Abstract
Chronic obstructive pulmonary disease (COPD) is conventionally thought of as a disease of adult smokers, related to airway inflammation and structural airway changes (remodeling). However, there is important epidemiological evidence, from a series of studies with overlapping age groups from birth to late middle age that early life events, including antenatal influences on lung growth, program the child to be at increased risk for future COPD. This paper reviews the evidence for potential gene: environment interactions in this process, in particular with respect to the maternal genotype of the COPD patient. It explores the hypothesis that genes important in early lung development are also important in determining adult risk for COPD. Although the major preventable factor adversely impacting on child lung health is maternal smoking, the effects of viral infection, nutrition, and indoor and outdoor pollution are reviewed. The survivors of preterm birth are another important cohort who may develop premature COPD in adult life. Early life events provide the substrate for COPD, with later cigarette smoking, and occasionally other exposures, pulling the trigger to produce COPD. Although a rigorous anti-smoking program is necessary to halt this spiral of lung destruction leading to COPD, a focus on early (including prenatal) lung health is also important. Any model of COPD which does not take into account early life influences is likely to be fatally flawed.
INTRODUCTION
In the adult literature, there is a huge focus on chronic obstructive pulmonary disease (COPD) as an inflammatory process, related to active cigarette smoking in the main. There are a small number of COPD patients with a minimal or no smoking history, in whom it is thought that environmental pollution, perhaps including passive smoking, may be responsible. Furthermore, not all smokers develop COPD (Citation[1]). There are clearly reasons for the discrepancies between smoking history and COPD outcome, and this article will argue that many can be found very early in life. Lip service may be paid to “childhood infections” (whatever that means; has anyone ever seen a child who did not have recurrent respiratory infections?), but otherwise there is often little concept that childhood factors may be important. The purpose of this article is to demonstrate that, without an understanding of normal and abnormal antenatal and postnatal lung development, there is exactly no chance of fully understanding adult COPD. This is important, not least because despite a huge amount of research, in reality the only really effective treatment offered to patients with COPD is to ‘quit smoking’, something they have usually tried and failed to do many times. Furthermore, the sources of genetic susceptibility for COPD may be in maternal DNA, and genes relating to lung growth and development, and not merely in polymorphisms relating to susceptibility to oxidant stress and the inflammatory response.
Asthma and COPD: It is not the purpose of this review to discuss the relationship in later life between chronic asthma with persistent airflow limitation, and COPD. The cohort studies described below are relevant to transient wheezers as well as those who go on to develop persistent asthma, and it is artificial to ignore the results in asthmatics in this context. As will be seen, both phenotypes have reduced lung function before age 6 years, which then tracks into middle age. Both have accelerated deterioration of lung function, putting them at risk for COPD. Whatever is thought to be the relationship between chronic asthma and COPD in mid- to late-adult life, it is antenatally and in early life that the roots of both problems are to be found.
A pivotal epidemiological observation
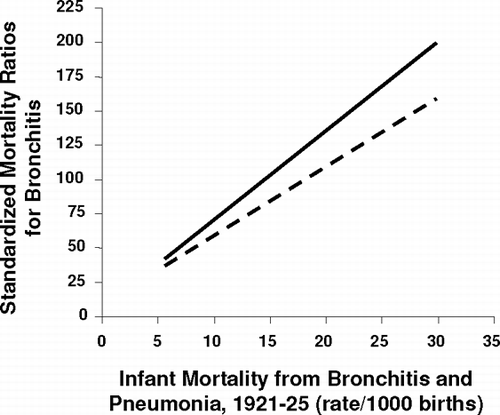
Barker's group (Citation[2]), in a series of seminal observations, analyzed death certificate data in a large number of individuals in widely differing communities, which were classified as county or London boroughs, or urban and rural areas. We know that death certificate data are often unreliable, but if a parameter of poor individual accuracy is measured thousands of times, inaccuracies will cancel out and important correlations which are robust, may emerge. Barker's group found that, across all these communities, there was an extraordinarily close correlation between infant mortality from pneumonia and other respiratory disease, and standardized mortality rate (SMR) for COPD 50 years later (). An area with high infant mortality rate would also have a high SMR for COPD 50 years later. Thus any hypothesis about COPD has to take in the importance of early life events if it is to hold up. This observation has been taken forward in a series of prospectively recruited cohort studies.
Figure 1 Relationship between early death from infant respiratory disease and subsequent standardized mortality rate for chronic bronchitis and emphysema. Solid line gives data for men. Dashed line gives data for women. A high infant mortality rate over a range of communities is associated with a high subsequent mortality in the same area from chronic obstructive airway disease. Adapted from Barker DP. Fetal and infant origins of adult disease. BMJ Books 1992; 343:52–53.
Recent epidemiological work
There have been a number of cohort studies, none obviously having run long enough to track the same patients through from prenatally to death in old age. Therefore, a composite picture must be constructed from overlapping cohort studies. Unsurprisingly, there are some differences between the results in different studies, in part related to the different populations studied (e.g., recruitment of all-comers versus selected, at risk populations). Nonetheless, it is possible to draw broad general conclusions, not the least of which is the prolonged influence of early changes in lung function. In this section, aspects of these studies which are particularly relevant to adult lung disease are highlighted, and I have not discussed the important details of their relevance to the understanding and treatment of childhood wheezing phenotypes.
As a general point, it is important to have some awareness of the difficulties and limitations of techniques used in the study of infant and pre-school lung function. For those not familiar with them, a brief summary is provided:
Rapid thoraco-abdominal compression (RTC) technique: the sleeping or sedated baby is placed in an inflatable jacket, and breathes through a close fitting face mask, sealed to the skin with therapeutic putty. Rapid inflation is performed at the end of a tidal inspiration. Expiratory flow rate is measured using a pneumotachygraph at functional residual capacity (VmaxFRC).
The raised volume RTC (RVRTC): the apparatus is the same, but the baby's respiration is taken over by bag and mask ventilation; the baby is passively inflated to total lung capacity, and the squeeze is then applied. This generates a full expiratory flow volume curve. Typically the forced expired volume (FEV) measured is at 0.4 or 0.5 not one second, and expressed as a ratio of forced vital capacity (FVC), because lung emptying is much more efficient in young children, and is often complete before one second. Note that the corollary of this is that FEVt does not measure flow in the same generation of airways across the entire age range. FEV1 is typically 80% of FVC in mid-childhood, but often 100% FVC in pre-school children.
Flow limited tidal breathing occurs when expiratory flow in tidal breathing cannot be increased by increasing expiratory effort, or increasing the squeeze pressure, and is associated with severe lung function impairment.
Lung clearance index (LCI) is a measure of gas mixing efficiency, currently most commonly measured after an initial wash-in of an inert gas such as sulphur hexafluoride, during the subsequent wash out phase (Citation[3], Citation[4]). It has the advantage of having a normal range that is the same from the first year of life to adulthood, independent of size. It is probably the most sensitive test of distal airway disease, except possibly in the first few months of life (Citation[5]), but has been little studied in birth cohorts.
Airway resistance (sRAW) is measured in an appropriate sized body plethysmograph, after sedation or natural sleep in a baby, or in toddlers, sitting on the mother's lap. It is less sensitive to disease, and in particular distal airway abnormalities, than LCI.
Z-scores (standard deviation (SD) scores) allow comparison of different measurements across ages. A normal Z score is therefore zero at all ages, provided the SD has been calculated on an appropriate age, sex and gender matched population. Z score is calculated as (measured value – mean value)/Population SD. It is a better way of expressing measurements than percent predicted, which now should be obsolete. For example, depending on the variance in the population, 80% predicted may be normal or abnormal, but a Z-score of −2.5 can be seen at once always to be abnormal.
In this article, ‘tracking’ of lung function over a time period is taken to mean that lung function over the period of study is very substantially determined by lung function at the beginning of the study; for example, poor lung function at the start of the study is strongly predictive of poor lung function at the end.
Spirometry and related lung function tests in early childhood: One of the most important series of papers has come from Tucson (Citation[6], Citation[7]). They recruited over 1200 babies, and were able to divide wheezing phenotypes in the first 6 years of life into 3 categories (). They found that transient wheezers had reduced lung function at birth, and although there was catch-up, they still had evidence of airflow obstruction age 6 years. The persistent wheezers had normal lung function at birth, and by age 6 had evidence of airflow obstruction. The late onset wheeze group had no impairment of lung function. This group has recently published data which shows that lung function patterns established at age 6 track to age 16 at least (Citation[7]). Thus the first 6 years are a pivotal time for the prevention of changes in lung function (). It has to be said that their physiological findings in the transient wheezers have not always been confirmed by other groups (later).
Figure 2 Tracking of lung function; data from the Tucson cohort. The transient early wheezers are born with low lung function, which has improved, albeit not to normal, by age 6; thereafter, there is no catch-up growth. The persistent wheezers are normal at birth, have airflow obstruction by age 6, and thereafter lung function tracks. The late onset wheezers do not show any decrement of lung function. Reproduced with permission from Am J Respir Crit Care Med. 2005; 172: 1253–8
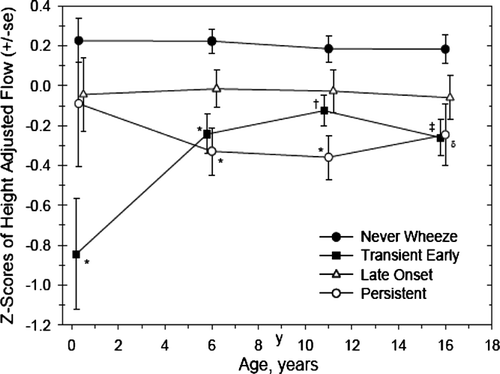
Table 1 Wheezing phenotypes in the Tucson study
The Perth birth cohort was smaller than Tucson (n = 243 vs. 1246), but more detailed physiological measurements were made, including bronchial hyper-reactivity in the newborn period (below) (Citation[8]). Their detailed conclusions, based on smaller numbers, were different, but the same broad theme of the importance of early life events emerged. The discrepancies between the two cohorts have yet to be resolved. The Perth group found that the group wheezing between one and three years of life (n = 17, transient wheeze, in the Tucson nomenclature; and note that Tucson followed up more than 100 transient wheezers into mid-childhood) had normal lung function at birth (using a tidal breathing, indirect measure of airway obstruction, or, in a subgroup, VmaxFRC), but forced expiratory flow (FEF)25 − 75 (which is probably the nearest equivalent in spirometry to VmaxFRC) was reduced at age 11.
The persistent wheezers (n = 12) had abnormal lung function shortly after birth, and it remained persistently lower than the never wheeze group. By contrast, and in this case similar to the Tucson findings, the group that only wheezed after age three had no impairment of lung function. In a study of more than 11,000 Sydney schoolchildren, those with a history of wheeze before two years of age had small decrements in lung function, supportive of the Tucson findings (Citation[9]). The Perth cohort also established that, in children admitted to hospital with acute bronchiolitis, premorbid VmaxFRC was reduced compared to a non-bronchiolitis control group, and that age eleven years, FEF25 − 75 was persistently lower (Citation[10]). They also showed that a group who were flow limited in the neonatal period during tidal breathing had a trend towards a reduced FEF25 − 75 at age 11 years, again supporting the concept of the tracking of lung function (Citation[11], Citation[12]).
The Manchester Birth Cohort (Citation[13]) used the same subdivisions of wheezing phenotypes as Tucson. They measured sRAW at age 3 and 5 years. By age three years, persistent wheezers had reduced lung function, and this tracked to age 5 years. Transient wheezers had normal lung function. sRAW at age three years could not predict which infants would develop new wheeze in the next 2 years of life. The conclusions were the same irrespective of whether pre- or post-bronchodilator sRAW was studied. It should be noted that sRAW may not be the most discriminatory test available (below). These findings have been confirmed in a Dutch cohort, where airway resistance measured by the interrupter technique was higher in persistent wheezers than in normals (Citation[14]). Interestingly, this group also could find no effect of transient wheeze on lung function, similar to Perth, but contrary to the Tucson and Sydney findings; this may reflect the relative insensitivity of the interrupter technique.
Most studies have focused on airway obstruction, with the implication that this is due to airway maldevelopment. However, alternative mechanisms may also be relevant. It is well known that there is interdependence between the airways and the lung parenchyma, with alveolar tethering to the airways being responsible in part for maintaining airway caliber. Alveolar attachment points have been shown to be reduced in number by maternal smoking (Citation[15]). This could lead to airflow obstruction without there being a direct effect on the airway itself. Another group, using measurements of input impedance, have suggested that airway wall compliance may be abnormal in wheezing infants. They suggested that compliance, rather than actual caliber, may be the reason for the described lung function abnormalities (Citation[16]).
Recently, an extended follow-up of a Norwegian birth cohort has been published (Citation[17]). Lung function was measured soon after birth by analysis of tidal flow in 664 infants, and 616 were restudied at the age of 10 years. Those with early tidal breathing lung function below the median were more likely to have a history of asthma, and severe airway hyper-responsiveness (AHR) at age 10. There was some evidence of tracking, with early tidal volume indices weakly predicting mid-expiratory flows at age 10. Studies in adults have extended these observations, also showing a relationship between lung function age 45–50 years and birth weight (Citation[18]).
Methodological issues in early childhood: Some of the differences between the cohort studies could be as a result of methodological issues. The use of Z scores to compare different techniques such as RTC and later spirometry may be helpful, but it is important to realize that the two techniques may be measuring subtly different things. It has long been known that spirometry is relatively insensitive, in particular to distal airway disease. RTC, used in the initial cohort studies, is less sensitive at discriminating cystic fibrosis (CF) from normals than RVRTC (Citation[19]). In the context of CF, LCI has been shown to be more sensitive to disease than either spirometry or sRAW (Citation[20], Citation[21], Citation[22]), but there is no long-term prospective study in infant wheeze using this technique. So it must be concluded that we may have introduced noise into the signal because of the imperfections of the instruments used. Nonetheless, the large numbers studied in different cohorts allow us to conclude that lung function does track from early life into mid-childhood.
Pathological counterparts of physiological studies: Ethical and practical constraints have limited the number of airway biopsy studies which can be performed in young children. However, recent pathological data have confirmed the importance of the first 3 years of life. We and others have confirmed the safety of endobronchial biopsy in infants and children (Citation[23], Citation[24], Citation[25], Citation[26]), so if a clinically indicated bronchoscopy is being formed, an airway biopsy for research is legitimate, subject to the approval of the institutional review board, and the agreement of the family (Citation[27]). What is not legitimate is the performance of a bronchoscopy just for research purposes (Citation[28]). Reticular basement membrane (rbm) thickening is one of the pathological hallmarks of asthma. We have shown that in children with severe asthma, rbm thickening is greater than adult and child controls, and equal to that seen in adults with both mild and severe asthma (Citation[29]).
Until recently, it was not known at what age the changes developed. We have recently shown in a cross-sectional study that even infants (median age one year) who are atopic and have bronchodilator responsiveness have no evidence of eosinophilic inflammation and rbm thickening (Citation[30]). However, in a further cross-sectional study in a different group of infants, median age 3 years, we have shown that the presence of wheeze confirmed on a videotape is associated with both eosinophilic inflammation and rbm thickening (Citation[31]). This pathological data confirms epidemiology in showing the likely long term importance of events in the first three years of life.
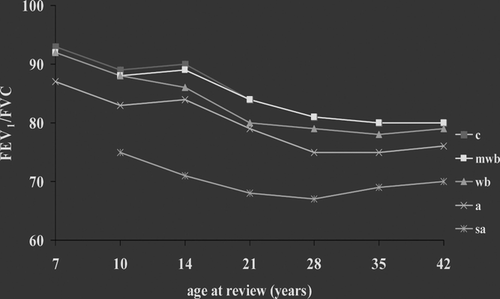
Lung function studies in later childhood and adult life: The Tucson and Perth data (Citation[6], Citation[7], Citation[8]) overlap a number of cohorts spanning childhood and early adult life. The Dunedin cohort (Citation[32]) followed over 600 people between the ages of 9 and 26. Their data clearly showed tracking of lung function over this period, perhaps more in males than females (). Pulmonary function was consistently lower in those who wheezed compared with those who did not, and the level of function was clearly set by events prior to recruitment, as the authors themselves recognized. The primary purpose of the CAMP study was the treatment of childhood asthma, not lung function tracking, but nonetheless, their data is supportive of this concept (Citation[33]). The Melbourne group recruited children age 7 who had either no wheeze, “wheezy bronchitis” (WB, what we would now call virus induced wheeze (VIW)), or persistent asthma. At age 10, the cohort was enriched by a group of really severe asthmatics. They showed that from recruitment until just below 50 years of age, lung function tracked in all groups, and that the level of lung function was set by early life events ().
Figure 3 Tracking of lung function, independent of wheeze phenotype, between age 9 and 26 years, in males and females. Reproduced with permission from N Engl J Med. 2003; 349: 1414–22
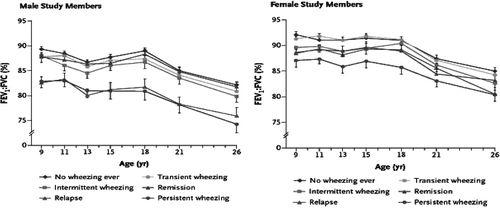
Figure 4 Data from the Melbourne cohort. Tracking of lung function from childhood into middle-age, irrespective of wheeze phenotype. C, control; mwb, mild wheezy bronchitis (wheezy bronchitis would today probably be called virus associated wheeze); wb, wheezy bronchitis; a, asthma; sa, severe asthma. Adapted from Pediatr Pulmonol 1997; 23; 14–20
Events during lung aging: The next consideration is the rate of lung aging. It has long been known that in the mid-40s, lung function starts to decline from a plateau. The plateau (highest lung function ever) is lower in asthmatics, and the slope of rate of decline is greater in smokers (Citation[35]). The rate of decline in lung function can be slowed by quitting smoking. Recently, patients from the Aberdeen cohort, recruited in 1964 were restudied (Citation[36]). They were divided into normals, asthmatics and “wheezy bronchitics,” who would now more likely be described as virus associated wheezers. In middle age, the plateau FEV1 is the same in normals and WB, and lower in asthmatics, as expected. However, when rate of decline of FEV1 was studied, both the asthmatics and WB groups were declining at the same accelerated rate. This finding persisted after controlling for smoking habit, and socioeconomic class. Further strong support for this hypothesis comes from a study which shows an increased risk of COPD in people with an accelerated decline in lung function (Citation[37]).
Lung function testing across the years – conclusions: Putting these data together, I suggest the following model. The future COPD patients are born with airflow obstruction, as a result of gene: environment interactions before birth (see later). They develop VIW and wheeze transiently in childhood. Very subtle evidence of airflow obstruction persists, too subtle to be detected by spirometry in early adult life, and tracks into middle age. As the patient grows, the effects of minor degrees of obstruction become less important in causing symptoms (a degree of airway narrowing in a tiny airway causes much greater proportional reduction in flow than the same absolute degree of narrowing in a large airway), and also, much harder to detect with crude tools like FEV1. However, they show accelerated lung aging, particularly if they smoke, and thus develop premature airflow obstruction and COPD, in particular if exposed to other risk factors.
The overwhelming conclusion must be that lifetime lung function is determined by the end of the preschool years. Although the precise details are still not clear, by the time the child goes to school, lung function is running on predetermined tracks which extend until death. There is scope for deterioration due to adverse events in childhood (below), but likely not for improvement. Any intervention must therefore be early on in life. If as seems likely, a key factor in COPD is level of lung function, an understanding of risk factors, such as gene polymorphisms, must be sought in early life, and preventive strategies must be focused early on also. To summarize, a series of over-lapping, cohort studies have provided a complete series of links between the seminal observations by Barker's group (Citation[2]), and the pre-natal events which account for that observation.
What have measurements of airway reactivity taught us?
Airway reactivity (AHR) can be measured in small infants, but this is technically very difficult, and only two cohorts have attempted this (Citation[38], Citation[39]), thus data are far scantier than for conventional lung function. Furthermore, just as FEV1 may mean different things at different ages, so does AHR. In older children and adults, the concept of ‘direct’ and ‘indirect’ challenges, which measure different things, is well established (Citation[40]). It is likely but unproven that AHR in babies may predominantly be determined by distal airway caliber rather than inflammation, in other words, ‘reactive’ airways are in fact smaller ones, the same degree of constriction causing a bigger signal in terms of airflow obstruction in an airway that started smaller than one of normal starting caliber. Unlike in asthmatic adults, AHR is not necessarily a feature of wheezy infants (Citation[41]), underscoring that AHR has different meanings at different ages.
There have been two major studies in which AHR was measured in the neonatal period. The Perth cohort of babies had AHR to histamine measured shortly after birth, and increased responsiveness was associated with reduced spirometry, a physician diagnosis of asthma, and increased respiratory symptoms at age 6 (Citation[38]). In the second (UK) study (Citation[39]), wheezing was categorized as occurring (1) only before the 4th birthday, (2) after the 4th birthday, or (3) never. No relation was seen between neonatal and later lung function. However, neonatal bronchial responsiveness predicted subsequent FEV1 (p = 0.03). Increased neonatal AHR was associated with transient wheeze < 4 years but not with later wheeze. Wheeze after 4 years of age was typical of classical asthma, as it was strongly related to atopy and AHR at age 10. Neonatal AHR was associated with both early wheezing and later lung function. The group also showed an influence of beta-receptor polymorphisms at both aa16 and aa27 on neonatal lung function. Wheezing beyond 4 years, typical of classical asthma, was unrelated to early measurements of lung function or bronchial responsiveness
The Groningen study has also provided many useful insights into the evolution of lung function (Citation[42], Citation[43]). They studied 119 allergic asthmatics for the first time between ages 5 and 19 years, and followed them up into their 40s. They showed that, at the first visit, low FEV1 and bronchial responsiveness were independent risk factors for low lung function in early adult life. Lower lung function in childhood, and less good evolution of lung function over time, was associated with worse AHR in adult life. Unsurprisingly, young adult asthmatics who smoked had a higher rate of decline in lung function. They too stressed that early interventions, in childhood, would be necessary to improve adult lung function.
In summary, early AHR appears to be an independent predictor of later lung function. It would be important to know whether AHR is merely a surrogate for distal airway function, and the association would disappear if LCI was measured also, or whether there are other factors which account for this association. Nonetheless, it is clear that AHR is another early marker for later impaired lung health and function.
CONCLUSIONS: WHAT ARE THE IMPLICATIONS OF THESE EPIDEMIOLOGICAL AND PATHOLOGICAL DATA?
Do we then require the setting aside of the COPD inflammation and thus consider remodeling studies as irrelevant? I think not; antenatal development and early environment creates the substrate for COPD, and in adult life other factors, including smoking and airway inflammation, are required to “pull the trigger” and cause the disease. The experience with respiratory syncytial virus (RSV) bronchiolitis demonstrates how airflow obstruction may interact with inflammation. Blind bronchoalveolar lavage was performed in three groups of patients, term babies ventilated with RSV bronchiolitis, bronchopulmonary dysplasia (BPD, chronic lung disease of prematurity) babies with RSV, and a group of controls, term babies who had undergone surgery (Citation[44], Citation[45]). The term babies with RSV had a higher neutrophil count and Interleukin (IL)-9 levels than the BPD RSV babies, and both were higher than non-infected controls.
The group had no lung function data on any patient, but it is safe to assume from other studies (Citation[46], Citation[47]) that the BPD babies would have airflow obstruction as a result of their prematurity and its treatment. The conclusion (which the authors failed to draw) is that if there is pre-existing airflow obstruction, lesser degrees of inflammation are needed to precipitate decompensation, and, in the case of RSV bronchiolitis, the need for mechanical ventilation. This is intuitively obvious, and could also account for the interaction of airflow obstruction and inflammation in the production of COPD in adulthood. Obviously, targeting the cause of inflammation (smoking, indoor pollution) is worthy, although this strategy has not met with uniform success. However, there is surely scope to re-focus preventive efforts by trying to ensure that people enter adult life with normal airway function.
What follows from the argument that the foundations for later COPD are laid antenatally and in early life? I suggest the following are important, and each will be discussed in turn:
A good understanding of normal lung development, in particularly antenatally, is essential;
Adverse influences on lung development antenatally and in the early postnatal years are likely to predict later COPD, and need to be studied, not just for mechanistic interest, but also as an opportunity for prevention;
The maternal genotype may influence fetal exposure to adverse environmental influences, and may therefore be important in adult life COPD risk;
Polymorphisms in genes important in fetal lung development may constitute risk factors for adult COPD.
Normal lung development
If, as argued here, prenatal events may be important, then knowledge of lung development is essential. Staggeringly, textbooks of respiratory disease for adult physicians may not even have a chapter on this crucial subject. Perhaps their Editors believe that, like the Greek Goddess Pallas Athene, the daughter of Olympian Zeus, we emerge from our father's heads as fully formed adults!
The periods of lung development most relevant to this article are the second half of pregnancy, and the first 2 years of life (Citation[48], Citation[49]). In the first half of pregnancy, the fetal airway branching pattern is established, and thereafter the airways increase in size, not number. It is therefore in the second half of pregnancy that they are most vulnerable to the effects of, for example, maternal smoking. The time course of alveolar development is controversial, but probably the vast majority is laid down by secondary septation in the first 18–24 months of life. This is particularly relevant to the effects of prematurity and its treatment (later). However, since alveolar tethering to the airway is an important mechanism for the maintenance of normal airway caliber, failure of normal alveolarisation may cause airflow obstruction. Further discussion of normal lung development can be found in recent reviews.
What are the important antenatal adverse events?
It therefore follows that, if we are to prevent the airflow obstruction which will be the substrate for later cigarette smoke induced inflammatory damage and airway remodeling to produce COPD, we must study events before birth, and intervene antenatally. Probably the major issues are the effects of maternal smoking; maternal atopy; and obstetric factors which lead to being small for gestational age (SGA), including maternal hypertension of pregnancy. Some but not all are amenable to intervention. In considering studies on both ante- and post-natal effects, it is important to consider whether the magnitude is very small, but of interest in generating hypotheses as to mechanisms, or large enough to be the focus of a public health intervention that will be clinically useful in that context.
Maternal smoking: A number of studies have demonstrated the deleterious effects of smoking during pregnancy. The Boston cohort (Citation[50]) showed reductions in VmaxFRC in the offspring of smoking mothers, more marked in girls than boys, and with diminishing effect over the next 18 months. Other studies have also confirmed an adverse effect of maternal smoking (Citation[51], Citation[52], Citation[53]). Maternal smoking may cause AHR in the newborn (Citation[54]), especially in those with a maternal history of asthma. It is important to note that smoking cessation in the second half of pregnancy may improve the outcome for the fetus (Citation[55]).
There are some pathological correlates of the effects of cigarette smoke on the developing lungs. The offspring of smoke exposed guinea pigs have reduced numbers of alveolar attachment points to the airway (Citation[56]). This is likely to result in a reduction in airway stability, and increased collapsibility. A study of the lungs of sudden infant death syndrome victims, whose mothers smoked in pregnancy, revealed increased inner airway wall thickness and increased airway smooth muscle (Citation[57], Citation[58]). Whether this is an effect of smoke, or related to another factor which caused sudden death, is not known.
Maternal smoking may have immunological as well as physiological effects. There is recent evidence that, in the infants of mothers that smoke, cord blood mononuclear cell responses to some, but not all, toll-like receptor ligands is down-regulated in the infants of mothers who smoked during pregnancy (Citation[59], Citation[60]). Thus smoking may be the link between (a) reduced airway caliber; (b) reduced alveolar tethering of airways; and (c) altered cord blood immune function, which subsequently interacts with effects of respiratory viral infections in infancy.
Maternal atopy: Infants of asthmatic mothers have a lower VmaxFRC (Citation[61]). Other studies have confirmed an adverse effect of maternal atopy. These findings are difficult to reconcile with the Tucson study reports of normal lung function at birth in those babies who will become persistent wheezers (Citation[6], Citation[7]), many of whom would be expected to have an atopic mother. Whether the differences between the studies are real, or a manifestation of population or methodological differences is unknown. However, it is clearly not possible to prevent atopic mothers having babies!
SGA effects: Two factors complicate the assessment of being SGA per se on lung function. The first is the choice of an appropriate control group, since forced expired flows are usually normalized to body length. This problem disappears if an age-independent variable such as LCI can be used. The second is the confounding effects of smoking, probably one of the commonest causes of SGA delivery. Impaired lung function soon after birth has been reported in SGA infants (Citation[62]), but it seemed that in the subgroup of non-smoking mothers, the changes related only to reduced body length.
The subsequent evolution of lung function changes suggested they were independent of somatic growth (Citation[63]). A follow-up study in SGA infants born to non-smoking mothers using the RVRTC technique showed that there was a consistent 9% reduction in forced expiratory flow and FVC, with no evidence of “catch-up” growth. Although no longitudinal study has followed these infants into adult life, cross-sectional studies have reported reduced lung function in adults and children who were born SGA, suggestive that the effect of SGA delivery, independent of smoking, persists long term (Citation[18], Citation[46], Citation[64]).
Other possible antenatal adverse effects: One study suggested that maternal hypertension was a risk factor for airflow obstruction (Citation[53]). First trimester invasive procedures (chorionic villus sampling, amniocentesis) may be associated with hyperinflation suggestive of airflow obstruction in the infant (Citation[65]), although another study suggested no influence of these procedures on wheeze (Citation[66]). This study identified maternal diabetes as a factor for persistent wheeze, antibiotics in pregnancy for maternal urinary infection as associated with transient early wheeze, and antibiotics at delivery for both phenotypes. It should be noted that antibiotic administration around delivery may be a marker of maternal chorioamnionitis, which accelerates lung maturation and may thus have long term consequences on lung function (Citation[67]).
Another maternal medication effect was identified by the ALSPAC study. Maternal paracetamol (Tylenol), but not aspirin, intake during pregnancy was associated with (Citation[68]) asthma, wheezing, and elevated IgE in school age children. The role of diet is unclear; early work has surprisingly suggested that high maternal vitamin C intake in pregnancy may actually increase the prevalence of wheeze in the second, but not the first year of life. High vitamin E intake may be protective (Citation[69]). No intervention study has determined that adjusting maternal diet affects outcomes in the baby.
Summary–Maternal adverse effects: Maternal smoking is the most important and obvious effect amenable to intervention. Paracetamol and antibiotics should be discouraged in pregnancy. Whether dietary manipulation in pregnancy will be beneficial is contentious.
What are the important early postnatal adverse events?
Postnatal smoke exposure: The adverse effects of tobacco smoke continue post-natally. The adverse effects have been well described previously (Citation[70]). Environmental smoke exposure in childhood has effects extending well into adulthood (Citation[71]). Childhood environmental tobacco smoke exposure in relation to chronic cough, phlegm, and asthma diagnosis was studied in never smokers from a cohort of Singaporeans of Chinese ethnicity aged 45–74 years at enrollment. Subjects were interviewed regarding environmental tobacco smoke exposure before and after the age of 18 and the presence and duration of current symptoms of chronic cough and phlegm production and asthma diagnosis. Staggeringly, nearly two thirds reported living with a daily smoker before the age of 18 years.
Living with a smoker before the age of 18 increased the odds of chronic dry cough and, to a lesser extent, phlegm, after adjustment for age, sex, dialect group, and current and past exposure to smokers at home and at work after the age of 18. Associations strengthened with higher numbers of smokers in childhood. There was no association with asthma or chronic bronchitis. In summary, for lifetime non-smokers who lived with a smoker in childhood, there was an associated with chronic dry cough and phlegm in adulthood, independent of later exposures to environmental tobacco smoke.
There is also new evidence of important gene-environment interactions with regard to smoking. Wenten et al. studied the effect of tumor necrosis factor (TNF)-308 variants on a marker of respiratory illness, respiratory-related school absences, in healthy children (Citation[72]). They found that the combination of exposure to second hand smoke doubled the risk of respiratory related school absences in children with the TNF-308 A compared with the G variant, and lead to a four-fold increased risk of lower respiratory tract infections causing school absence. The mechanisms are still speculative, but this paper again makes clear that genetic association studies neglecting the environment, in the hope that environmental effects will cancel themselves out, are not a good approach.
Respiratory viral infections: There is a huge literature on the possible effects of viral infections on the TH2-TH1 switch, the so-called hygiene hypothesis, and the extent to which early viral infection is a manifestation of an immune problem present at birth. A review of these important topics is beyond the scope of this paper. The focus here is, whether severe viral or other respiratory infections cause airway obstruction, or are a manifestation of it. As a general point, the question cannot be answered by studies recruiting children after their severe infection.
We lack a large study with pre-pneumonia lung function. For example, a Boston cohort study which recruited children between 4 and 10 years of age showed that those who had had preceding pneumonia or hospital admission with respiratory illness had worse lung function, but could not disentangle which came first (Citation[73]). Another group followed patients who had pneumonia or pertussis into middle age, and found that the both groups, decades after their infection, still had small decrements in lung function (greater in the pneumonia patients) (Citation[74]).
The best study, from Perth, did confirm decrements in lung function precede bronchiolitis (above). A study from Tucson also showed impaired lung function preceded pneumonia (Citation[75]). On the other hand, there is no doubt that, for example, adenovirus infection leads to chronic severe airway obstruction due to obliterative bronchiolitis in putatively previously normal babies, or at least babies with at worst only mild airflow obstruction (Citation[76]). There is accumulating evidence that the ill-studied non-atopic late onset wheeze phenotype may be related to viral bronchiolitis (Citation[77]). Thus although severe viral infection may certainly cause de novo airflow obstruction, it would be a mistake to forget the antecedent insults that may increase the child's susceptibility to respiratory infection. These include immunological effects on the unborn baby (previous discussion).
Diet: If oxidative stress is important in lung growth and development, then it might be predicted that increasing dietary anti-oxidants might be beneficial. In one study, vitamin C, fruit and magnesium intake was associated with better lung-function in mid-childhood (Citation[78]). However, whether intervening to alter diet would benefit lung function has not been shown. A healthy diet is desirable in itself, but is perhaps not likely to be hugely beneficial to lung development.
Medication administration: This is still controversial. One study showed that frequent paracetamol (Citation[79]) administration to children lead to increased wheezing, and possibly rhinitis and eczema. Studies of the effects of antibiotic administration for respiratory infection are dogged by reverse causation, i.e., the antibiotics may have been erroneously given for asthmatic symptoms. Studies of antibiotics for non-respiratory infections are inconclusive, but recent evidence suggests that any association between antibiotic use and subsequent wheeze is weak at best (Citation[80]). On general principles, antibiotics should be avoided in childhood unless really necessary, but I doubt that such a policy will have much effect on population lung health.
Air pollution: Pollution may be indoor (domestic heating, aeroallergens) or outdoor (particulates, ozone etc.). Clearly any pollution is undesirable; but whether either indoor or outdoor pollution has a long-term effect on lung function and COPD requires further studies. One study aimed to determine the association between level of lung function in pre-adolescence and indoor air quality in the postnatal period in a retrospective cohort design (Citation[81]). They reported on 1036 pre-adolescent school children (9 years of age) in Poland. Indoor air quality was based on reported environmental tobacco smoke and type of household heating. In addition, the number of winter months that occurred during the first 6 months of life was included as a key independent variable. Findings were adjusted for potential confounders such as maternal smoking during pregnancy and parental education as a proxy of social class. The results of this study suggested that a lower level of lung function in pre-adolescent children can be related to postnatal exposure to indoor emissions in the winter. The findings must be extrapolated cautiously to communities with different indoor heating exposures, but does underscore the importance of the air that we breathe in our homes for future outcomes.
There is challenging evidence for the role of allergens in airway development. The Manchester group showed that high house dust mite exposure was associated with airflow obstruction in infancy and stringent mite avoidance was beneficial (Citation[82]). The same group showed in adults that house dust mite and dog, but not cat, allergen levels, if high, had adverse effects on airway function and inflammation even if the patient is not sensitized to the allergen, as least on IgE mediated criteria (Citation[83]). The implication is that allergens have effects other than by stimulating an allergic response. The obvious mechanism is by proteolytic damage to the airway epithelium; and indeed, many allergens are also proteolytic enzymes (Citation[84]). The effects of allergen reduction are sometimes unpredictable; but this may be one mechanism whereby early exposure to pollutants may influence long-term outcome.
Outside air pollution is important in childhood lung function. Sixty-four children had induced sputum studied, and the carbon content of airway macrophages determined. There was a tight relationship between primary particulate matter (PM10) in the environment, carbon content of alveolar macrophages, and spirometry (Citation[85]). A longitudinal study of 3677 children over 8 years in California showed that exposure to traffic pollution lead to marked decrements in spirometry. Regional air pollution, and exposure to traffic fumes, had independent and detrimental effects on lung growth (Citation[86]). However, the evidence that changing the flow of traffic can improve outcomes for childhood respiratory health is at best contentious (Citation[87], Citation[88]).
Summary: Postnatal adverse effects: There is clearly scope for improvement in lung health, and reducing the prevalence of COPD in adult life, by improving indoor air quality (not least by reducing environmental tobacco smoke) and reducing out door pollution. Dietary manipulations, and alteration in prescribing habits, are likely to be far less significant public health measures.
The potential role of the maternal genotype
Before any genetic studies are planned, it must be appreciated that the manifestation of genes depends critically on the environment in which they are expressed. Polymorphisms may show a biphasic influence, being protective in some environments, but detrimental in others. For example, the CD 14/-260 C → T polymorphism in the CD 14 promoter was associated with higher levels of both total and specific IgE to aeroallergens in children in regular contact with domestic pets, but the opposite relationship, not explained by endotoxin levels, in children in contact with stable animals (Citation[89]). Thus it is essential to start with a focussed hypothesis, in which both genetic and environmental factors are considered. Blindly trawling through the genome in the Micawber-like hope that, with the aid of ever more powerful computers, something will turn up, is unlikely to pay dividends. At the moment, very few studies are characterised by focussed hypotheses about gene-environment interactions.
Maternal smoking illustrates an important gene: environment interaction. Genes do not exist in isolation, but they interact with the environment; for example, the glutathione-S-transferase gene is one of those responsible for protecting the organism against oxidative damage. Polymorphisms of this gene in the mother but not the father are important determinant of the effects of maternal smoking during smoking on fetal airway development (Citation[90], Citation[91]); the null polymorphism exposes the fetus to the threat of oxidant damage, and is associated with airflow obstruction in the newborn period. Maternal exposure to tobacco smoke negatively affects neonatal birth weight, and the adverse effect of maternal exposure on neonatal birth weight could be modified by the maternal metabolic genotypes, GSTM1 and GSTT1 (Citation[92]). Maternal smoke exposure also resulted in reduced birth weight in mothers who had any of the aryl hydrocarbon receptor wild type genotype, the CYP1A1 variant genotype or the GSTM1 null genotype (Citation[93]). The relationship of birth weight and long-term lung function was discussed previously.
Clearly fetal genotype is also important. In a large American study, GSTM1 null children exposed to smoke in utero had more early and persistent asthma and wheeze than those with the GSTM+ genotype (Citation[94]). This important study confirms the medium-term adverse effects of antenatal smoke exposure. These findings were confirmed in a large study of German school children (Citation[95]), in which GSTT1 null children exposed to smoke antenatally had decrements in lung function, and GSTM1 null children exposed to environmental tobacco smoke had more asthma and respiratory symptoms.
In conclusion, at least some of the roots of COPD may lie in the proband's mother's genotype. These definitely include genes determining fetal susceptibility to the effects of maternal smoking in pregnancy, but perhaps also, genes related to the development of maternal hypertension in pregnancy? This area is fraught with difficulty, and at the moment much remains conjectural, but the collection of DNA from the mothers of COPD sufferers, in particular if the proband has only a relatively minor smoking history, could be a fruitful avenue for research.
COPD genes: are important clues to be found in antenatal lung development?
The importance of early life events has considerable implications for the hunt for COPD susceptibility genes. It is likely that early genetic effects will be increasingly masked by environmental influences, only to re-surface at the time of lung aging. The obvious targets have long been genes related to protection against oxidative stress, and inflammatory pathways. The hypothesis advanced here would open up a completely different avenue to explore, namely genetic polymorphisms related to lung growth and development. There are, however, some data connecting genes important in lung growth with those affecting the susceptibility to COPD. This will be illustrated by reviewing the ADAM33, the beta-receptor, the fibroblast growth factor (FGF) and GSTM genes.
ADAM33 was identified as a potential gene for asthma by positional cloning, and has numerous putative effects (Citation[96]). Different splice variants may be concerned in cell adhesion, cell signalling, cell fusion and proteolysis. The role of ADAM33 in intra-uterine lung development has been studied in mice (Citation[97]). ADAM33 is expressed in the developing lung during branching morphogenesis, increases throughout gestation, and persists into adult life, suggesting that it may have a role in the developmental regulation of airway calibre. Polymorphisms in the ADAM33 have been associated with impaired early life lung function (Citation[98]). The Manchester group showed that the SNP F+1 was a determinant of lung function age 3 years.
Carriers of the A allele had poorer lung function at three years of age, and AA homozygotes had worse lung function at age 5 years. (In both cases, sRAW was the measurement made). The association was independent of sex, smoking, and allergen exposure. Polymorphisms in ADAM33 are known to be associated with asthma and bronchial hyper-responsiveness in different adult populations (Citation[99]). Now it has been shown that polymorphisms in ADAM33 are implicated in the severity of COPD. Adults with the AA genotype for the SNP ST+5 had more severe AHR, greater numbers of sputum inflammatory cells, and more CD8 positive cells on bronchial biopsy, than those with the GG genotype. Some more minor ADAM33 alleles had the opposite effects (Citation[100]). It must be conceded that the two studies suggested influences at the opposite ends of the long and complex ADAM33 gene (5′ for F+1, 3′ for ST+5), but nonetheless, the data suggest a common genetic factor at the start and end of life, suggesting that further probing of this theme might be rewarding.
Another gene linking early lung function, asthma and COPD is the beta-receptor. Although there have been many studies of beta-receptor function in asthma, much less is known about any role in early life; only two studies have explored this. In a UK cohort in whom VmaxFRC and AHR had been measured in the first month of life, the possession of any Glyn 27 allele was predictive of airflow obstruction (Citation[39]). The same result was found with any Arg16 allele. In both cases, the association was independent of maternal smoking or atopy. There was no effect of genotype on AHR in either the newborn period, or when re-studied at age 11 years. Nor was there any effect of beta-receptor genotype and lung function at age 11; this presumably reflects the small numbers studied, and a lack of power to show a marginal effect. It should be noted that this was a high-risk population (> 80% with a parental history of asthma), and may not be applicable to the general population.
The Perth cohort (Citation[101]) found an association between the haplotype arg16gln27 and the prevalence of positive AHR at age 6 years. The gly16gln27 haplotype was associated with better spirometry at age 6 and age 11. In contrast, arg16gln27 was associated with worse spirometry at age 11. Children with the gly16gln27 haplotype were less likely to have asthma-ever or doctor-diagnosed asthma at age 11. They concluded that haplotypes of beta(2)-adrenoceptor polymorphisms are associated with lung function, AHR, and asthma susceptibility in childhood. A recent study genotyped the polymorphisms at positions 16 and 27 in more than 1100 people with a variety of respiratory problems, including asthma and COPD, as well as a control population (Citation[102]). The Arg 16 homozygotes had an increased risk of COPD, asthma, and wheeze. The Glyn 27 homozygotes had a greater risk of asthma and AHR. The Arg16/Glyn27 haplotype was predictive of asthma and COPD, and more respiratory symptoms. Beta- receptor polymorphisms have also been found to be a predictor of persistence of asthma symptoms from childhood into adult life (Citation[103]), but predict only a small component of long term prognosis. This is not to say, however, that they may not be of interest in the study of biological mechanisms.
Another illustration of an association between genes important in lung growth and also later susceptibility to COPD concerns FGF-1, FGF-2 and their receptor FGF1-R. FGFs are important in lung growth (reviewed in 104), and bind to one of four transmembrane, tyrosine kinase receptors FGFR-1 to 4. In an in vitro study, human airway smooth muscle cells were shown to proliferate when exposed to FGF1 and 2. In vivo, there was cytoplasmic expression of FGF-2 in epithelium, and nuclear localization in airway smooth muscle cells, in COPD patients when compared with controls. There were also elevated levels of FGF-1 in airway smooth muscle cells, and FGF-1 and FGFR-1 in bronchial epithelial cells, in COPD patients as compared with controls (Citation[105]). In all cases, the lung tissue was from surgical resection specimens. In terms of functional correlations, there were inverse associations between FEV1/FVC ratio and both FGF-2 and FGFR-1 expression in airway smooth muscle. Epithelial FGF-1 correlated with smoking history. These authors reviewed the roles of the FGF system in a number of adult diseases, but the relevance of FGFs to early life events, and the relationship between this and COPD, was not discussed.
The effects of GSTM polymorphisms in mother and child, and early lung function and symptoms, have been discussed. A study in mid-childhood showed that (Citation[106]) GSTM1 and GSTP1 polymorphisms are associated with defects in lung growth, particularly in children with asthma. GSTT1 deficiency is also associated with an accelerated decline in lung function, a known risk factor for COPD, in middle aged men, and in another study, a combination of polymorphisms in all three GST genes were associated with accelerated decline in lung function (Citation[107]). There is a significant interaction with active smoking. However, the effect of the polymorphisms is independent of smoking history, and thus presumably reflects not later smoke exposure but the antenatal and immediate post-natal effects of this gene on airway growth and development.
This section is not intended to be comprehensive; other genes are undoubtedly important. The aim of this section was to establish the principle that important COPD genes may be found in processes important in very early life.
The ‘new’ COPD – what happens to the survivors of preterm birth?
This is a very important growth area. No area has seen the frontiers of the possible pushed back further and faster than neonatal intensive care. Babies weighing less than 500 gm at birth are now surviving to go home, after periods of often heroic intensive care. Most often, these tiny preterm babies will have a prolonged period of home oxygen, but eventually oxygenation normalizes. Common experience is that as well as coming out of oxygen, survivors of CLD of prematurity, although initially experiencing recurrent episodes of cough and wheeze, gradually become less symptomatic as the years go by. Should we be reassured that the lung is recovering from the early insults? And in any case, given that very low birth weight is very uncommon, and as such is unlikely to make a huge impact on the population burden of disease, does it matter anyway?
Studies have shown that impairment of lung function occurs even in those born prematurely who were never ventilated; it is clear that prematurity per se is a risk factor for at least mild impairment of lung function in mid-childhood (Citation[46], Citation[108], Citation[109], Citation[110]). Summarizing the studies of airway function, in mid-childhood, survivors of CLD of prematurity exhibit reduced indices of expiratory airway function (FEV1 and MEF50); increased bronchodilator responsiveness; and bronchial hyper-reactivity to histamine and methacholine. Abnormalities of exercise tolerance may be found, but these are usually subtle, and most children appear functionally normal for the purposes of normal activity and exercise in daily life (reviewed in 111).
It is, of course, true that many of the abnormalities described are quite subtle and do not appear to impact on the child very much. Our own studies, following up a previous cohort, are that by early adult life, lung function is essentially normal for the cohort as a whole (Citation[112]). However, we found that in adult life low birth weight or preterm babies who were small for gestational age, but not those who were appropriate for gestational age, had airflow obstruction (reduced FEV1 and flows at low lung volumes) which were strongly correlated with birth weight (Citation[113]). Although the absolute deficits may not be too profound, the situation may be analogous to VIW (above). The concern in finding these mild changes is not so much that they cause present trouble, but that they are a sign of reduced reserve. This is likely to be of significance when the reserves are stretched, most notably when the lungs start to age; even if there is no accelerated rate of aging (and this is not known) early onset of airflow obstruction and chronic obstructive pulmonary disease or a variant thereof is likely.
By definition, there are no studies of CLD of prematurity into old age because CLD of prematurity was unknown before the 1960s. Prior to this time, preterm babies either survived with no apparent long-term morbidity, or died. On this basis, it seems reasonable to suggest that infants who have survived CLD of prematurity may be at high risk of respiratory failure in adulthood. The pathology is of course different, but more severe, encompassing pulmonary hypoplasia as well as airway obstruction. We do not know what effect these changes will have on the rate of lung aging in the survivors of CLD of prematurity; but there is no doubt in my mind that there will be new health problems in this group as they reach adult life. There is a real need for studies of the effects (if any) of CLD of prematurity on the rate and nature of lung aging.
It could be argued that modern neonatal practice will mean that CLD of prematurity will become only a disease of the past. Surprisingly, however, the incidence of unexpected preterm delivery is rising, not falling, in low risk women. All the evidence is that CLD of prematurity will not decline, and certainly that premature babies, no matter how superb their neonatal care, will have impaired lung function in mid-childhood. Likewise, there seems no realistic prospect that placental insufficiency leading to babies being born small for gestational age will disappear. There is compelling evidence that prematurity per se, even with no iatrogenic influences, is associated with detectable problems in mid-childhood; hence until prematurity ceases to be a problem, these cohorts will keep appearing.
There was one small and admittedly incomplete follow-up study that compared evidence of airflow obstruction in preterm infants who had been in a randomized, placebo controlled, double blind trial of surfactant. There was no difference in the long-term pulmonary outcome in the two groups (Citation[114]). Clearly surfactant is a major benefit in the acute management of premature infants, but there is no reason to believe it will impact on long-term outcome. In summary, the survivors of preterm birth are likely to be a cohort prone to COPD in later life; that better neonatal care can reduce the risk is likely, but that prematurity and reduced weight for gestational age will disappear seems unlikely, and thus the airway risk factors will remain.
Prevention of COPD – what should we do now?
Smoking in the young: The most obviously beneficial measure would be the prevention of smoking in young people. Elsewhere I have criticized strongly the ineffectual public health measures of most governments (Citation[115], Citation[116]). If we are not to continue to jeopardize the future of our children, we must now:
Have a total ban on cigarette smoking in ALL public places;
Totally ban the display of tobacco products in any commercial site;
Have a total ban on advertising tobacco, and the depiction of smoking under any circumstances;
Purchase of tobacco requiring the production of photo-identity;
Introduce really severe penalties, including closing down of businesses and imprisonment for those who sell cigarettes to children.
In addition, a campaign needs to be targeted in antenatal clinics:
If you smoke during pregnancy, your unborn baby is smoking, too;
This will affect the baby throughout their life;
Even if you became pregnant without having given up smoking it is not too late; if you give up early in pregnancy, you can avoid harming the baby;
(Preferably) link maternity benefits to quitting smoking, with objective checks on urine or salivary cotinine levels.
Viral infections? I have argued previously that severe RSV infection is a marker for previous damage, not a cause of subsequent problems. Admittedly there is some evidence for latent RSV infection, analogous to latent adenovirus infection (Citation[117], Citation[118]), but whether this is clinically significant awaits further study. At the moment, the likelihood that RSV immunization (if it could safely be performed) would prevent later COPD is not great. Since RSV infection is virtually universal in the first 2 years of life, and COPD is not universal even in adults who smoke, one would have to postulate other factors in a model whereby RSV was causative of COPD. However, there may be genetic interactions with RSV (polymorphisms in pro-inflammatory genes for example (Citation[119], Citation[120])), and it is possible that there are subgroups of children with anatomical damage, who also have polymorphisms in particular genes, in whom RSV infection may cause an increased likelihood of later COPD, over and above the effects of antenatal factors and the polymorphisms themselves.
Severe adenoviral infections are rare in childhood, but the prevention of more mild infections, which may possibly become latent with long term deleterious effects, may contribute to a reduction in subsequent COPD. The role of other childhood viruses, and Chlamydia, also needs further study. In summary, although there may be a role for viral infection over and above that of airflow obstruction due to antenatal factors, at the moment it seems likely to be minor. We need studies with a clearly focused hypothesis to delineate any possible interactions between genes, environment, antenatal airway damage, and viral infections to move the field forwards.
Nutrition? Obesity seems likely to be the next childhood epidemic. The relationship between obesity and respiratory disease is not clear – if you are fat, you will become breathless on exertion, while not necessarily having a lung problem. However, it is possible that obesity, which may be a pro-inflammatory state, may affect airway reactivity (Citation[121]). There have also been reports on adverse effects of eating “fast food” and the benefits of other dietary manipulations (Citation[122], Citation[123]). Clearly healthy eating is a good thing in its own right, but it is likely only to make a minor contribution to the prevention of COPD.
Ensuring optimal obstetric outcomes: It may seem even more bizarre to urge that COPD is an obstetric as well as a pediatric disease, but, as discussed above, maternal obstetric problems may have long-term effects. Optimal obstetric care is clearly desirable in its own right, but it may have the spin-off of also preventing prematurity and its consequences, and promoting good lung health in term infants.
SUMMARY AND CONCLUSIONS
There is no question but that to understand COPD, one needs to understand antenatal lung growth, and postnatal disease. Lung growth offers hope for recovery in the short and medium term from childhood insults if suitable strategies to enhance it can be found in the future, but any childhood problem may potentially come back to haunt the patient in adult life and in old age, as lung function exhibits a physiological decline. The best way of preventing the substrate for COPD lies in optimizing antenatal conditions by preventing maternal smoking, both for its effects on placental function and foetal airway growth; and energetically preventing the effects of postnatal influences which might cause airflow obstruction, such as adenoviral infection, and indoor and outdoor pollution. Research into COPD genes will also benefit from an understanding of lung growth and its genetic control, with focused, hypothesis driven studies of gene-environment interactions in early life; and this may involve the maternal genotype, as well as that of the proband. Clearly the substrate for COPD, airflow obstruction, will never be wholly prevented, and obviously, prevention of later smoking ‘pulling the trigger’ is very important. Ultimately, prevention of smoking will benefit all ages; but babies and children have even more to gain than adults.
REFERENCES
- Løkke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax 2006; 61: 935–939
- Barker D JP. Fetal and Neonatal Origins of Adult Disease. BMJ, London 1992
- Gustafsson P M, Ljungberg H K, Kjellman B. Peripheral airway involvement in asthma assessed by single breath SF6 and He washout. Eur RespirJ 2003; 21: 1033–1039
- Gustafsson P M, Aurora P, Lindblad A. Evaluation of ventilation maldistribution as an early indicator of lung disease in children with cystic fibrosis. Eur RespirJ 2003; 22: 972–979
- Lum S, Gustafsson P, Ljungberg H, Hulskamp G, Bush A, Carr S B, Castle R, Hoo A F, Price J F, Ranganathan S, Stroobant J, Wade A, Wallis C, Wyatt H, Stocks J. Early detection of cystic fibrosis lung disease: multiple-breath washout vs. raised volume tests. Thorax 2007; 62: 341–347
- Martinez F D, Morgan W J, Wright A L, et al. Diminished lung function as a predisposing factor for wheezing respiratory illness in infants. N EnglJ Med 1988; 319: 1112–1117
- Morgan W J, Stern D A, Sherrill D L, Guerra S, Holberg C J, Guilbert T W, Taussig L M, Wright A L, Martinez F D. Outcome of asthma and wheezing in the first 6 Years of life: Follow-up through adolescence. AmJ Respir Crit Care Med 2005; 172: 1253–1258
- Turner S W, Palmer L J, Rye P J, Gibson N A, Judge P K, Cox M, Young S, Goldblatt J, Landau L I, Le Souef P N. The relationship between infant airway function, childhood airway responsiveness, and asthma. AmJ Respir Crit Care Med 2004; 169: 921–927
- Woolcock A J, Leeder S R, Peat J K, Blackburn C R. The influence of lower respiratory illness in infancy and childhood and subsequent cigarette smoking on lung function in Sydney schoolchildren. Am Rev Respir Dis. 1979; 120: 5–14
- Turner S W, Young S, Landau L I, Le Souef P N. Reduced lung function both before bronchiolitis and at 11 years. Arch Dis Child. 2002; 87: 417–420
- Turner S W, Palmer L J, Rye P J, Gibson N A, Judge P K, Young S, Landau L I, Le Souef P N. Infants with flow limitation at 4 weeks: outcome at 6 and 11 years. AmJ Respir Crit Care Med 2002; 165: 1294–1298
- Young S, Arnott J, Le Souef P N, Landau L I. Flow limitation during tidal expiration in of asthma symptom-free infants and the subsequent development. J Pediatr 1994; 124: 681–688
- Lowe L A, Simpson A, Woodcock A, Morris J, Murray C S, Custovic A NAC, Manchester Asthma and Allergy Study Group. Wheeze phenotypes and lung function in preschool children. AmJ Respir Crit Care Med 2005; 171: 231–237
- Brussee J E, Smit H A, Koopman L P, et al. Interrupter resistance and wheezing phenotypes at 4 years of age. AmJ Respir Crit Care Med 2004; 169: 209–213
- Elliot J G, Carroll N G, James A L, Robinson P J. Airway alveolar attachment points and exposure to cigarette smoke in utero. AmJ Respir Crit Care Med 2003; 167: 45–49
- Frey U, Makkonen K, Wellman T, Beardsmore C, Silverman M. Alterations in airway wall properties in infants with a history of wheezing disorders. AmJ Respir Crit Care Med 2000; 161: 1825–1829
- Haland G, Lodrup-Carlsen K C, Sandvik L, et al. Reduced lung function at birth and the risk of asthma at 10 years of age. N EnglJ Med 2006; 355: 1682–1689
- Edwards C A, Osman L M, Godden D J, Campbell D M, Douglas J G. Relationship between birth weight and adult lung function: controlling for maternal factors. Thorax 2003; 58: 1061–106
- Ranganathan S C, Bush A, Dezateux C, Carr S B, Hoo A F, Lum S, Madge S, Price J, Stroobant J, Wade A, Wallis C, Wyatt H, Stocks J. Relative ability of full and partial forced expiratory maneuvers to identify diminished airway function in infants with cystic fibrosis. AmJ Respir Crit Care Med 2002; 166: 1350–1357
- Aurora P, Gustafsson P M, Bush A, et al. Multiple breath inert gas washout as a measure of ventilation distribution in children with cystic fibrosis. Thorax 2004; 59: 1068–1073
- Aurora P, Bush A, Gustafsson P, et al. Multiple-breath washout as a marker of lung disease in preschool children with cystic fibrosis. AmJ Respir Crit Care Med 2005; 171: 249–256
- Kraemer R, Blum A, Schibler A, et al. Ventilation inhomogeneities in relation to standard lung function in patients with cystic fibrosis. AmJ Respir Crit Care Med 2005; 171: 371–378
- Payne D NR, McKenzie S A, Stacey S, Misra D, Haxby E, Bush A. Safety and ethics of bronchoscopy and endobronchial biopsy in difficult asthma. Arch Dis Child 2001; 84: 423–426
- Saglani S, Payne D N, Nicholson A G, Scallan M, Haxby E, Bush A. The safety and quality of endobronchial biopsy in children under five years old. Thorax 2003; 58: 1053–1057
- Salva P S, Theroux C, Schwartz D. Safety of endobronchial biopsy in 170 children with chronic respiratory symptoms. Thorax. 2003; 58: 1058–1060
- Molina-Teran A, Hilliard T N, Saglani S, Haxby E, Scallan M, Bush A, Davies J C. Safety of endobronchial biopsy in children with cystic fibrosis. Pediatr Pulmonol 2006; 41: 1021–1024
- Bush A, Davies J C. Rebuttal: You are wrong, Dr. Mallory … Pediatr Pulmonol 2006; 41: 1017–1020
- McIntosh N, Bates P, Brykczynska G, et al. Guidelines for the ethical conduct of medical research involving children. Arch Dis Child 2000; 82: 177–182
- Payne D N, Rogers A V, Adelroth E, Bandi V, Guntupalli K K, Bush A, Jeffery P K. Early thickening of the reticular basement membrane in children with difficult asthma. AmJ Respir Crit Care Med 2003; 167: 78–82
- Saglani S, Malmstrom K, Pelkonen A S, Malmberg L P, Lindahl H, Kajosaari M, Turpeinen M, Rogers A V, Payne D N, Bush A, et al. Airway remodeling and inflammation in symptomatic infants with reversible airflow obstruction. AmJ Respir Crit Care Med 2005; 171: 722–727
- Saglani S, Payne D N, Zhu J, Wang Z, Nicholson A G, Bush A, Jeffery P K. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. AmJ Respir Crit Care Med 2007; 176: 858–864
- Sears M R, Greene J M, Willan A R, Wiecek E M, Taylor D R, Flannery E M, Cowan J O, Herbison G P, Silva P A, Poulton R. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. N EnglJ Med 2003; 349: 1414–1422
- CAMP study. Long-term effects of budesonide or nedocromil in children with asthma. The Childhood Asthma Management Program Research Group. N EnglJ Med 2000; 343: 1054–1063
- Oswald H, Phelan P D, Lanigan A, et al. Childhood asthma and lung function in mid-adult life. Pediatr Pulmonol 1997; 23: 14–20
- Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br MedJ 1977; 1: 1645–1648
- Edwards C A, Osman L M, Godden D J, Douglas J G. Wheezy bronchitis in childhood: a distinct clinical entity with lifelong significance. Chest 2003; 124: 18–24
- Mannino D M, Davis K J. Lung function decline and outcomes in an elderly population. Thorax 2006; 61: 472–477
- Palmer L J, Rye P J, Gibson N A, Burton P R, Landau L I, LeSouef P N. Airway responsiveness in infancy predicts asthma, lung function, and respiratory symptoms by school age. AmJ Respir Crit Care Med 2001; 163: 37–42
- Wilson N M, Lamprill J R, Mak J C, Clarke J R, Bush A, Silverman M. Symptoms, lung function, and beta2-adrenoceptor polymorphisms in a birth cohort followed for 10 years. Pediatr Pulmonol. 2004; 38: 75–81
- Joos G F, O'Connor B, Anderson S D, et al. Indirect airway challenges. Eur RespirJ 2003; 21: 1050–1068
- Stick S, Arnott J, Turner D, et al. Bronchial responsiveness and lung function in recurrently wheezy infants. Am Rev Respir Dis 1991; 144: 1012–1015
- Grol M H, Gerritsen J, Vonk J M, et al. Risk factors for growth and decline of lung function in asthmatic individuals up to 42 years. AmJ Respir Crit Care Med 1999; 160: 1830–1837
- Grol M H, Postma D, Vonk J M, et al. Risk factors from childhood to adulthood for bronchial responsiveness aged 32–42 years. AmJ Respir Crit Care Med 1999; 160: 150–156
- McNamara P S, Ritson P, Selby A, Hart C A, Smyth R L. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch Dis Child 2003; 88: 922–926
- McNamara P S, Flanagan B F, Baldwin L M, Newland P, Hart C A, Smyth R L. Interleukin 9 production in the lungs of infants with severe respiratory syncytial virus bronchiolitis. Lancet 2004; 363: 1031–1037
- Chan K N, Noble-Jamieson C M, Elliman A, Bryan E M, Silverman M. Lung function in children of low birth weight. Arch Dis Child 1989; 64: 1284–1293
- Lebourges F, Moriette G, Boule M, Delaperche M F, Relier J P, Gaultier C. Pulmonary function in infancy and in childhood following mechanical ventilation in the neonatal period. Pediatr Pulmonol 1990; 9: 34–40
- Hislop A. Developmental biology of the pulmonary circulation. Paediatr Respir Rev 2005; 6: 35–43
- Rosenthal M, Bush A. The growing lung: normal development, and the long-term effects of pre- and post-natal insults. In: Bush A, Zach M, Carlsen K-H, Growing up with lung disease: the lung in transition to adult life. Euro Respir Monogr 2002; 7: 1–24
- Tager I B, Ngo L, Hanrahan J P. Maternal smoking during pregnancy: effects on lung function during the first 18 months of life. AmJ Respir Crit Care Med 1995; 152: 977–983
- Martinez F D, Wright A L, Taussig L M, et al. Asthma and wheezing in the first six years of life. N EnglJ Med 1995; 132: 133–138
- Lodrup-Carlsen K C, Jaakkola J J, Nafstad P, Carlsen K H. In utero exposure to cigarette smoking influences lung function at birth. Eur RespirJ 1997; 10: 1774–1779
- Stick S M, Burton P R, Gurrin L, Sly P D, LeSouef P N. Effects of maternal smoking during pregnancy and a family history of asthma on respiratory function in newborn infants. Lancet 1996; 348: 1060–1064
- Young S, LeSouef P N, Geelhoed G C, et al. The influence of a family history of asthma and parental smoking on airway responsiveness in early infancy. N EnglJ Med 1991; 324: 1166–1173
- Dezateux C, Stocks J. Lung development and early origins of childhood respiratory illness. Br Med Bull 1997; 53: 40–57
- Elliot J, Carroll N, Bosco M, McCrohan M, Robinson P. Increased airway responsiveness and decreased alveolar attachment points following in utero smoke exposure in the guinea pig. AmJ Respir Crit Care Med 2001; 163: 140–144
- Elliot J, Vullermin P, Carroll N, James A, Robinson P. Increased airway smooth muscle in sudden infant death syndrome. AmJ Respir Crit Care Med 1999; 160: 313–316
- Elliot J, Vullermin P, Robinson P. Maternal cigarette smoking is associated with increased inner airway wall thickness in children who die from sudden infant death syndrome. AmJ Respir Crit Care Med 1998; 158: 802–806
- Noakes P S, Hale J, Thomas R, Lane C, Devadason S G, Prescott S L. Maternal smoking is associated with impaired neonatal toll-like-receptor-mediated immune responses. Eur RespirJ 2006; 28: 721–729
- Le Souef P N. Adverse effects of maternal smoking during pregnancy on innate immunity in infants. Eur RespirJ 2006; 28: 675–677
- Clarke J R, Salmon B, Silverman M. Bronchial responsiveness in the neonatal period as a risk factor for wheezing in infancy. AmJ Respir Crit Care Med 1995; 151: 1434–1440
- Lum S, Hoo A F, Dezateux C, et al. The association between birthweight, sex and lung function in the infants of nonsmoking mothers. Am Rev Respir Crit Care Med 2001; 164: 2078–2084
- Hoo A F, Stocks J, Lum S, et al. Development of lung function in early life. Influence of birthweight in infants of nonsmokers. Am Rev Respir Crit Care Med 2004; 170: 527–533
- Rona R J, Gulliford M C, Chinn S. Effects of prematurity and intrauterine growth on respiratory health and lung function in childhood. Br MedJ 1993; 306: 817–820
- Yuksel B, Greenough A, Naik S, Cheeseman P, Nicolaides K H. Perinatal lung function and invasive antenatal procedures. Thorax 1997; 52: 181–184
- Rusconi F, Galassi C, Forastiere F, et al. Maternal complications and procedures in pregnancy and at birth and wheezing phenotypes in children. AmJ Respir Crit Care Med 2007; 175: 16–21
- Bush A, Annesi-Maesano I. Beam me up, Scotty. AmJ Respir Crit Care Med 2007; 175: 1–2
- Shaheen S O, Newson R B, Henderson A J, et al. Prenatal paracetamol exposure and risk of asthma and elevated immunoglobulin E in childhood. Clin Exp Allergy 2005; 35: 18–25
- Martindale S, McNeill G, Devereux G, Campbell D, Russell G, Seaton A. Antioxidant intake in pregnancy in relation to wheeze and eczema in the first two years of life. AmJ Respir Crit Care Med 2005; 171: 121–128
- Cook D G, Strachan D P. Health effects of passive smoking-10: Summary of effects of parental smoking on the respiratory health of children and implications for research. Thorax. 1999; 54: 357–366
- David G L, Koh W P, Lee H P, Yu M C, London S J. Childhood exposure to environmental tobacco smoke and chronic respiratory symptoms in non-smoking adults: the Singapore Chinese Health Study. Thorax 2005; 60: 1052–1058
- Wenten M, Berhane K, Rappaport E B, Avol E, Tsai W-W, Gauderman W J, McConnell R, Dubeau L, Gilliland F D. TNF-308 Modifies the Effect of Second-Hand Smoke on Respiratory Illness-Related School Absences. AmJ Respir Crit Care Med 2005; 172: 1563–1568
- Gold D, Tager I, Weiss S, et al. Acute lower respiratory illness in childhood as a predictor of lung function and respiratory symptoms. Am Rev Respir Dis 1989; 140: 877–884
- Johnston I D, Strachan D P, Anderson H R. Effect of pneumonia and whooping cough in childhood on adult lung function. N EnglJ Med 1998; 338: 581–587
- Castro-Rodriguez J, Holberg C, Wright T A, et al. Association of radiologically ascertained pneumonia before age 3 years and asthmalike symptoms and pulmonary function in childhood: a prospective study. AmJ Respir Crit Care Med 1999; 159: 1891–1897
- Mistchenko A S, Diez R A, Mariani A L, et al. Cytokines in adenoviral disease in children: association of interleukin-6, interleukin-8, and tumor necrosis factor alpha levels with clinical outcome. J Pediatr 1994; 124: 714–720
- Stein R T, Holberg C J, Morgan W J, Wright A L, Lombardi E, Taussig L, Martinez F D. Peak flow variability, methacholine responsiveness and atopy as markers for detecting different wheezing phenotypes in childhood. Thorax 1997; 52: 946–952
- Tujague J, Bastaki M, Holland N, Balmes J R, Tager I B. Antioxidant intake, GSTM1 polymorphism and pulmonary function in healthy young adults. Eur RespirJ 2006; 27: 282–288
- Barragan-Meijueiro M M, Morfin-Maciel B, Nava-Ocampo A A. A Mexican population-based study on exposure to paracetamol and the risk of wheezing, rhinitis, and eeczema in childhood. J Investig Allergol Clin Immunol 2006; 16: 247–252
- Harris J M, Mills P, White C, Moffat S, Newman Taylor A J, Cullinan P. Recorded infections and antibiotics in early life; associations with allergy in UK children and their parents. Thorax. 2007; 62: 631–637
- Jedrychowski W, Maugeri U, Jedrychowska-Bianchi I, Flak E. Effect of indoor air quality in the postnatal period on lung function in pre-adolescent children: a retrospective cohort study in Poland. Public Health 2005; 119: 535–541
- Woodcock A, Lowe L A, Murray C S, Simpson B M, Pipis S D, Kissen P, Simpson A, Custovic A NAC, Manchester Asthma and Allergy Study Group. Early life environmental control: effect on symptoms, sensitization, and lung function at age 3 years. AmJ Respir Crit Care Med 2004; 170: 433–439
- Langley S J, Goldthorpe S, Craven M, Woodcock A, Custovic A. Relationship between exposure to domestic allergens and bronchial hyperresponsiveness in non-sensitised, atopic asthmatic subjects. Thorax 2005; 60: 17–21
- Bush A. Coughs and wheezes spread diseases: but what about the environment?. Thorax 2006; 61: 367–369
- Kulkarni N, Pierse N, Rushton L, Grigg J. Carbon in airway macrophages and lung function in children. N EnglJ Med 2006; 355: 21–30
- Gauderman W J, Vora H, McConnell R, et al. Effect of exposure to traffic on lung development from 10 to 18 years of age: a cohort study. Lancet 2007; 369: 571–577
- Friedman M S, Powell K E, Hutwagner L, Graham L M, Teague W G. Impact of changes in transportation and commuting behaviors during the 1996 Summer Olympic Games in Atlanta on air quality and childhood asthma. JAMA. 2001; 285: 897–905
- Burr M L, Karani G, Davies B, Holmes B A, Williams K L. Effects on respiratory health of a reduction in air pollution from vehicle exhaust emissions. Occup Environ Med 2004; 61: 212–218
- Eder W, Klimecki W, Yu L, et al. Opposite effects of CD 14/-260 on serum IgE levels in children raised in different environments. J Allergy Clin Immunol 2005; 116: 601–607
- Flamant C, Henrion-Caude A, Boelle P Y, et al. Glutathione-S-transferase M1, M3, P1 and T1 polymorphisms and severity of lung disease in children with cystic fibrosis. Pharmacogenetics. 2004; 14: 295–301
- Gilliland F D, Li Y F, Dubeau L, et al. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. AmJ Respir Crit Care Med 2002; 166: 457–463
- Hong Y C, Lee K H, Son B K, Ha E H, Moon H S, Ha M. Effects of the GSTM1 and GSTT1 polymorphisms on the relationship between maternal exposure to environmental tobacco smoke and neonatal birth weight. J Occup Environ Med 2003; 45: 492–498
- Sasaki S, Kondo T, Sata F, et al. Maternal smoking during pregnancy and genetic polymorphisms in the Ah receptor, CYP1A1 and GSTM1 affect infant birth size in Japanese subjects. Mol Hum Reprod. 2006; 12: 77–83
- Gilliland F D, Li Y-F, Dubeau L, et al. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. AmJ Respir Crit Care Med 2002; 166: 457–463
- Kabesch M, Hoefler C, Carr D, Leupold W, Weiland S K, von Mutius E. GlutathioneS Transferase deficiency and maternal smoking increase childhood asthma. Thorax 2004; 59: 59–73
- van Eerdewigh P, Little R D, Dupuis J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 2002; 418: 426–430
- Haitchi H M, Powell R M, Shaw T J, et al. ADAM33 expression in human lungs and asthmatic airways. AmJ Respir Crit Care Med 2005; 171: 958–965
- Simpson A, Maniatis M, Jury F, et al. Polymorphisms in a disintegrin and metalloproteinase 33 (ADAM33) predict impaired early lung function. AmJ Respir Crit Care Med 2005; 172: 55–60
- Lee J-Y, Park S-W, Chang H K, et al. A disintegrin and metalloproteinase 33 in patients with asthma. Relationship to airflow limitation. AmJ Respir Crit Care Med 2006; 173: 729–735
- Gosman M ME, Boezen H M, van Dieman C C, et al. A disintegrin and metalloproteinase 33 and chronic obstructive pulmonary disease pathophysiology. Thorax 2007; 62: 242–247
- Zhang G, Hayden C M, Khoo S K, et al. Association of haplotypes of beta2-adrenoceptor polymorphisms with lung function and airway responsiveness in a pediatric cohort. Pediatr Pulmonol. 2006; 41: 1233–1241
- Matheson M C, Ellis J A, Raven J, Johns D P, Walters E H, Abramson M J. Beta2-adrenergic receptor polymorphisms are associated with asthma and COPD in adults. J Hum Genet. 2006; 51: 943–951
- Hall I P, Blakey J D, Al Balushi K A, et al. β 2-adrenoceptor polymorphisms and asthma from childhood to middle age in the British 1958 birth cohort: a genetic association study. Lancet 2006; 368: 771–779
- Warburton D, Gauldie J, Bellusci S, Shi W. Lung development and susceptibility to chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006; 3: 668–672
- Kranenburg A R, Willems-Widyastuti A, Mooi W J, et al. Chronic obstructive pulmonary disease is associated with enhanced bronchial expression of FGF-1, FGF-2 and FGFR-1. J Pathol 2005; 206: 28–38
- Gilliland F D, Gauderman J, Vora H, Rappaport E, Dubeau L. Effects of glutathione-S-transferase M1, P1 and T1 on childhood lung function growth. AmJ Respir Crit Care Med 2002; 166: 710–716
- He J Q, Ruan J, Connett J E, Anthonisen N R, Pare P D, Sandford A J. Antioxidant gene polymorphisms and susceptibility to a rapid decline in lung function in smokers. AmJ Respir Crit Care Med 2002; 166: 323–328
- Pelkonen A S, Hakulinen A L, Turpeinen M. Bronchial lability and responsiveness in school children born very preterm. AmJ Respir Crit Care Med 1997; 156: 1178–1184
- Mitchell S H, Teague W G. Reduced gas transfer at rest and during exercise in school-age survivors of bronchopulmonary dysplasia. AmJ Respir Crit Care Med 1998; 157: 1406–1412
- Chan K N, Wong Y C, Silverman M. Relationship between infant lung mechanics and childhood lung function in children of very low birth weight. Pediatr Pulmonol 1990; 8: 74–81
- Narang I, Baraldi E, Silverman M, Bush A. Airway function measurements and the long-term follow-up of survivors of preterm birth with and without chronic lung disease. Pediatr Pulmonol 2006; 41: 497–508
- Narang I, Cremonesini D, Rosenthal M, Bush A. Longitudinal evaluation of respiratory morbidity 21 years after low birthweight and premature birth. Eur RespirJ 2003; 22: S140, (Suppl 45)
- Narang I, Rosenthal M, Cremonesini D, Silverman M, Bush A. Evidence of programming in preterm, small for gestational age subjects 21 years after birth. Proc Am Thorac Soc 2005; 2: A606
- Gappa M, Berner M M, Hohenschild S, Dammann C E, Bartmann P. Pulmonary function at school-age in surfactant-treated preterm infants. Pediatr Pulmonol 1999; 27: 191–198
- Bush A. Update in pediatrics 2005. Am J Respir Crit Care Med 2006; 173: 585–592
- Bush A. Update in pediatric lung disease 2006. Am J Respir Crit Care Med 2007; 175: 532–540
- Hogg J C. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. AmJ Respir Crit Care Med 2001; 164: S71–75
- Schwarze J, O'Donnell D R, Rohwedder A, Openshaw P J. Latency and persistence of respiratory syncytial virus despite T cell immunity. AmJ Respir Crit Care Med 2004; 169: 801–805
- Wilson J, Rowlands K, Rockett K, Moore C, Lockhart E, Sharland M, Kwiatkowski D, Hull J. Genetic variation at the IL10 gene locus is associated with severity of respiratory syncytial virus bronchiolitis. J Infect Dis. 2005; 191: 1705–1709
- Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, Oron A, Itskovich S, Ballin A, Houri S, Beigelman A, Lider O, Rechavi G, Amariglio N. Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. Infect Dis. 2004; 189: 2057–2063
- Guerra S, Wright A L, Morgan W J, Sherrill D L, Holberg C J, Martinez F D. Persistence of Asthma Symptoms during Adolescence: Role of Obesity and Age at the Onset of Puberty. AmJ Respir Crit Care Med 2004; 170: 78–85
- Hijazi N, Abalkhail B, Seaton A. Diet and childhood asthma in a society in transition: a study in urban and rural Saudi Arabia. Thorax 2000; 55: 775–779
- Farchi S, Forastiere F, Agabiti N, Corbo G, Pistelli R, Fortes C, Dell'Orco V, Perucci C A. Dietary factors associated with wheezing and allergic rhinitis in children. Eur RespirJ 2003; 22: 772–780