of FEV1 to determine the etiology of exertional dyspnea found that, in normocapnic individuals, the mechanical load and length-tension inappropriateness on ventilatory muscles were the main determinants of exertional dyspnea. As soon as hypercapnia occurs, the CO2 retention seemed to override all other inputs for dyspnea (Citation[152]).
A similar study in patients with severe COPD (FEV1 < 1 L) found that hypercapnia is associated with shallow breathing and inspiratory muscle weakness (Citation[153]). The rapid and shallow breathing pattern observed in some of these patients appeared to be linked to both marked increase in the pressure required for breathing relative to inspiratory muscle strength and to the severity of their dyspnea. There is also evidence that patients can improve blood gas tensions towards normal by hyperventilation during exercise, suggesting a possible concomitant role of central respiratory abnormalities in the development of CO2 retention in conjunction with severe airflow limitation (Citation[154], Citation[155]). Additional work has shown that clinical rating of dyspnea is associated with muscle weakness and increase in respiratory drive, which is modulated into a rapid and shallow pattern of breathing, which then leads to hypercapnia (Citation[156]).
These studies suggest that hypercapnia is not a distinct phenotype in COPD, but represents the complicated synergistic effects of variable physiologic factors. While these studies matched patients for FEV1, we have found in our cohort that degree of airflow limitation strongly correlates with resting hypercapnia (unpublished data). Interestingly, hypercapnia was also associated with a significantly lower BMI, supporting the clinical observation that the “blue bloaters” no longer exist, likely from the early identification and treatment of hypoxemia and obstructive sleep apnea. However, a small cohort study recently showed that, once hypercapnia develops in a patient with COPD, other prognostic factors (such as FEV1, age, and BMI) do not independently affect survival (Citation[157]).
Impaired Exercise Tolerance
Exercise tolerance has become an important patient-centered outcome in COPD since it is a major determinant of an impaired health status (Citation[158]). Indeed, the patient with COPD and a low exercise tolerance, independent of other physiologic variables, represents an important and distinct phenotype, and many patients with COPD complain of exercise intolerance (typically with exertional dyspnea) that may not correlate with their degree of airflow limitation (Citation[159]). With this, the improvement of exercise capacity is one of the major goals for the management of COPD (Citation[159]).
Exercise capacity has been shown to be an important predictor of mortality in COPD (Citation[13], Citation[160],Citation[161],Citation[162]), and current guidelines recommend performing direct cardiopulmonary exercise testing since no resting physiologic measurements can predict exercise capacity (Citation[160]). Exercise testing in COPD can range from simple field tests (such as the 6-minute walk or the incremental shuttle walk) to a maximal cardiopulmonary laboratory test. Maximal tests are designed to measure peak exercise capacity and/or peak oxygen uptake, whereas field tests reflect functional capacity (Citation[163],Citation[164],Citation[165],Citation[166]).
In the National Emphysema Treatment Trial (NETT), maximal exercise capacity was assessed by incremental cycle ergometry (Citation[167]). The most widely used field test is the 6-minute walk (6MWT), given its simplicity and no need for exercise equipment (Citation[165]). While the strongest indication for the 6MWT is measuring the response to medical intervention in patients with moderate to severe heart or lung disease, it has also been used as a one-time measure of functional status, as well as a predictor of morbidity and mortality in patients with COPD (Citation[165], Citation[168],Citation[169],Citation[170]). The fact that investigators have used the 6MWT in these settings does not prove that the test is clinically useful (or the best test) for determining functional capacity, either at one time point or to assess changes in response to therapy (Citation[165]). The incremental shuttle walking test (ISWT) is a field test that is similar to the laboratory tests as it is externally paced and progressive; although it is more complex than the 6MWT, there is evidence that peak exercise capacity measured by cycle ergometry could be estimated from an ISWT with similar accuracy in patients with COPD (Citation[163], Citation[171])
Until recently, there were no studies investigating how exercise intolerance in COPD progresses over time. Oga et al. studied 131 male patients with moderate-to-very-severe COPD longitudinally over 5 years to determine what factors contribute to the long-term deterioration in exercise capacity (Citation[162]). Although there was clear evidence of a measurable and progressive deterioration in exercise capacity over time, they found that no static pulmonary function measures significantly predicted the change in peak VO2. In addition, baseline exercise indexes also did not correlate with subsequent deterioration in exercise capacity. Most notable was the finding that the decline in peak VO2 was more rapid than the decline in FEV1, reflecting the multidimensional nature of exercise capacity as a more sensitive marker of overall clinical deterioration.
The results from NETT, which showed differential effects on mortality and exercise capacity at 2 years when comparing medical therapy to lung-volume-reduction surgery (LVRS), provide the most clinically relevant application of assessing exercise capacity in patients with COPD and severe emphysema (Citation[165]). Patients with upper-lobe predominant emphysema and a low baseline exercise capacity (defined as ≤ 25 watts for women and ≤ 40 watts for men) who underwent LVRS had a lower risk of death than patients in the medical-therapy group (RR, 0.47; p = 0.005) (Citation[165]). In addition, these patients were more likely to have an improvement of > 10 watts in the maximal workload at 2 years (Citation[165]). In contrast, in patients with upper-lobe disease and a high baseline exercise capacity, there was no mortality benefit to LVRS over medical therapy.
Hyperinflation
The lungs in patients with COPD can be hyperinflated, either at rest (“static”) or during exercise (“dynamic”), causing several detrimental effects, including impairment of respiratory muscle function and gas exchange, and increases in the work of breathing (Citation[172]). Hyperinflation is commonly considered to be an elevation above normal of the resting functional residual capacity (FRC) or end-expiratory lung volume (EELV) (Citation[172]). Hyperinflation often remains undetected in the absence of detailed physiology analysis, and it is postulated that most (if not all) patients with COPD have some degree of hyperinflation (Citation[172]). Both static and dynamic effects of breathing contribute to lung hyperinflation in COPD.
Static hyperinflation results from decreased elasticity of the lung parenchyma; the destruction of elastic tissue seen with emphysema alters lung recoil, so that a given lung volume produces a lesser recoil pressure than in the healthy lung (Citation[172]). This shift in lung recoil and its pressure-volume curve alters the balance between the opposing recoil pressures of the chest wall and the lung; the reduced lung recoil pressure requires a greater volume to balance the chest wall recoil, increasing FRC (Citation[172], Citation[173]). However, emphysema-associated static hyperinflation is a modest contributor to hyperinflation in all but the most severe patients (Citation[172]).
Dynamic hyperinflation is the physiologic correlate of exertional dyspnea in COPD patients, and is due to the inability to expand their tidal volume in the face of increased inspiratory effort during exercise (Citation[174]), leading to “unsatisfied inspiration.” Dynamic hyperinflation occurs either independent of or in addition to static hyperinflation, and can be observed in patients across all severities of COPD (Citation[172]). Dynamic hyperinflation occurs when a patient commences inhalation before full exhalation has been achieved; consequently, air is trapped within the lungs with each successive breath. The development of dynamic hyperinflation is a function of expiratory airflows and expiratory time; in COPD, expiratory airflows are limited by airway narrowing resulting from increases in cholinergic (vagal) tone, inflammation, and mucus plugging (Citation[172], Citation[175]). As airflow limitation progresses, the time available for exhalation can become insufficient whenever demand for a greater minute ventilation increases tidal and respiratory rate, resulting in an increase in EELV and reduced inspiratory capacity (IC) (Citation[172], Citation[175]) (). Onset of dynamic hyperinflation will occur at even lower minute ventilations as disease severity limiting exhalation worsens, even occurring during quiet breathing in severe patients (Citation[172]).
Figure 9 Dynamic hyperinflation in COPD. Source: (Citation[175]), , p. 774. Changes in operational lung volumes are shown as ventilation increases with exercise in patients with COPD and in normal subjects. “Restrictive” constraints on tidal volume (VT, solid area) expansion during exercise are significantly greater in the COPD group from both below (reduced IC) and above (minimal IRV, open area).
![Figure 9 Dynamic hyperinflation in COPD. Source: (Citation[175]), Fig. 3, p. 774. Changes in operational lung volumes are shown as ventilation increases with exercise in patients with COPD and in normal subjects. “Restrictive” constraints on tidal volume (VT, solid area) expansion during exercise are significantly greater in the COPD group from both below (reduced IC) and above (minimal IRV, open area).](/cms/asset/cb012d3f-0cdf-4f88-a8c2-52762125b06f/icop_a_262814_uf0009_b.gif)
Oga et al. confirmed that dynamic ventilatory constraints progressed significantly over time, and that the resultant restrictive ventilatory defect due to dynamic hyperinflation is the most important mechanical consequence of this (Citation[160], Citation[176]). The worsening of dynamic volume constraints leads to a compensatory increase in the respiratory rate of COPD patients, which worsens dynamic hyperinflation and results in a deterioration of exertional dyspnea and breathing patterns. Once hyperinflated, a patient's ability to breath is significantly compromised due to mechanical restrictions resulting from a reduced IC, an increase in the work of breathing, and deleterious effects on inspiratory muscles and respiration (Citation[172]).
The resultant hyperinflation seen in many patients with COPD correlates better with their exercise limitation than measurements of airflow obstruction, likely from a resultant decrease in IC (Citation[144], Citation[177]). Long-acting bronchodilators (such as tiotropium and salmeterol) reduce hyperinflation and thereby increase IC, 6-minute walk distance and cycle endurance time (Citation[144], Citation[178]). Thus, the phenotypic characterization of “long-acting bronchodilator-responsiveness” may actually reflect the degree of hyperinflation and, with that, exercise limitation.
Low DLCO
Single-breath diffusing capacity of the lung for carbon monoxide (DLCO) represents the best pulmonary function test to assess the integrity of the pulmonary capillary bed (Citation[179]). DLCO correlates strongly with the presence and severity of emphysema, and correlates much better with reduced functional capacity and dyspnea that the FEV1. A recent study characterizing COPD clinical phenotypes based purely upon the severity of emphysema in 274 patients found a much better correlation between emphysema score and DLCO than between emphysema score and percent predicted FEV1 (Citation[180]). Data from the National Emphysema Treatment Trial (NETT) demonstrated that patients with a reduced DLCO (particularly when ≤ 20% of predicted) are more likely to have resting hypoxemia and are more likely to require supplemental oxygen with low levels of activity (Citation[181]). Notably, this is the group that has a higher mortality after lung volume reduction surgery.
The specificity and sensitivity of the DLCO is limited by numerous technical and physiologic factors that affect the test (Citation[179]). However, there remains consensus that the DLCO should be measured in all patients with chronic airflow limitation. In a similar manner to the wide range of other physiologic abnormalities seen in many patients with COPD, a low DLCO does not represent a unique phenotype but reflects the complex interaction of emphysema, airflow obstruction and pulmonary capillary bed destruction.
Pulmonary hypertension
Pulmonary hypertension (PH) represents a range of clinical and physiologic abnormalities, from cor pulmonale (clinical right heart failure from an elevated pulmonary arterial pressure) to hemodynamically defined resting mean pulmonary arterial pressure (PAP) greater than 25 mm Hg with a normal pulmonary capillary or left arterial pressure (Citation[182], Citation[183]). In patients with COPD, clinically diagnosed cor pulmonale has long been known to be an important negative prognostic sign, even after correction for FEV1 (Citation[184], Citation[185]). Early studies from the 1960s and 1970s, although prospective, were limited to subgroups of patients with COPD, and thus could not estimate the overall prevalence and effect of cor pulmonale on mortality (Citation[184]). When pulmonary hemodynamics have been measured, an increased mean pulmonary arterial pressure (PAP) at rest also imparts a poor prognosis, with a increase in the mean PAP of 10 mm Hg associated with a greater than 4-fold increase in mortality (Citation[185], Citation[186]). This suggests that certain COPD patients may be prone to the development of cor pulmonale in association with PH independent of severity of airflow limitation.
Given limitations in the standard measurement of pulmonary hemodynamics in patients with COPD, the exact prevalence and severity of PH in this population is unknown. It is estimated that 6% of patients with COPD will develop cor pulmonale each year (Citation[185]). The largest prospective study (N = 131) investigating the incidence of PH in patients with COPD found that 25% of all patients developed a resting PAP > 20 mm Hg at a mean follow-up of 6.8 years (Citation[187]). The development of PH was more common in subjects who had exercise-induced PH at their first assessment. Notably, this study was conducted on patients who did not require long-term oxygen therapy with a wide range of airflow limitation (28.2% with FEV1 < 35% predicted; 26.5% with FEV1 > 50% predicted). Recent data suggests that 10–30% of patients with COPD and at least one previous hospitalization have clinically significant PH (Citation[188],Citation[189],Citation[190]). Extrapolation from COPD-related hospitalization and mortality data suggest that the prevalence of significant cor pulmonale is 2–6/1,000, with an incidence of 1–3/10,000, which is 100 times the incidence of idiopathic PH (Citation[189], Citation[191]).
Historically, PH in patients with COPD was generally believed to be a result of long-standing hypoxemia and associated with only mild-to-moderate increases in mean PAP (25–35 mm Hg) (Citation[192]). However, there is ample evidence that PH in COPD often is not (or only minimally) reversible by supplemental oxygen (Citation[192],Citation[193],Citation[194]). It is now well understood that pulmonary vascular remodeling in COPD is more than just medial hypertrophy from chronic hypoxic vasoconstriction (Citation[192], Citation[195], Citation[196]). Histological specimens from pulmonary arteries in COPD patients with secondary PH show abnormal changes in all layers of the vessel wall, with intimal changes being the most prominent (Citation[192]) (). It is postulated that the cause of vascular remodeling in COPD-related PH is from injury to the endothelial cells and subsequent “endothelial dysfunction” (Citation[185]). Endothelial dysfunction and vascular remodeling might result from the direct action of cigarette smoke on the vessel, acting to alter cytokine or vasoactive mediator balance (Citation[185], Citation[197]) (). Thus, COPD patients who manifest clinically significant PH may have an underlying genetic predisposition to altered inflammatory mediators and oxidant/antioxidant balance in response to cigarette smoke.
Figure 10 Section of a pulmonary arteriole from a patient with PH secondary to COPD. Source: (Citation[192]), , p. 287. All three vessel wall layers are remodeled, with prominent intimal thickening.
![Figure 10 Section of a pulmonary arteriole from a patient with PH secondary to COPD. Source: (Citation[192]), Fig. 1, p. 287. All three vessel wall layers are remodeled, with prominent intimal thickening.](/cms/asset/76a0c874-05c3-47ba-898f-d92f84ac48cf/icop_a_262814_uf0010_b.gif)
Figure 11 Mechanisms of endothelial dysfunction and vascular remodeling in the small pulmonary arteries of patients with COPD. Source: (Citation[185]), , p. 607. Diagram showing various cigarette smoke-related processes that drive vasoconstriction, vasodilation, and cell proliferation in the walls of the small pulmonary arteries. Most of the effects shown are potentially directly mediated by oxidants in the smoke acting on the vessel wall. The vessel is shown in the centre with muscular layer and adventitial layers highlighted. Oxidants in the smoke cause production of the vasoconstrictor endothelin and the vasoproliferative agents endothelin and vascular endothelial growth factor (VEGF). Smoke interferes with nitric oxide mediated vasodilation by decreasing nitric oxide bioactivity. Air trapping as part of chronic airflow obstruction appears to play an early role by inducing vascular compression, and hypoxia causes vasoconstriction, but only when the oxygen tension is extremely low.
![Figure 11 Mechanisms of endothelial dysfunction and vascular remodeling in the small pulmonary arteries of patients with COPD. Source: (Citation[185]), Fig. 2, p. 607. Diagram showing various cigarette smoke-related processes that drive vasoconstriction, vasodilation, and cell proliferation in the walls of the small pulmonary arteries. Most of the effects shown are potentially directly mediated by oxidants in the smoke acting on the vessel wall. The vessel is shown in the centre with muscular layer and adventitial layers highlighted. Oxidants in the smoke cause production of the vasoconstrictor endothelin and the vasoproliferative agents endothelin and vascular endothelial growth factor (VEGF). Smoke interferes with nitric oxide mediated vasodilation by decreasing nitric oxide bioactivity. Air trapping as part of chronic airflow obstruction appears to play an early role by inducing vascular compression, and hypoxia causes vasoconstriction, but only when the oxygen tension is extremely low.](/cms/asset/f746f7a3-fda4-435a-911c-deb3f2eebc5d/icop_a_262814_uf0011_b.gif)
RADIOLOGIC PHENOTYPES IN COPD
As high-resolution computed tomography (HRCT) technology has matured, the ability to define normal and abnormal lung findings has markedly improved. In COPD patients, the abnormalities are typically found in both lung parenchyma and airways, and have been described both qualitatively and quantitatively. A large number of studies have been done to detect and quantify pulmonary emphysema using computed tomography (CT), and low attenuation areas on CT scans in vivo have been shown to represent macroscopic or microscopic emphysematous changes in the lungs of patients (Citation[100], Citation[198],Citation[199],Citation[200],Citation[201],Citation[202],Citation[203],Citation[204],Citation[205],Citation[206],Citation[207],Citation[208],Citation[209],Citation[210],Citation[211],Citation[212],Citation[213],Citation[214]). Qualitative phenotypic features include presence, pattern and severity of emphysema, and airway abnormalities, such as increased thickness and bronchiectasis.
Quantitative assessments include measurements of lung parenchyma density (Hounsfield Units) and, more recently, airway diameter of large and intermediate size airways (Citation[198], Citation[215]) (). A limitation of many HRCT studies of COPD patients is that they do not include adequate control patients (smokers) and that airway abnormalities (but not emphysema) are not specific (especially for asthma). For instance, general population HRCT screenings of smokers without a history of respiratory disease and normal FEV1% report that emphysema (62% in current smokers versus 0% in non-smokers) and airway abnormalities (55% in current smokers versus 35% in non-smokers) are common, but are typically very mild (Citation[216]).
Figure 12 Quantification of airways disease. Source: (Citation[198]), , p. 1103. A representative case showing the process of airway analysis. Using a helical CT image containing the apical bronchus to the upper lobe (A), the algorithm defines the luminal area (B) and wall thickness (C).
![Figure 12 Quantification of airways disease. Source: (Citation[198]), Fig. 1, p. 1103. A representative case showing the process of airway analysis. Using a helical CT image containing the apical bronchus to the upper lobe (A), the algorithm defines the luminal area (B) and wall thickness (C).](/cms/asset/a407b526-0660-48d0-9244-a438fbcfc8a3/icop_a_262814_uf0012_b.gif)
Emphysema
The major pathologic forms of emphysema are centriacinar emphysema (CAE) and panacinar emphysema (PAE). The radiographic correlate of CAE and PAE are centrilobular and panlobular emphysema, respectively, and can be distinguished by HRCT ( and ). CAE occurs as a result of dilation and loss of the respiratory bronchioles, typically in the upper lobes of the lung. It is recognized on CT by the presence of small poorly defined areas of low lung attenuation, often transversed by a centrilobular vessel (Citation[217],Citation[218],Citation[219]). PAE results from a uniform loss of the acini and occurs in upper and lower lobes. On CT this is characterized by diffuse decrease in lung attenuation, sometimes associated with linear structures thought to represent the walls of over-inflated secondary pulmonary lobules (Citation[219], Citation[220]).
Figure 13 Centrilobular emphysema. (A) a CT through the upper lobes shows multiple poorly defined “black holes”, some of which contain a central vessel, indicating centrilobular emphysema. (B) a similar pattern is seen in the lower lobes of the same patient.
Figure 14 Comparison of centrilobular to panlobular emphysema. (A) the scan through the upper lobes shows some areas of centrilobular emphysema, but there are also some larger areas of decreased attenuation indicating confluent panlobular emphysema. (B) a scan through the lower lobes (same patient as A) shows larger areas of confluent panlobular emphysema.
Other forms of emphysema, such as distal acinar emphysema (DAE), are less common. DAE, also called paraseptal or localized emphysema, is characterized by well-demarcated large or small focal subpleural areas of decreased attenuation, with well-defined walls () (Citation[221]). These areas of emphysema, which typically occur in the upper lobes, often occur in rows, and may mimic honeycombing. DAE does not usually cause physiologic impairment unless it progresses to form large bullae (vanishing lung) (Citation[222]). However, it may predispose to pneumothorax. Paracicatricial emphysema (Citation[219]) is not pertinent to radiographic phenotypes of COPD since it is secondary to underlying lung fibrosis (such as pneumoconiosis).
Figure 15 Distal acinar emphysema. Distal acinar emphysema in a tall, thin, 42-year-old cigarette smoker. CT through the lung apices shows predominantly subpleural lucencies indicating distal acinatr (paraseptal) emphysema.
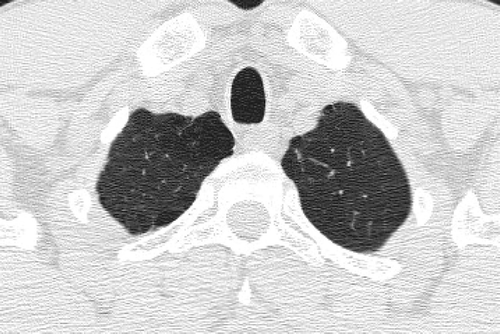
The majority of patients with emphysema have a predominantly PAE or CAE phenotype; however, only 44%–59% have pure CAE or PAE (Citation[223],Citation[224],Citation[225]). The pathologic phenotypes also have functional associations: CAE patients tend to have more small airways disease, whereas PAE patients tend to have high lung compliance (Citation[223], Citation[225], Citation[226]). PAE is associated most strongly with α1-antitrypsin (AAT) deficiency; however, the majority of patients with PAE do not have AAT. Multiple independent studies have shown good correlation between the grading of emphysema on CT and the pathology “gold standard” (Citation[207], Citation[217], Citation[218], Citation[227],Citation[228],Citation[229]).
HRCT is the best method to detect emphysema in vivo (Citation[230]) and is also substantially more sensitive than spirometry (Citation[231], Citation[232]). The extent of HRCT-detected emphysema has been reported to be associated with decreasing FEV1 (Citation[198]); however, other investigators (Citation[233], Citation[234]) have demonstrated that the correlation between the severity of airflow obstruction and emphysema is poor. In particular, there are some patients with severe airflow obstruction and minimal emphysema and some patients with severe emphysema and no to mild airflow obstruction ( and ).
Figure 16 Variable degree of airflow limitation (FEV1) with similar emphysema severity. (A) and (B) are from the same patient, and demonstrate severe panlobular and centrilobular upper lobe emphysema (A), with a predominant centrilobular pattern in the lower lobes (B) but a preserved FEV1. (C) and (D) are from a different patient with a comparable pattern of diffuse upper and lower lobe emphysema. Although there is only slightly worse lower lobe emphysema (D) in the second patient, there is much more severe airflow limitation and loss of DLCO.
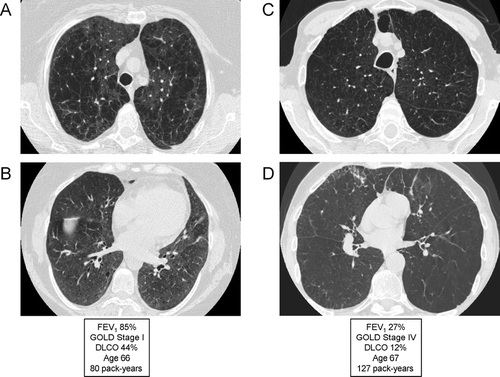
Figure 17 Variable emphysema severity with similar degree of airflow limitation (FEV1). (A) and (B) are from the same patient, and demonstrate mild-to-moderate centrilobular emphysema (CLE) with an upper lobe predominance (A). (C) and (D) are from a different patient, demonstrating moderate-to-severe CLE with an upper lobe predominance (C). Despite the marked visual differences in emphysema severity, the patients are matched for degree of airflow limitation; note that the DLCO correlates better with emphysema severity than the FEV1.
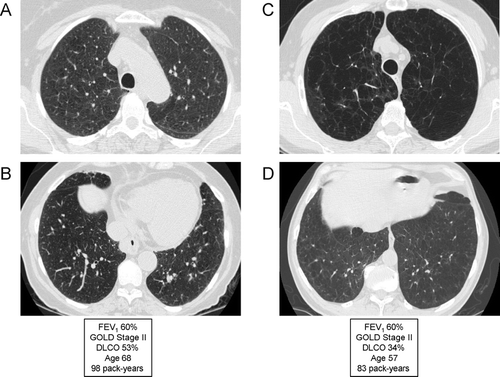
Additionally, severe airflow limitation may be seen in the absence of emphysema when significant airways disease in present (). As the previous examples illustrate, compared to the FEV1, the diffusing capacity for carbon monoxide (DLco) correlates much better with emphysema (Citation[235]) and can be used as a surrogate for emphysema when HRCT or pathologic data are not available. However, Gelb et al. reported that severe small airways disease could cause significant hyperinflation and a low DLco without emphysema (Citation[236]). Evaluation of air trapping on spiral CT data sets obtained at functional residual capacity (FRC) has been helpful in assessing air trapping due to small airway disease (Citation[237],Citation[238],Citation[239],Citation[240],Citation[241]). Availability of scans performed at TLC and FRC with multi-channel CT spiral data sets will also enable evaluation of airway compliance, by comparing airway geometry at the two different lung volumes.
The significance of the phenotype of centrilobular emphysema is illustrated by Remy-Jardin and others (Citation[242]), who have shown that asymptomatic smokers will have a more rapid decline in lung function if their HRCT has evidence of emphysema. Matsuoka et al., using quantitative CT techniques, showed that, in persistent smokers, centrilobular emphysema tends to progress by enlargement of existing low attenuation areas, but not by development of new areas of low attenuation (Citation[243]). Patients with CAE tend to do better with lung volume reduction surgery (LVRS) compared to PAE (Citation[226]) and the National Emphysema Treatment Trial showed that patients with upper lobe predominant emphysema had better outcomes compared to those with non-upper lobe predominant emphysema (Citation[167]).
In a study of 84 Japanese subjects with COPD (GOLD II, III, and IV), patients were qualitatively scored as having either emphysema or airway wall thickening (Citation[244]). Despite comparable FEV1, subjects without emphysema had a significantly lower BMI, later age of onset of dyspnea, were more likely to wheeze, and were less likely to receive long-term oxygen therapy. The subjects without emphysema were also less likely to have hyperinflation, low DLco, and more likely to have β2-agonist reversibility on PFTs and sputum eosinophilia. In particular, airway wall thickening was associated with dyspnea score, sputum eosinophilia and reversibility of airflow limitation in response to ICS. This suggests that patients with airway wall thickening by HRCT show more of the clinical characteristics of patients who respond to bronchodilators and ICS. Although airway wall thickness is assessed in intermediate and large airways by HRCT, this correlates with small airway diameter histologically (Citation[215]). Furthermore, this group has shown that airway dimensions are related to severity of airflow obstruction and gas trapping, but not related to the DLco.
A recently published study characterized clinical phenotypes of COPD based purely on severity of emphysema (Citation[180]). The visual assessment of emphysema on HRCT in 274 patients with COPD confirmed that the severity of emphysema is varied widely even among the same degree of airflow limitation. Chronic bronchitic symptoms, however, were equally distributed irrespective of emphysema severity. These observations continue to lend weight to modifications of COPD classification based upon objective measurements of emphysema and airways diseases (and away from classical descriptions of “pink puffers” and “blue bloaters”). In this study, the phenotype with emphysema in predominance had lower BMI and poorer health-related quality of life (Citation[180]).
α1-antitrypsin deficiency (AAT) is one of the earliest described genetic diseases and is strongly associated with emphysema (Citation[245]). The two major abnormal alleles are Z and S; however, patients with AAT only account for about 3% of COPD patients (Citation[105], Citation[246]). AAT is classically associated with panacinar emphysema (Citation[247]); however, centriacinar emphysema, particularly of the upper lobes, is the most common type of emphysema found in this condition. A single nucleotide polymorphism in the promoter of the TNF-α gene has also been associated with emphysema severity (Citation[248]). Ito et al. found that polymorphism of MMP-9 (C-1562T) was associated with upper lung dominant emphysema in patients with COPD (Citation[249]).
Minematsu et al. found that the CYP2A6del allele, involved in nicotine metabolism, was associated with both nicotine dependence and a higher percentage of emphysema on CT (Citation[250]). There has been no evaluation of the relationship between gene polymorphisms and other markers of COPD such as small airways abnormality and airway wall thickening. Variants in other candidate genes have also been studied in COPD case-control genetic association studies, and variants in several candidate genes have been significantly associated with COPD in multiple studies, including glutathione S-transferase P1 (GSTP1), glutathione S-transferase M1 (GSTM1), α 1-antichymotrypsin (SERPINA3), surfactant protein B (SFTPB), tumor necrosis factor-α (TNF-α), microsomal epoxide hydrolase (EPHX1), and vitamin D binding protein (GC) (Citation[251]); however, many of these findings are not replicated and the authors rarely include sub-analysis of HRCT phenotypes. The reader is referred to multiple other recent reviews for additional information on the genetics of COPD (Citation[252],Citation[253],Citation[254],Citation[255],Citation[256],Citation[257]).
Airways disease
There is renewed interest in the airway wall pathology of COPD. Hogg et al. (Citation[258]) reported that the progression of COPD was strongly associated with an increase in the volume of tissue in the wall. The number and percentage of inflammatory cells was particularly associated with severity of airflow obstruction. Recently, advancements in image analysis have permitted airway diameter to be assessed by volumetric CT. Increased thickness of airway walls is associated with the presence of COPD (Citation[259]), with reversibility of airway obstruction (Citation[244]), and with symptoms of chronic bronchitis (Citation[260]). In patients with COPD, bronchial wall thickening is an important independent predictor of FEV1 (Citation[261], Citation[262]). Airway wall thickening can be assessed quantitatively by measuring the percentage wall area as a proportion of total bronchial area. Evaluation of the airways on axial CT images is compromised by the fact that the airways are often not imaged orthogonal to their course, resulting in varying degrees of obliquity of section. With the use of volumetric CT, it is possible to develop a three-dimensional model of the airways, allowing measurement of the airways in a plane orthogonal to the long axis of the airway (Citation[263]). Using curved multiplanar reconstruction, bronchi with internal diameters down to about 2 mm may be routinely evaluated.
Recently, Hasegawa and colleagues used curved multiplanar reconstruction to obtain longitudinal airway reconstructions and accurate measurements of airway wall area with airways as small as 2 mm in luminal diameter (Citation[262]). Of interest, they found stronger correlations between airway wall area measurements (using the “half-max” method) of smaller rather than larger airways with FEV1. This work indicates the potential value of airway wall measurements in dissecting the heterogeneity of COPD, as well as the importance of studying small airways in such analyses. Novel automated approaches for airway wall measurement, which have recently been developed, will be essential for the dissecting of the COPD syndrome into emphysema and airway components that should be more etiologically homogeneous.
An important, and often neglected, phenotypic manifestation of smoking-related lung disease is small airways abnormality. Respiratory bronchiolitis is commonly found in smokers with moderate or heavy tobacco exposures, and may be manifested on CT by centrilobular nodularity on high-resolution evaluation, and/or by air trapping if expiratory CT is performed. Remy-Jardin et al. (Citation[242]) showed that individual foci of centrilobular nodularity may progress to centrilobular emphysema on follow-up over several years. Some heavy smokers with respiratory bronchiolitis may develop the clinical phenotype of respiratory bronchiolitis interstitial lung disease (Citation[264],Citation[265],Citation[266]), characterized by substantial physiologic impairment, and associated with CT findings of patchy ground glass abnormality and centrilobular nodularity ().
Figure 18 Radiographic appearance of airways disease predominant COPD associated with severe airflow limitation. (A) demonstrates mild upper lobe centrilobular emphysema, while (B) shows very minimal emphysema in the lower lobes but moderate airway wall thickening indicating bronchitis (white arrows) and centrilobular thickening suggesting respiratory bronchiolitis (black arrows).
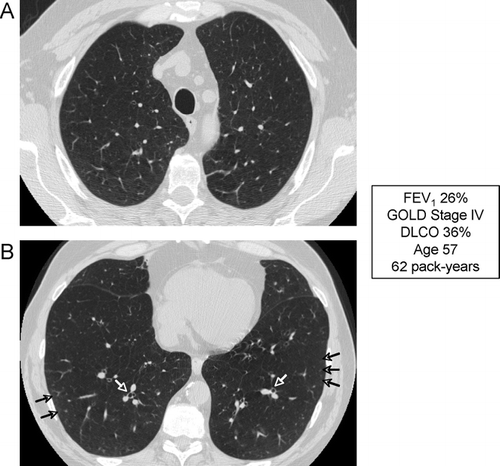
Figure 19 Radiographic appearance of respiratory bronchiolitis-interstitial lung disease (RB-ILD). A 35-year-old heavy smoker with RB-ILD. CT images through through upper (A) and lower (B) lobes show mild centrilobular emphysema, and widespread poorly defined centrilobular nodularity and ground glass abnormality. Focus of decreased attenuation in the right lower lobe is due to air trapping (arrow).
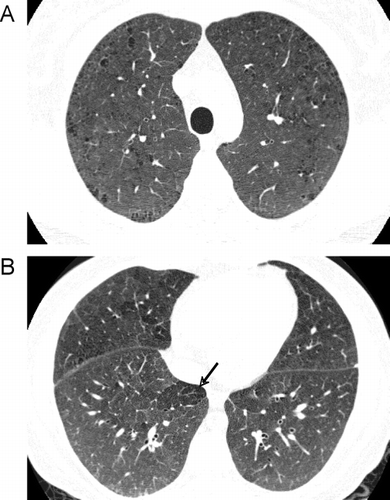
CONCLUSION
Although exposure to tobacco smoke is a predominant risk factor for COPD, we have described several different COPD phenotypes with distinct clinical outcomes. These phenotypes include frequent exacerbators, pulmonary cachectic, ICS responsive, rapid decliners, and emphysema/airways disease phenotypes. Thus, the paradigm that COPD is one disease may be incorrect, and suggests that COPD should be considered as a spectrum of smoking-related diseases. Failure to consider COPD phenotypes is likely to limit the power of therapeutic trials since not all COPD patients are likely to benefit from each therapy. The challenge to future COPD researchers is to better characterize these phenotypes and identify their risk factors.
REFERENCES
- Pauwels R. Global initiative for chronic obstructive lung diseases (gold): Time to act. Eur Respir J 2001; 18: 901–902
- Pauwels R A, Buist A S, Calverley P M, Jenkins C R, Hurd S S. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Nhlbi/who global initiative for chronic obstructive lung disease (gold) workshop summary. Amer J Respir Crit Care Med 2001; 163: 1256–1276
- American Thoracic Society. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. American thoracic society. Amer J Respir Criti Care Med 1995; 152: S77–121
- Anthonisen N R, Wright E C, Hodgkin J E. Prognosis in chronic obstructive pulmonary disease. Amer Rev Respir Dis 1986; 133: 14–20
- Nocturnal Oxygen Therapy Trial Group. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: A clinical trial. Nocturnal oxygen therapy trial group. Ann Intern Med 1980; 93: 391–398
- Dolan S, Varkey B. Prognostic factors in chronic obstructive pulmonary disease. Curr Opin Pulmon Med 2005; 11: 149–152
- Wolkove N, Dajczman E, Colacone A, Kreisman H. The relationship between pulmonary function and dyspnea in obstructive lung disease. Chest 1989; 96: 1247–1251
- Hajiro T, Nishimura K, Tsukino M, Ikeda A, Koyama H, Izumi T. Comparison of discriminative properties among disease-specific questionnaires for measuring health-related quality of life in patients with chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 1998; 157: 785–790
- Hajiro T, Nishimura K, Tsukino M, Ikeda A, Oga T, Izumi T. A comparison of the level of dyspnea vs disease severity in indicating the health-related quality of life of patients with COPD. Chest 1999; 116: 1632–1637
- Fletcher C M, Elmes P C, Fairbairn A S, Wood C H. The significance of respiratory symptoms and the diagnosis of chronic bronchitis in a working population. Br Med J 1959; 2: 257–266
- BTS. Guidelines for the management of chronic obstructive pulmonary disease. The copd guidelines group of the standards of care committee of the BTS. Thorax 1997; 52: S1–28, Suppl 5
- Nishimura K, Izumi T, Tsukino M, Oga T. Dyspnea is a better predictor of 5-year survival than airway obstruction in patients with COPD. Chest 2002; 121: 1434–1440
- Celli B R, Cote C G, Marin J M, Casanova C, Montes de Oca M, Mendez R A, Pinto Plata V, Cabral H J. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. New Engl J Med 2004; 350: 1005–1012
- Fan V S, Curtis J R, Tu S P, McDonell M B, Fihn S D. Using quality of life to predict hospitalization and mortality in patients with obstructive lung diseases. Chest 2002; 122: 429–436
- Garcia-Aymerich J, Farrero E, Felez M A, Izquierdo J, Marrades R M, Anto J M. Risk factors of readmission to hospital for a COPD exacerbation: A prospective study. Thorax 2003; 58: 100–105
- Garcia-Aymerich J, Monso E, Marrades R M, Escarrabill J, Felez M A, Sunyer J, Anto J M. Risk factors for hospitalization for a chronic obstructive pulmonary disease exacerbation. Efram study. Amer J Respir Crit Care Med 2001; 164: 1002–1007
- Niewoehner D E, Lokhnygina Y, Rice K, Kuschner W G, Sharafkhaneh A, Sarosi G A, Krumpe P, Pieper K, Kesten S. Risk indexes for exacerbations and hospitalizations due to COPD. Chest 2007; 131: 20–28
- Roberts C M, Lowe D, Bucknall C E, Ryland I, Kelly Y, Pearson M G. Clinical audit indicators of outcome following admission to hospital with acute exacerbation of chronic obstructive pulmonary disease. Thorax 2002; 57: 137–141
- Skrepnek G H, Skrepnek S V. Epidemiology, clinical and economic burden, and natural history of chronic obstructive pulmonary disease and asthma. Amer J Manag Care 2004; 10: S129–138
- Parshall M B. Adult emergency visits for chronic cardiorespiratory disease: Does dyspnea matter?. Nurs Res 1999; 48: 62–70
- Patil S P, Krishnan J A, Lechtzin N, Diette G B. In-hospital mortality following acute exacerbations of chronic obstructive pulmonary disease. Arch Intern Med 2003; 163: 1180–1186
- Groenewegen K H, Schols A M, Wouters E F. Mortality and mortality-related factors after hospitalization for acute exacerbation of COPD. Chest 2003; 124: 459–467
- Goel A, Pinckney R G, Littenberg B. Apache ii predicts long-term survival in copd patients admitted to a general medical ward. J Gen Intern Med 2003; 18: 824–830
- Almagro P, Calbo E, Ochoa de Echaguen A, Barreiro B, Quintana S, Heredia J L, Garau J. Mortality after hospitalization for COPD. Chest 2002; 121: 1441–1448
- Decramer M, Gosselink R, Bartsch P, Lofdahl C G, Vincken W, Dekhuijzen R, Vestbo J, Pauwels R, Naeije R, Troosters T. Effect of treatments on the progression of COPD: Report of a workshop held in leuven, 11–12 march 2004. Thorax 2005; 60: 343–349
- Mapel D W. Treatment implications on morbidity and mortality in COPD. Chest 2004; 126: 150S–158S, Discussion 159S–161S
- Sin D D, Wu L, Anderson J A, Anthonisen N R, Buist A S, Burge P S, Calverley P M, Connett J E, Lindmark B, Pauwels R A, et al. Inhaled corticosteroids and mortality in chronic obstructive pulmonary disease. Thorax 2005; 60: 992–997
- Schols A M, Slangen J, Volovics L, Wouters E F. Weight loss is a reversible factor in the prognosis of chronic obstructive pulmonary disease. Amer J Respir Criti Care Med 1998; 157: 1791–1797
- Schols A M, Broekhuizen R, Weling-Scheepers C A, Wouters E F. Body composition and mortality in chronic obstructive pulmonary disease. Amer J Clin Nutr 2005; 82: 53–59
- Prescott E, Almdal T, Mikkelsen K L, Tofteng C L, Vestbo J, Lange P. Prognostic value of weight change in chronic obstructive pulmonary disease: Results from the copenhagen city heart study. Eur Respir J 2002; 20: 539–544
- Kotler D P. Cachexia. Ann Intern Med 2000; 133: 622–634
- Chailleux E, Laaban J P, Veale D. Prognostic value of nutritional depletion in patients with COPD treated by long-term oxygen therapy: Data from the antadir observatory. Chest 2003; 123: 1460–1466
- Schols A M. Nutrition in chronic obstructive pulmonary disease. Curr Opin Pulmon Med 2000; 6: 110–115
- Schols A M, Soeters P B, Dingemans A M, Mostert R, Frantzen P J, Wouters E F. Prevalence and characteristics of nutritional depletion in patients with stable copd eligible for pulmonary rehabilitation. Amer Rev Respir Dis 1993; 147: 1151–1156
- Engelen M P, Wouters E F, Deutz N E, Does J D, Schols A M. Effects of exercise on amino acid metabolism in patients with chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2001; 163: 859–864
- Engelen M P, Schols A M, Does J D, Gosker H R, Deutz N E, Wouters E F. Exercise-induced lactate increase in relation to muscle substrates in patients with chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2000; 162: 1697–1704
- Debigare R, Cote C H, Maltais F. Peripheral muscle wasting in chronic obstructive pulmonary disease. Clinical relevance and mechanisms. Amer J Respir Crit Care Med 2001; 164: 1712–1717
- Vestbo J, Prescott E, Almdal T, Dahl M, Nordestgaard B G, Andersen T, Sorensen T I, Lange P. Body mass, fat-free body mass, and prognosis in patients with chronic obstructive pulmonary disease from a random population sample: Findings from the copenhagen city heart study. Amer J Respir Crit Care Med 2006; 173: 79–83
- Agusti A G, Noguera A, Sauleda J, Sala E, Pons J, Busquets X. Systemic effects of chronic obstructive pulmonary disease. Eur Respir J 2003; 21: 347–360
- Balasubramanian V P, Varkey B. Chronic obstructive pulmonary disease: Effects beyond the lungs. Curr Opin Pulmon Med 2006; 12: 106–112
- Wouters E F, Creutzberg E C, Schols A M. Systemic effects in COPD. Chest 2002; 121: 127S–130S
- Jagoe R T, Engelen M P. Muscle wasting and changes in muscle protein metabolism in chronic obstructive pulmonary disease. Eur Respir J 2003; 46: 52s–63s
- Gosker H R, Kubat B, Schaart G, van der Vusse G J, Wouters E F, Schols A M. Myopathological features in skeletal muscle of patients with chronic obstructive pulmonary disease. Eur Respir J 2003; 22: 280–285
- Jakobsson P, Jorfeldt L, Brundin A. Skeletal muscle metabolites and fibre types in patients with advanced chronic obstructive pulmonary disease (copd), with and without chronic respiratory failure. Eur Respir J 1990; 3: 192–196
- Hildebrand I L, Sylven C, Esbjornsson M, Hellstrom K, Jansson E. Does chronic hypoxaemia induce transformations of fibre types?. Acta Physiol Scand 1991; 141: 435–439
- Sato Y, Asoh T, Honda Y, Fujimatsu Y, Higuchi I, Oizumi K. Morphologic and histochemical evaluation of muscle in patients with chronic pulmonary emphysema manifesting generalized emaciation. Euro Neurol 1997; 37: 116–121
- Maltais F. LeBlanc P Simard C Jobin J Berube C Bruneau J Carrier L Belleau R. Skeletal muscle adaptation to endurance training in patients with chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 1996; 154: 442–447
- Serres I, Varray A, Vallet G, Micallef J P, Prefaut C. Improved skeletal muscle performance after individualized exercise training in patients with chronic obstructive pulmonary disease. J Cardiopul Rehab 1997; 17: 232–238
- Vallet G, Ahmaidi S, Serres I, Fabre C, Bourgouin D, Desplan J, Varray A, Prefaut C. Comparison of two training programmes in chronic airway limitation patients: Standardized versus individualized protocols. Eur Respir J 1997; 10: 114–122
- Couillard A, Prefaut C. From muscle disuse to myopathy in copd: Potential contribution of oxidative stress. Eur Respir J 2005; 26: 703–719
- Agusti A G, Sauleda J, Miralles C, Gomez C, Togores B, Sala E, Batle S, Busquets X. Skeletal muscle apoptosis and weight loss in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2002; 166: 485–489
- Swallow E B, Reyes D, Hopkinson N S, Man W D, Porcher R, Cetti E J, Moore A J, Moxham J, Polkey M I. Quadriceps strength predicts mortality in patients with moderate to severe chronic obstructive pulmonary disease. Thorax 2007; 62: 115–120
- Li X, Moody M R, Engel D, Walker S, Clubb F J, Jr, Sivasubramanian N, Mann D L, Reid M B. Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation 2000; 102: 1690–1696
- Creutzberg E C, Schols A M, Weling-Scheepers C A, Buurman W A, Wouters E F. Characterization of nonresponse to high caloric oral nutritional therapy in depleted patients with chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2000; 161: 745–752
- Agusti A, Morla M, Sauleda J, Saus C, Busquets X. Nf-kappab activation and inos upregulation in skeletal muscle of patients with COPD and low body weight. Thorax 2004; 59: 483–487
- Rabinovich R A, Bastos R, Ardite E, Llinas L, Orozco-Levi M, Gea J, Vilaro J, Barbera J A, Rodriguez-Roisin R, Fernandez-Checa J C, et al. Mitochondrial dysfunction in copd patients with low body mass index. Eur Respir J 2007; 29: 643–650
- Couillard A, Koechlin C, Cristol J P, Varray A, Prefaut C. Evidence of local exercise-induced systemic oxidative stress in chronic obstructive pulmonary disease patients. Eur Respir J 2002; 20: 1123–1129
- Rahman I, Skwarska E. MacNee W. Attenuation of oxidant/antioxidant imbalance during treatment of exacerbations of chronic obstructive pulmonary disease. Thorax 1997; 52: 565–568
- Palazzetti S, Rousseau A S, Richard M J, Favier A, Margaritis I. Antioxidant supplementation preserves antioxidant response in physical training and low antioxidant intake. Br J Nutr 2004; 91: 91–100
- Budweiser S, Heinemann F, Meyer K, Wild P J, Pfeifer M. Weight gain in cachectic copd patients receiving noninvasive positive-pressure ventilation. Respir Care 2006; 51: 126–132
- Donohue J F. Minimal clinically important differences in copd lung function. COPD 2005; 2: 111–124
- Petty T L. COPD in perspective. Chest 2002; 121: 116S–120S
- Paggiaro P L, Dahle R, Bakran I, Frith L, Hollingworth K, Efthimiou J. Multicentre randomised placebo-controlled trial of inhaled fluticasone propionate in patients with chronic obstructive pulmonary disease. International copd study group. Lancet 1998; 351: 773–780
- Vestbo J, Sorensen T, Lange P, Brix A, Torre P, Viskum K. Long-term effect of inhaled budesonide in mild and moderate chronic obstructive pulmonary disease: A randomised controlled trial. Lancet 1999; 353: 1819–1823
- Pauwels R A, Lofdahl C G, Laitinen L A, Schouten J P, Postma D S, Pride N B, Ohlsson S V. Long-term treatment with inhaled budesonide in persons with mild chronic obstructive pulmonary disease who continue smoking. European respiratory society study on chronic obstructive pulmonary disease. New Engl J Med 1999; 340: 1948–1953
- Burge P S, Calverley P M, Jones P W, Spencer S, Anderson J A, Maslen T K. Randomised, double blind, placebo controlled study of fluticasone propionate in patients with moderate to severe chronic obstructive pulmonary disease: The isolde trial. BMJ (Clin Res Ed) 2000; 320: 1297–1303
- The Lung Health Study Research Group. Effect of inhaled triamcinolone on the decline in pulmonary function in chronic obstructive pulmonary disease. New Engl J Med 2000; 343: 1902–1909
- Mahler D A, Wire P, Horstman D, Chang C N, Yates J, Fischer T, Shah T. Effectiveness of fluticasone propionate and salmeterol combination delivered via the diskus device in the treatment of chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2002; 166: 1084–1091
- Calverley P, Pauwels R, Vestbo J, Jones P, Pride N, Gulsvik A, Anderson J, Maden C. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: A randomised controlled trial. Lancet 2003; 361: 449–456
- Szafranski W, Cukier A, Ramirez A, Menga G, Sansores R, Nahabedian S, Peterson S, Olsson H. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. Eur Respir J 2003; 21: 74–81
- Hanania N A, Darken P, Horstman D, Reisner C, Lee B, Davis S, Shah T. The efficacy and safety of fluticasone propionate (250 microg)/salmeterol (50 microg) combined in the diskus inhaler for the treatment of copd. Chest 2003; 124: 834–843
- Sutherland E R, Allmers H, Ayas N T, Venn A J, Martin R J. Inhaled corticosteroids reduce the progression of airflow limitation in chronic obstructive pulmonary disease: A meta-analysis. Thorax 2003; 58: 937–941
- Vestbo J, Pauwels R, Anderson J A, Jones P, Calverley P. Early onset of effect of salmeterol and fluticasone propionate in chronic obstructive pulmonary disease. Thorax 2005; 60: 301–304
- Calverley P M, Anderson J A, Celli B, Ferguson G T, Jenkins C, Jones P W, Yates J C, Vestbo J. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. New Engl J Med 2007; 356: 775–789
- Soriano J B, Sin D D, Zhang X, Camp P G, Anderson J A, Anthonisen N R, Buist A S, Burge P S, Calverley P M, Connett J E, et al. A pooled analysis of fev1 decline in COPD patients randomized to inhaled corticosteroids or placebo. Chest 2007; 131: 682–689
- Barnes P J, Ito K, Adcock I M. Corticosteroid resistance in chronic obstructive pulmonary disease: Inactivation of histone deacetylase. Lancet 2004; 363: 731–733
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur Respir J 2003; 22: 672–688
- Cully J A, Graham D P, Stanley M A, Ferguson C J, Sharafkhaneh A, Souchek J, Kunik M E. Quality of life in patients with chronic obstructive pulmonary disease and comorbid anxiety or depression. Psychosomatics 2006; 47: 312–319
- Dahlen I, Janson C. Anxiety and depression are related to the outcome of emergency treatment in patients with obstructive pulmonary disease. Chest 2002; 122: 1633–1637
- Kim H F, Kunik M E, Molinari V A, Hillman S L, Lalani S, Orengo C A, Petersen N J, Nahas Z, Goodnight-White S. Functional impairment in copd patients: The impact of anxiety and depression. Psychosomatics 2000; 41: 465–471
- van de E L, Yzermans C J, Brouwer H J. Prevalence of depression in patients with chronic obstructive pulmonary disease: A systematic review. Thorax 1999; 54: 688–692
- Smoller J W, Simon N M, Pollack M H, Kradin R, Stern T. Anxiety in patients with pulmonary disease: Comorbidity and treatment. Semin Clin Neuropsych 1999; 4: 84–97
- Brenes G A. Anxiety and chronic obstructive pulmonary disease: Prevalence, impact, and treatment. Psychosom Med 2003; 65: 963–970
- Prigatano G P, Wright E C, Levin D. Quality of life and its predictors in patients with mild hypoxemia and chronic obstructive pulmonary disease. Arch Intern Med 1984; 144: 1613–1619
- Kvaal K, Macijauskiene J, Engedal K, Laake K. High prevalence of anxiety symptoms in hospitalized geriatric patients. Inter J Geriatr Psych 2001; 16: 690–693
- Kunik M E, Roundy K, Veazey C, Souchek J, Richardson P, Wray N P, Stanley M A. Surprisingly high prevalence of anxiety and depression in chronic breathing disorders. Chest 2005; 127: 1205–1211
- van Manen J G, Bindels P J, Dekker F W, CJ I J, van der Zee J S, Schade E. Risk of depression in patients with chronic obstructive pulmonary disease and its determinants. Thorax 2002; 57: 412–416
- Di Marco F, Verga M, Reggente M. Maria Casanova F Santus P Blasi F Allegra L Centanni S. Anxiety and depression in copd patients: The roles of gender and disease severity. Respir Med 2006; 100: 1767–1774
- Felker B, Katon W, Hedrick S C, Rasmussen J, McKnight K, McDonnell M B, Fihn S D. The association between depressive symptoms and health status in patients with chronic pulmonary disease. Gen Hosp Psych 2001; 23: 56–61
- Neuman A, Gunnbjornsdottir M, Tunsater A, Nystrom L, Franklin K A, Norrman E, Janson C. Dyspnea in relation to symptoms of anxiety and depression: A prospective population study. Respir Med 2006; 100: 1843–1849
- Agle D P, Baum G L. Psychological aspects of chronic obstructive pulmonary disease. Med Clin N Amer 1977; 61: 749–758
- Norwood R, Balkissoon R. Current perspectives on management of co-morbid depression in copd. COPD 2005; 2: 185–193
- Borson S, Claypoole K, McDonald G J. Depression and chronic obstructive pulmonary disease: Treatment trials. Semin Clin Neuropsych 1998; 3: 115–130
- Wamboldt F S. Anxiety and depression in COPD: A call (and need) for further research. COPD 2005; 2: 199–201
- Clark D M. A cognitive approach to panic. Behav Res Ther 1986; 24: 461–470
- Moore M C, Zebb B J. The catastrophic misinterpretation of physiological distress. Behav Res Ther 1999; 37: 1105–1118
- Behrendt C E. Mild and moderate-to-severe copd in nonsmokers: Distinct demographic profiles. Chest 2005; 128: 1239–1244
- Birring S S, Brightling C E, Bradding P, Entwisle J J, Vara D D, Grigg J, Wardlaw A J, Pavord I D. Clinical, radiologic, and induced sputum features of chronic obstructive pulmonary disease in nonsmokers: A descriptive study. Amer J Respir Crit Care Med 2002; 166: 1078–1083
- Pena V S, Miravitlles M, Gabriel R, Jimenez-Ruiz C A, Villasante C, Masa J F, Viejo J L, Fernandez-Fau L. Geographic variations in prevalence and underdiagnosis of copd: Results of the iberpoc multicentre epidemiological study. Chest 2000; 118: 981–989
- Ekici A, Ekici M, Kurtipek E, Akin A, Arslan M, Kara T, Apaydin Z, Demir S. Obstructive airway diseases in women exposed to biomass smoke. Environ Res 2005; 99: 93–98
- Matheson M C, Benke G, Raven J, Sim M R, Kromhout H, Vermeulen R, Johns D P, Walters E H, Abramson M J. Biological dust exposure in the workplace is a risk factor for chronic obstructive pulmonary disease. Thorax 2005; 60: 645–651
- Tashkin D P, Detels R, Simmons M, Liu H, Coulson A H, Sayre J, Rokaw S. The ucla population studies of chronic obstructive respiratory disease: Xi. Impact of air pollution and smoking on annual change in forced expiratory volume in one second. Amer J Respir Crit Care Med 1994; 149: 1209–1217
- Silva G E, Sherrill D L, Guerra S, Barbee R A. Asthma as a risk factor for copd in a longitudinal study. Chest 2004; 126: 59–65
- O'Connor G T, Sparrow D, Weiss S T. A prospective longitudinal study of methacholine airway responsiveness as a predictor of pulmonary-function decline: The normative aging study. Amer J Respir Crit Care Med 1995; 152: 87–92
- Dahl M, Tybjaerg-Hansen A, Lange P, Vestbo J, Nordestgaard B G. Change in lung function and morbidity from chronic obstructive pulmonary disease in alpha1-antitrypsin mz heterozygotes: A longitudinal study of the general population. Ann Intern Med 2002; 136: 270–279
- Kraft M, Mortenson R L, Colby T V, Newman L, Waldron J A, Jr, King T E, Jr. Cryptogenic constrictive bronchiolitis. A clinicopathologic study. Amer Rev Respir Dis 1993; 148: 1093–1101
- Turton C W, Firth G, Grundy E, Rigden B G, Smyth J T, Turner-Warwick M. Raised enzyme markers of chronic inflammation in asymptomatic farmers' lung. Thorax 1981; 36: 122–125
- Homma H, Yamanaka A, Tanimoto S, Tamura M, Chijimatsu Y, Kira S, Izumi T. Diffuse panbronchiolitis. A disease of the transitional zone of the lung. Chest 1983; 83: 63–69
- Aguayo S M, Miller Y E, Waldron J A, Jr, Bogin R M, Sunday M E, Staton G W, Jr, Beam W R, King T E, Jr. Brief report: Idiopathic diffuse hyperplasia of pulmonary neuroendocrine cells and airways disease. New Engl J Med 1992; 327: 1285–1288
- Global Initiative for Chronic Obstrcutive Lung Disease. Executive summary: Global strategy for the diagnosis, management, and prevention of chronic obstrcutive pulmonary disease. 2006, http://wwwgoldcopdcom(accessed January 2007)
- Siafakas N M, Vermeire P, Pride N B, Paoletti P, Gibson J, Howard P, Yernault J C, Decramer M, Higenbottam T, Postma D S, et al. Optimal assessment and management of chronic obstructive pulmonary disease (COPD). The European Respiratory Society Task Force. Eur Respir J 1995; 8: 1398–1420
- Speizer F E, Fay M E, Dockery D W, Ferris B G, Jr. Chronic obstructive pulmonary disease mortality in six U.S. Cities. Amer Rev Respir Dis 1989; 140: S49–55
- Tager I B, Segal M R, Speizer F E, Weiss S T. The natural history of forced expiratory volumes. Effect of cigarette smoking and respiratory symptoms. Amer Rev Respir Dis 1988; 138: 837–849
- Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J 1977; 1: 1645–1648
- Mannino D M, Davis K J. Lung function decline and outcomes in an elderly population. Thorax 2006; 61: 472–477
- Mannino D M, Reichert M M, Davis K J. Lung function decline and outcomes in an adult population. Amer J Respir Crit Care Med 2006; 173: 985–990
- Anthonisen N R, Connett J E, Kiley J P, Altose M D, Bailey W C, Buist A S, Conway W A, Jr, Enright P L, Kanner R E, O'Hara P, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of fev1. The lung health study. JAMA 1994; 272: 1497–1505
- Tashkin D P, Altose M D, Connett J E, Kanner R E, Lee W W, Wise R A. Methacholine reactivity predicts changes in lung function over time in smokers with early chronic obstructive pulmonary disease. The lung health study research group. Amer J Respir Crit Care Med 1996; 153: 1802–1811
- Anthonisen N R. Lessons from the lung health study. Proc Amer Thor Soc 2004; 1: 143–145
- Kanner R E, Anthonisen N R, Connett J E. Lower respiratory illnesses promote fev(1) decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: Results from the lung health study. Amer J Respir Critical Care Med 2001; 164: 358–364
- Fletcher C, Peto R, Tinkeer C, Speizer F E. The natural history of chornic bronchitis and emphsema. 1976
- Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with fev1 decline and chronic obstructive pulmonary disease morbidity. Copenhagen City Heart Study Group. Amer J Respir Crit Care Med 1996; 153: 1530–1535
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. New Engl J Med 1998; 339: 1194–1200
- Almind M, Viskum K, Evald T, Dirksen A, Kok-Jensen A. A seven-year follow-up study of 343 adults with bronchial asthma. Dan Med Bull 1992; 39: 561–565
- Sherrill D, Guerra S, Bobadilla A, Barbee R. The role of concomitant respiratory diseases on the rate of decline in FEV1 among adult asthmatics. Eur Respir J 2003; 21: 95–100
- Sandford A J, Chagani T, Weir T D, Connett J E, Anthonisen N R, Pare P D. Susceptibility genes for rapid decline of lung function in the lung health study. Amer J Respir Crit Care Med 2001; 163: 469–473
- Joos L, Weir T D, Connett J E, Anthonisen N R, Woods R, Pare P D, Sandford A J. Polymorphisms in the beta2 adrenergic receptor and bronchodilator response, bronchial hyperresponsiveness, and rate of decline in lung function in smokers. Thorax 2003; 58: 703–707
- Ogawa E, Ruan J, Connett J E, Anthonisen N R, Pare P D, Sandford A J. Transforming growth factor-beta1 polymorphisms, airway responsiveness and lung function decline in smokers. Respir Med 2006
- Boyd G, Morice A H, Pounsford J C, Siebert M, Peslis N, Crawford C. An evaluation of salmeterol in the treatment of chronic obstructive pulmonary disease (copd). Eur Respir J 1997; 10: 815–821
- Oberbeck R, Dahlweid M, Koch R, van Griensven M, Emmendorfer A, Tscherne H, Pape H C. Dehydroepiandrosterone decreases mortality rate and improves cellular immune function during polymicrobial sepsis. Critical Care Med 2001; 29: 380–384
- Wadbo M, Lofdahl C G, Larsson K, Skoogh B E, Tornling G, Arwestrom E, Bengtsson T, Strom K. Effects of formoterol and ipratropium bromide in copd: A 3-month placebo-controlled study. Eur Respir J 2002; 20: 1138–1146
- Dahl R, Greefhorst L A, Nowak D, Nonikov V, Byrne A M, Thomson M H, Till D, Della Cioppa G. Inhaled formoterol dry powder versus ipratropium bromide in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2001; 164: 778–784
- Jones P W, Bosh T K. Quality of life changes in copd patients treated with salmeterol. Amer J Respir Crit Care Med 1997; 155: 1283–1289
- Calverley P M, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J 2003; 22: 912–919
- Liesker J J, Van De Velde V, Meysman M, Vincken W, Wollmer P, Hansson L, Kerstjens H A, Qvint U, Pauwels R A. Effects of formoterol (oxis turbuhaler) and ipratropium on exercise capacity in patients with COPD. Respir Med 2002; 96: 559–566
- Soriano J B, Vestbo J, Pride N B, Kiri V, Maden C, Maier W C. Survival in copd patients after regular use of fluticasone propionate and salmeterol in general practice. Eur Respir J 2002; 20: 819–825
- van Schayck C P, Dompeling E, van Herwaarden C L, Folgering H, Akkermans R P, van den Broek P J, van Weel C. Continuous and on demand use of bronchodilators in patients with non-steroid dependent asthma and chronic bronchitis: Four-year follow-up randomized controlled study. Br J Gen Pract 1995; 45: 239–244
- Anthonisen N R, Lindgren P G, Tashkin D P, Kanner R E, Scanlon P D, Connett J E. Bronchodilator response in the lung health study over 11 yrs. Eur Respir J 2005; 26: 45–51
- Calverley P M, Burge P S, Spencer S, Anderson J A, Jones P W. Bronchodilator reversibility testing in chronic obstructive pulmonary disease. Thorax 2003; 58: 659–664
- Celedon J C, Speizer F E, Drazen J M, Weiss S T, Campbell E J, Carey V J, Reilly J J, Ginns L, Silverman E K. Bronchodilator responsiveness and serum total ige levels in families of probands with severe early-onset COPD. Eur Respir J 1999; 14: 1009–1014
- Palmer L J, Celedon J C, Chapman H A, Speizer F E, Weiss S T, Silverman E K. Genome-wide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary disease. Human Mol Gen 2003; 12: 1199–1210
- Vincken W, van Noord J A, Greefhorst A P, Bantje T A, Kesten S, Korducki L, Cornelissen P J. Improved health outcomes in patients with COPD during 1 yr's treatment with tiotropium. Eur Respir J 2002; 19: 209–216
- Casaburi R, Mahler D A, Jones P W, Wanner A, San P G, Zu Wallack R L, Menjoge S S, Serby C W, Witek T, Jr. A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J 2002; 19: 217–224
- O'Donnell D E, Fluge T, Gerken F, Hamilton A, Webb K, Aguilaniu B, Make B, Magnussen H. Effects of tiotropium on lung hyperinflation, dyspnoea and exercise tolerance in copd. Eur Respir J 2004; 23: 832–840
- Casaburi R, Kukafka D, Cooper C B, Witek T J. Jr., Kesten S. Improvement in exercise tolerance with the combination of tiotropium and pulmonary rehabilitation in patients with copd. Chest 2005; 127: 809–817
- Tashkin D P, Altose M D, Bleecker E R, Connett J E, Kanner R E, Lee W W, Wise R. The lung health study: Airway responsiveness to inhaled methacholine in smokers with mild to moderate airflow limitation. The lung health study research group. Amer Rev Respir Dis 1992; 145: 301–310
- Wise R A, Kanner R E, Lindgren P, Connett J E, Altose M D, Enright P L, Tashkin D P. The effect of smoking intervention and an inhaled bronchodilator on airways reactivity in COPD: The lung health study. Chest 2003; 124: 449–458
- Brutsche M H, Downs S H, Schindler C, Gerbase M W, Schwartz J, Frey M, Russi E W, Ackermann-Liebrich U, Leuenberger P. Bronchial hyperresponsiveness and the development of asthma and COPD in asymptomatic individuals: Sapaldia cohort study. Thorax 2006; 61: 671–677
- Postma D S, Kerstjens H A. Characteristics of airway hyperresponsiveness in asthma and chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 1998; 158: S187–192
- Meyers D A, Larj M J, Lange L. Genetics of asthma and COPD. Similar results for different phenotypes. Chest 2004; 126: 105S–110S, discussion 159S–161S
- Robinson R W, White D P, Zwillich C W. Relationship of respiratory drives to dyspnea and exercise performance in chronic obstructive pulmonary disease. Amer Rev Respir Dis 1987; 136: 1084–1090
- Cloosterman S G, Hofland I D, van Schayck C P, Folgering H T. Exertional dyspnoea in patients with airway obstruction, with and without CO2 retention. Thorax 1998; 53: 768–774
- Gorini M, Misuri G, Corrado A, Duranti R, Iandelli I, De Paola E, Scano G. Breathing pattern and carbon dioxide retention in severe chronic obstructive pulmonary disease. Thorax 1996; 51: 677–683
- Johnson M A, Woodcock A A, Rehahn M, Geddes D M. Are “Pink puffers” more breathless than “Blue bloaters”?. Br Med J (Clin Res Ed) 1983; 286: 179–182
- Marin J M, Montes de Oca M, Rassulo J, Celli B R. Ventilatory drive at rest and perception of exertional dyspnea in severe copd. Chest 1999; 115: 1293–1300
- Ferrari K, Goti P, Duranti R, Iandelli I, Misuri G, Mancini M, Rosi E, Scano G. Breathlessness and control of breathing in patients with copd Monaldi archives for chest disease = Archivio Monaldi per le malattie del torace / Fondazione clinica del lavoro, IRCCS [and] Istituto di clinica tisiologica e malattie apparato respiratorio,. Universita di Napoli, Secondo ateneo 1997; 52: 18–23
- Nizet T A, van den Elshout F J, Heijdra Y F, van de Ven M J, Mulder P G, Folgering H T. Survival of chronic hypercapnic COPD patients is predicted by smoking habits, comorbidity, and hypoxemia. Chest 2005; 127: 1904–1910
- Jones P W. Health status measurement in chronic obstructive pulmonary disease. Thorax 2001; 56: 880–887
- Celli B R, MacNee W. Standards for the diagnosis and treatment of patients with copd: A summary of the ats/ers position paper. Eur Respir J 2004; 23: 932–946
- Oga T, Nishimura K, Tsukino M, Sato S, Hajiro T, Mishima M. Exercise capacity deterioration in patients with COPD: Longitudinal evaluation over 5 years. Chest 2005; 128: 62–69
- Pinto-Plata V M, Cote C, Cabral H, Taylor J, Celli B R. The 6-min walk distance: Change over time and value as a predictor of survival in severe COPD. Eur Respir J 2004; 23: 28–33
- Oga T, Nishimura K, Tsukino M, Sato S, Hajiro T. Analysis of the factors related to mortality in chronic obstructive pulmonary disease: Role of exercise capacity and health status. Amer J Respir Crit Care Med 2003; 167: 544–549
- Arnardottir R H, Emtner M, Hedenstrom H, Larsson K, Boman G. Peak exercise capacity estimated from incremental shuttle walking test in patients with COPD: A methodological study. Respir Res 2006; 7: 127
- European Respiratory Society. Clinical exercise testing with reference to lung diseases: Indications, standardization and interpretation strategies. Ers task force on standardization of clinical exercise testing. Eur Respir J 1997; 10: 2662–2689
- ATS Statement. Guidelines for the six-minute walk test. Amer J Respir Crit Care Med 2002; 166: 111–117
- McGavin C R, Gupta S P, McHardy G J. Twelve-minute walking test for assessing disability in chronic bronchitis. Br Med J 1976; 1: 822–823
- Fishman A, Martinez F, Naunheim K, Piantadosi S, Wise R, Ries A, Weinmann G, Wood D E. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348: 2059–2073
- Bernstein M L, Despars J A, Singh N P, Avalos K, Stansbury D W, Light R W. Reanalysis of the 12-minute walk in patients with chronic obstructive pulmonary disease. Chest 1994; 105: 163–167
- Hajiro T, Nishimura K, Tsukino M, Ikeda A, Koyama H, Izumi T. Analysis of clinical methods used to evaluate dyspnea in patients with chronic obstructive pulmonary disease. AmerJ Respir Crit Care Med 1998; 158: 1185–1189
- Kessler R, Faller M, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 1999; 159: 158–164
- Singh S J, Morgan M D, Scott S, Walters D, Hardman A E. Development of a shuttle walking test of disability in patients with chronic airways obstruction. Thorax 1992; 1019–1024
- Ferguson G T. Why does the lung hyperinflate?. Proc Amer Thor Soc 2006; 3: 176–179
- Leith D E, Brown R. Human lung volumes and the mechanisms that set them. Eur Respir J 1999; 13: 468–472
- O'Donnell D E, Bertley J C, Chau L K, Webb K A. Qualitative aspects of exertional breathlessness in chronic airflow limitation: Pathophysiologic mechanisms. Amer J Respir Crit Care Med 1997; 155: 109–115
- O'Donnell D E, Revill S M, Webb K A. Dynamic hyperinflation and exercise intolerance in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 2001; 164: 770–777
- O'Donnell D E. Ventilatory limitations in chronic obstructive pulmonary disease. Med Sci Sports Exer 2001; 33: S647–655
- Cooper C B. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Amer J Med 2006; 119: 21–31
- O'Donnell D E, Voduc N, Fitzpatrick M, Webb K A. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24: 86–94
- Cotton D J, Soparkar G R, Grahan B L. Diffusing capacity in the clinical assessment of chronic airflow limitation. Med Clinics North Amer 1996; 80: 549–564
- Makita H, Nasuhara Y, Betsuyaku T, Onodera Y, Hizawa N, Nishimura M. Characterization of phenotypes based on severity of emphysema in chronic obstructive pulmonary disease. Thorax 2007, in press
- Mohsenifar Z, Lee S M, Diaz P, Criner G, Sciurba F, Ginsburg M, Wise R A. Single-breath diffusing capacity of the lung for carbon monoxide: A predictor of pao2, maximum work rate, and walking distance in patients with emphysema. Chest 2003; 123: 1394–1400
- Rubin L J. Diagnosis and management of pulmonary arterial hypertension: Accp evidence-based clinical practice guidelines. Chest 2004; 126: 4S–6S
- Rubin L J, Badesch D B. Evaluation and management of the patient with pulmonary arterial hypertension. Ann Intern Med 2005; 143: 282–292
- Weitzenblum E, Hirth C, Ducolone A, Mirhom R, Rasaholinjanahary J, Ehrhart M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax 1981; 36: 752–758
- Wright J L, Levy R D, Churg A. Pulmonary hypertension in chronic obstructive pulmonary disease: Current theories of pathogenesis and their implications for treatment. Thorax 2005; 60: 605–609
- Cooper R, Ghali J, Simmons B E, Castaner A. Elevated pulmonary artery pressure. An independent predictor of mortality. Chest 1991; 99: 112–120
- Kessler R, Faller M, Weitzenblum E, Chaouat A, Aykut A, Ducolone A, Ehrhart M, Oswald-Mammosser M. “Natural history” Of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Amer J Respir Crit Care Med 2001; 164: 219–224
- Eddahibi S, Chaouat A, Morrell N, Fadel E, Fuhrman C, Bugnet A S, Dartevelle P, Housset B, Hamon M, Weitzenblum E, et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation 2003; 108: 1839–1844
- Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Amer Thorac Soc 2005; 2: 20–22
- Scharf S M, Iqbal M, Keller C, Criner G, Lee S, Fessler H E. Hemodynamic characterization of patients with severe emphysema. Amer J Respir Critical Care Med 2002; 166: 314–322
- Rubin L J. Primary pulmonary hypertension. New Engl J Med 1997; 336: 111–117
- Naeije R, Barbera J A. Pulmonary hypertension associated with COPD. Crit Care (London, England) 2001; 5: 286–289
- Lejeune P, Mols P, Naeije R, Hallemans R, Melot C. Acute hemodynamic effects of controlled oxygen therapy in decompensated chronic obstructive pulmonary disease. Crit Care Med 1984; 12: 1032–1035
- Timms R M, Khaja F U, Williams G W. Hemodynamic response to oxygen therapy in chronic obstructive pulmonary disease. Ann Intern Med 1985; 102: 29–36
- Peinado V I, Barbera J A, Abate P, Ramirez J, Roca J, Santos S, Rodriguez-Roisin R. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 1999; 159: 1605–1611
- Wilkinson M, Langhorne C A, Heath D, Barer G R, Howard P. A pathophysiological study of 10 cases of hypoxic cor pulmonale. Quart J Med 1988; 66: 65–85
- Wright J L, Tai H, Churg A. Cigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteries. Amer J Respir Cell Mol Biol 2004; 31: 501–509
- Nakano Y, Muro S, Sakai H, Hirai T, Chin K, Tsukino M, Nishimura K, Itoh H, Pare P D, Hogg J C, et al. Computed tomographic measurements of airway dimensions and emphysema in smokers. Correlation with lung function. Amer J Respir Crit Care Med 2000; 162: 1102–1108
- Coxson H O, Rogers R M, Whittall K P, D'Yachkova Y, Pare P D, Sciurba F C, Hogg J C. A quantification of the lung surface area in emphysema using computed tomography. Amer J Respir Crit Care Med 1999; 159: 851–856
- Nishimura K, Murata K, Yamagishi M, Itoh H, Ikeda A, Tsukino M, Koyama H, Sakai N, Mishima M, Izumi T. Comparison of different computed tomography scanning methods for quantifying emphysema. J Thorac Imag 1998; 13: 193–198
- Sakai N, Mishima M, Nishimura K, Itoh H, Kuno K. An automated method to assess the distribution of low attenuation areas on chest ct scans in chronic pulmonary emphysema patients. Chest 1994; 106: 1319–1325
- Gevenois P A, de Maertelaer V, De Vuyst P, Zanen J, Yernault J C. Comparison of computed density and macroscopic morphometry in pulmonary emphysema. Amer J Respir Crit Care Med 1995; 152: 653–657
- Gevenois P A, De Vuyst P, de Maertelaer V, Zanen J, Jacobovitz D, Cosio M G, Yernault J C. Comparison of computed density and microscopic morphometry in pulmonary emphysema. Amer J Respir Crit Care Med 1996; 154: 187–192
- Gevenois P A, De Vuyst P, Sy M, Scillia P, Chaminade L, de Maertelaer V, Zanen J, Yernault J C. Pulmonary emphysema: Quantitative CT during expiration. Radiology 1996; 199: 825–829
- Gurney J W, Jones K K, Robbins R A, Gossman G L, Nelson K J, Daughton D, Spurzem J R, Rennard S I. Regional distribution of emphysema: Correlation of high-resolution CT with pulmonary function tests in unselected smokers. Radiology 1992; 183: 457–463
- Kinsella M, Muller N L, Abboud R T, Morrison N J. DyBuncio A. Quantitation of emphysema by computed tomography using a “Density mask” Program and correlation with pulmonary function tests. Chest 1990; 97: 315–321
- Kuwano K, Matsuba K, Ikeda T, Murakami J, Araki A, Nishitani H, Ishida T, Yasumoto K, Shigematsu N. The diagnosis of mild emphysema. Correlation of computed tomography and pathology scores. Amer Rev Respir Dis 1990; 141: 169–178
- Lamers R J, Thelissen G R, Kessels A G, Wouters E F, van Engelshoven J M. Chronic obstructive pulmonary disease: Evaluation with spirometrically controlled CT lung densitometry. Radiology 1994; 193: 109–113
- Mishima M, Hirai T, Itoh H, Nakano Y, Sakai H, Muro S, Nishimura K, Oku Y, Chin K, Ohi M, et al. Complexity of terminal airspace geometry assessed by lung computed tomography in normal subjects and patients with chronic obstructive pulmonary disease. Proc Natl Acad Sci USA 1999; 96: 8829–8834
- Mishima M, Hirai T, Jin Z, Oku Y, Sakai N, Nakano Y, Sakai H, Chin K, Ohi M, Kawakami K, et al. Standardization of low attenuation area versus total lung area in chest X-ray CT as an indicator of chronic pulmonary emphysema. Front Med Biol Eng 1997; 8: 79–86
- Mishima M, Itoh H, Sakai H, Nakano Y, Muro S, Hirai T, Takubo Y, Chin K, Ohi M, Nishimura K, et al. Optimized scanning conditions of high resolution CT in the follow-up of pulmonary emphysema. J Comput Assist Tomogr 1999; 23: 380–384
- Mishima M, Oku Y, Kawakami K, Sakai N, Fukui M, Hirai T, Chin K, Ohi M, Nishimura K, Itoh H, et al. Quantitative assessment of the spatial distribution of low attenuation areas on X-ray CT using texture analysis in patients with chronic pulmonary emphysema. Front Med Biol Eng 1997; 8: 19–34
- Muller N L, Staples C A, Miller R R, Abboud R T. “Density mask”. An objective method to quantitate emphysema using computed tomography. Chest 1988; 94: 782–787
- Nakano Y, Sakai H, Muro S, Hirai T, Oku Y, Nishimura K, Mishima M. Comparison of low attenuation areas on computed tomographic scans between inner and outer segments of the lung in patients with chronic obstructive pulmonary disease: Incidence and contribution to lung function. Thorax 1999; 54: 384–389
- Nakano Y, Wong J C, de Jong P A, Buzatu L, Nagao T, Coxson H O, Elliott W M, Hogg J C, Pare P D. The prediction of small airway dimensions using computed tomography. Amer J Respir Crit Care Med 2005; 171: 142–146
- Vikgren J, Boijsen M, Andelid K, Ekberg-Jansson A, Larsson S, Bake B, Tylen U. High-resolution computed tomography in healthy smokers and never-smokers: A 6-year follow-up study of men born in 1933. Acta Radiol 2004; 45: 44–52
- Hruban R H, Meziane M A, Zerhouni E A, Khouri N F, Fishman E K, Wheeler P S, Dumler J S, Hutchins G M. High resolution computed tomography of inflation-fixed lungs. Pathologic-radiologic correlation of centrilobular emphysema. Amer Rev Respir Dis 1987; 136: 935–940
- Foster W L, Jr, Pratt P C, Roggli V L, Godwin J D, Halvorsen R A, Jr, Putman C E. Centrilobular emphysema: Ct-pathologic correlation. Radiology 1986; 159: 27–32
- Foster W L, Jr, Gimenez E I, Roubidoux M A, Sherrier R H, Shannon R H, Roggli V L, Pratt P C. The emphysemas: Radiologic-pathologic correlations. Radiographics 1993; 13: 311–328
- Copley S J, Wells A U, Muller N L, Rubens M B, Hollings N P, Cleverley J R, Milne D G, Hansell D M. Thin-section CT in obstructive pulmonary disease: Discriminatory value. Radiology 2002; 223: 812–819
- Satoh K, Kobayashi T, Misao T, Hitani Y, Yamamoto Y, Nishiyama Y, Ohkawa M. Ct assessment of subtypes of pulmonary emphysema in smokers. Chest 2001; 120: 725–729
- Stern E J, Webb W R, Weinacker A, Muller N L. Idiopathic giant bullous emphysema (vanishing lung syndrome): Imaging findings in nine patients. AJR Am J Roentgenol 1994; 162: 279–282
- Kim W D, Eidelman D H, Izquierdo J L, Ghezzo H, Saetta M P, Cosio M G. Centrilobular and panlobular emphysema in smokers. Two distinct morphologic and functional entities. Am Rev Respir Dis 1991; 144: 1385–1390
- Saetta M, Kim W D, Izquierdo J L, Ghezzo H, Cosio M G. Extent of centrilobular and panacinar emphysema in smokers' lungs: Pathological and mechanical implications. Eur Respir J 1994; 7: 664–671
- Hogg J C, Senior R M. Chronic obstructive pulmonary disease—part 2: Pathology and biochemistry of emphysema. Thorax 2002; 57: 830–834
- Sugi K, Kaneda Y, Murakami T, Esato K. The outcome of volume reduction surgery according to the underlying type of emphysema. Surg Today 2001; 31: 580–585
- Goddard P R, Nicholson E M, Laszlo G, Watt I. Computed tomography in pulmonary emphysema. Clin Radiol 1982; 33: 379–387
- Bergin C J, Muller N L, Miller R R. Ct in the qualitative assessment of emphysema. J Thorac Imaging 1986; 1: 94–103
- Coddington R, Mera S L, Goddard P R, Bradfield J W. Pathological evaluation of computed tomography images of lungs. J Clin Pathol 1982; 35: 536–540
- Thurlbeck W M, Muller N L. Emphysema: Definition, imaging, and quantification. AJR Am J Roentgenol 1994; 163: 1017–1025
- Klein J S, Gamsu G, Webb W R, Golden J A, Muller N L. High-resolution ct diagnosis of emphysema in symptomatic patients with normal chest radiographs and isolated low diffusing capacity. Radiology 1992; 182: 817–821
- Spaggiari E, Zompatori M, Verduri A, Chetta A, Bna C, Ormitti F, Sverzellati N, Rabaiotti E. Early smoking-induced lung lesions in asymptomatic subjects. Correlations between high resolution dynamic ct and pulmonary function testing. Radiol Med (Torino) 2005; 109: 27–39
- Baldi S, Miniati M, Bellina C R, Battolla L, Catapano G, Begliomini E, Giustini D, Giuntini C. Relationship between extent of pulmonary emphysema by high-resolution computed tomography and lung elastic recoil in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164: 585–589
- Gelb A F, Hogg J C, Muller N L, Schein M J, Kuei J, Tashkin D P, Epstein J D, Kollin J, Green R H, Zamel N, et al. Contribution of emphysema and small airways in COPD. Chest 1996; 109: 353–359
- Sanders C, Nath P H, Bailey W C. Detection of emphysema with computed tomography. Correlation with pulmonary function tests and chest radiography. Invest Radiol 1988; 23: 262–266
- Gelb A F, Zamel N, Hogg J C, Muller N L, Schein M J. Pseudophysiologic emphysema resulting from severe small-airways disease. Am J Respir Crit Care Med 1998; 158: 815–819
- Zeidler M R, Kleerup E C, Goldin J G, Kim H J, Truong D A, Simmons M D, Sayre J W, Liu W, Elashoff R, Tashkin D P. Montelukast improves regional air-trapping due to small airways obstruction in asthma. Eur Respir J 2006; 27: 307–315
- Jain N, Covar R A, Gleason M C, Newell J D, Jr, Gelfand E W, Spahn J D. Quantitative computed tomography detects peripheral airway disease in asthmatic children. Pediatr Pulmonol 2005; 40: 211–218
- Grenier P A, Beigelman-Aubry C, Fetita C, Preteux F, Brauner M W, Lenoir S. New frontiers in ct imaging of airway disease. Eur Radiol 2002; 12: 1022–1044
- Goldin J G. Quantitative ct of the lung. Radiol Clin North Am 2002; 40: 145–162
- Newman K B, Lynch D A, Newman L S, Ellegood D, Newell J D. Jr. Quantitative computed tomography detects air trapping due to asthma. Chest 1994; 106: 105–109
- Remy-Jardin M, Edme J L, Boulenguez C, Remy J, Mastora I, Sobaszek A. Longitudinal follow-up study of smoker's lung with thin-section CT in correlation with pulmonary function tests. Radiology 2002; 222: 261–270
- Matsuoka S, Kurihara Y, Yagihashi K, Nakajima Y. Morphological progression of emphysema on thin-section ct: Analysis of longitudinal change in the number and size of low-attenuation clusters. J Comput Assist Tomogr 2006; 30: 669–674
- Kitaguchi Y, Fujimoto K, Kubo K, Honda T. Characteristics of COPD phenotypes classified according to the findings of hrct. Respir Med 2006; 100: 1742–1752
- Eriksson S. Studies in alpha 1-antitrypsin deficiency. Acta Med Scand Suppl 1965; 432: 1–85
- Lieberman J, Winter B, Sastre A. Alpha 1-antitrypsin pi-types in 965 COPD patients. Chest 1986; 89: 370–373
- Eriksson S. Discovery of alpha 1-antitrypsin deficiency. Lung 1990; 523–529, 168 Suppl
- Sakao S, Tatsumi K, Igari H, Watanabe R, Shino Y, Shirasawa H, Kuriyama T. Association of tumor necrosis factor-alpha gene promoter polymorphism with low attenuation areas on high-resolution CT in patients with COPD. Chest 2002; 122: 416–420
- Ito I, Nagai S, Handa T, Muro S, Hirai T, Tsukino M, Mishima M. Matrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysema. Am J Respir Crit Care Med 2005; 172: 1378–1382
- Minematsu N, Nakamura H, Iwata M, Tateno H, Nakajima T, Takahashi S, Fujishima S, Yamaguchi K. Association of cyp2a6 deletion polymorphism with smoking habit and development of pulmonary emphysema. Thorax 2003; 58: 623–628
- Silverman E K. Progress in chronic obstructive pulmonary disease genetics. Proc Am Thorac Soc 2006; 3: 405–408
- Sampsonas F, Karkoulias K, Kaparianos A, Spiropoulos K. Genetics of chronic obstructive pulmonary disease, beyond a1-antitrypsin deficiency. Curr Med Chem 2006; 13: 2857–2873
- Wood A M, Stockley R A. The genetics of chronic obstructive pulmonary disease. Respir Res 2006; 7: 130
- Cookson W O. State of the art. Genetics and genomics of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006; 3: 473–475
- Joos L. Copd and genetics–what's new?. Swiss Med Wkly 2004; 134: 437–439
- Molfino N A. Genetics of COPD. Chest 2004; 125: 1929–1940
- Hoidal J R. Genetics of COPD: Present and future. Eur Respir J 2001; 18: 741–743
- Hogg J C, Chu F, Utokaparch S, Woods R, Elliott W M, Buzatu L, Cherniack R M, Rogers R M, Sciurba F C, Coxson H O, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645–2653
- Pedersen S K, Sloane A J, Prasad S S, Sebastian L T, Lindner R A, Hsu M, Robinson M, Bye P T, Weinberger R P, Harry J L. An immunoproteomic approach for identification of clinical biomarkers for monitoring disease: Application to cystic fibrosis. Mol Cell Proteomics 2005; 4: 1052–1060
- Orlandi I, Moroni C, Camiciottoli G, Bartolucci M, Pistolesi M, Villari N, Mascalchi M. Chronic obstructive pulmonary disease: Thin-section ct measurement of airway wall thickness and lung attenuation. Radiology 2005; 234: 604–610
- Aziz Z A, Wells A U, Desai S R, Ellis S M, Walker A E, MacDonald S, Hansell D M. Functional impairment in emphysema: Contribution of airway abnormalities and distribution of parenchymal disease. AJR Am J Roentgenol 2005; 185: 1509–1515
- Hasegawa M, Nasuhara Y, Onodera Y, Makita H, Nagai K, Fuke S, Ito Y, Betsuyaku T, Nishimura M. Airflow limitation and airway dimensions in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 173: 1309–1315
- Tschirren J, Hoffman E A. McLennan G Sonka M. Intrathoracic airway trees: Segmentation and airway morphology analysis from low-dose ct scans. IEEE Trans Med Imaging 2005; 24: 1529–1539
- Heyneman L E, Ward S, Lynch D A, Remy-Jardin M, Johkoh T, Muller N L. Respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease, and desquamative interstitial pneumonia: Different entities or part of the spectrum of the same disease process?. AJR Am J Roentgenol 1999; 173: 1617–1622
- Moon J, du Bois R M, Colby T V, Hansell D M, Nicholson A G. Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax 1999; 54: 1009–1014
- Park J S, Brown K K, Tuder R M, Hale V A, King T E, Jr, Lynch D A. Respiratory bronchiolitis-associated interstitial lung disease: Radiologic features with clinical and pathologic correlation. J Comput Assist Tomogr 2002; 26: 13–20
- Afessa B, Morales I J, Scanlon P D, Peters S G. Prognostic factors, clinical course, and hospital outcome of patients with chronic obstructive pulmonary disease admitted to an intensive care unit for acute respiratory failure. Crit Care Med 2002; 30: 1610–1615
- Okada Y, Nagase H, Harris E D, Jr. Matrix metalloproteinases 1, 2, and 3 from rheumatoid synovial cells are sufficient to destroy joints. J Rheumatol 1987; 41–42, 14 Spec No
- Joos L, He J Q, Shepherdson M B, Connett J E, Anthonisen N R, Pare P D, Sandford A J. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Human Mol Genet 2002; 11: 569–576
- Maines M D. The heme oxygenase system: A regulator of second messenger gases. Ann Rev Pharmacol Toxicol 1997; 37: 517–554
- He J Q, Ruan J, Connett J E, Anthonisen N R, Pare P D, Sandford A J. Antioxidant gene polymorphisms and susceptibility to a rapid decline in lung function in smokers. Amer J Respir Crit Care Med 2002; 166: 323–328
- Horne S L, Cockcroft D W, Dosman J A. Possible protective effect against chronic obstructive airways disease by the gc2 allele. Human Hered 1990; 40: 173–176
- Kew R R, Fisher J A, Webster R O. Co-chemotactic effect of gc-globulin (vitamin d binding protein) for c5a. Transient conversion into an active co-chemotaxin by neutrophils. J Immunol 1995; 155: 5369–5374
- Yamamoto N, Homma S. Vitamin d3 binding protein (group-specific component) is a precursor for the macrophage-activating signal factor from lysophosphatidylcholine-treated lymphocytes. Proc Natl Acad Sci USA 1991; 88: 8539–8543
- Bouma G, Crusius J B, Oudkerk Pool M, Kolkman J J, von Blomberg B M, Kostense P J, Giphart M J, Schreuder G M, Meuwissen S G, Pena A S. Secretion of tumour necrosis factor alpha and lymphotoxin alpha in relation to polymorphisms in the tnf genes and hla-dr alleles. Relevance for inflammatory bowel disease. Scand J Immunol 1996; 43: 456–463
- Turki J, Pak J, Green S A, Martin R J, Liggett S B. Genetic polymorphisms of the beta 2-adrenergic receptor in nocturnal and nonnocturnal asthma. Evidence that gly16 correlates with the nocturnal phenotype. J Clin Invest 1995; 95: 1635–1641
- Weir T D, Mallek N, Sandford A J, Bai T R, Awadh N, Fitzgerald J M, Cockcroft D, James A, Liggett S B, Pare P D. β2-adrenergic receptor haplotypes in mild, moderate and fatal/ near fatal asthma. Amer J Respir Crit Care Med 1998; 158: 787–791
- Hall I P, Wheatley A, Wilding P, Liggett S B. Association of glu 27 beta 2-adrenoceptor polymorphism with lower airway reactivity in asthmatic subjects. Lancet 1995; 345: 1213–1214
- Ramsay C E, Hayden C M, Tiller K J, Burton P R, Goldblatt J, Lesouef P N. Polymorphisms in the beta2-adrenoreceptor gene are associated with decreased airway responsiveness. Clin Exp Allergy 1999; 29: 1195–1203
- D'Amato M, Vitiani L R, Petrelli G, Ferrigno L, di Pietro A, Trezza R, Matricardi P M. Association of persistent bronchial hyperresponsiveness with beta2-adrenoceptor (adrb2) haplotypes. A population study. Amer J Respir Crit Care Med 1998; 158: 1968–1973
- Lima J J, Thomason D B, Mohamed M H, Eberle L V, Self T H, Johnson J A. Impact of genetic polymorphisms of the beta2-adrenergic receptor on albuterol bronchodilator pharmacodynamics. Clin Pharmacol Therap 1999; 65: 519–525
- Martinez F D, Graves P E, Baldini M, Solomon S, Erickson R. Association between genetic polymorphisms of the beta2-adrenoceptor and response to albuterol in children with and without a history of wheezing. J Clin Invest 1997; 100: 3184–3188
- Ulbrecht M, Hergeth M T, Wjst M, Heinrich J, Bickeboller H, Wichmann H E, Weiss E H. Association of beta(2)-adrenoreceptor variants with bronchial hyperresponsiveness. Amer J Respir Crit Care Med 2000; 161: 469–474
- Summerhill E, Leavitt S A, Gidley H, Parry R, Solway J, Ober C. β(2)-adrenergic receptor arg16/arg16 genotype is associated with reduced lung function, but not with asthma, in the hutterites. Amer J Respir Crit Care Med 2000; 162: 599–602
- Mannervik B. The isoenzymes of glutathione transferase. Adv Enzymol Related Areas Mol Biol 1985; 57: 357–417
- Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Nakahara K, Umeda A. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Amer J Respir Crit Care Med 2001; 163: 1476–1483
- de Boer W I, van Schadewijk A, Sont J K, Sharma H S, Stolk J, Hiemstra P S, van Krieken J H. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med 1998; 158: 1951–1957
- Chung K F. Cytokines in chronic obstructive pulmonary disease. Eur Respir J 2001; 34: 50s–59s