Abstract
Chronic obstructive pulmonary disease (COPD) results in significant morbidity and mortality. Smoking has long been recognized as the primary risk factor for development of COPD, but factors determining the severity or pattern of disease in smokers are largely unknown. Recent interest has focused on the potential role of infectious agents and the associated host response in accelerating progression of airway obstruction or in perpetuating its progression following discontinuation of tobacco exposure. Pneumocystis jirovecii is a fungal pathogen that causes pneumonia in immunocompromised individuals. Recent evidence has linked this organism with COPD. Using sensitive molecular techniques, low levels of Pneumocystis have been detected in the respiratory tract of certain individuals and termed colonization. Several findings support the theory that colonization with Pneumocystis is involved in the “vicious circle” hypothesis of COPD in which colonization with organisms perpetuates an inflammatory and lung remodeling response. Pneumocystis colonization is more prevalent in smokers and in those with severe COPD. The presence of Pneumocystis in the lungs, even at low levels, produces inflammatory changes similar to those seen in COPD, with increases in numbers of neutrophils and CD8+ lymphocytes. HIV-infected subjects who have had PCP develop permanent airway obstruction, and HIV-infected patients have a high prevalence of both emphysema and Pneumocystis colonization. In addition, a non-human primate model of colonization shows development of airway obstruction and radiographic emphysema. Additional studies are needed to confirm the role of Pneumocystis in the pathogenesis of COPD, given that this agent might be a treatable co-factor in disease progression.
Key words: :
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in the United States (Citation[1]) and is expected to rank third in the world by 2020 (Citation[2]). In the year 2000, 2.7 million people died of the disease worldwide (Citation[3]). Twenty-four million adults in the United States have evidence of impaired lung function and experience 9.5 million office and emergency room visits, 726,000 hospitalizations, and 119,000 deaths each year (Citation[4]).
Despite efforts aimed at smoking cessation, little impact has been made on COPD incidence, and current treatments have a marginal impact in slowing progression of the disease. The enormous health burden and associated medical costs of COPD have prompted renewed interest in determining factors that alter disease progression. Although smoking is clearly the leading risk factor for COPD, the severity and patterns of disease expression vary widely (Citation[5]). In addition to the influences of genetics and the environment, there has been increased interest in the role of infections in the pathogenesis and progression of COPD (Citation[3]).
Recent evidence supports the hypothesis that infectious agents are critical in triggering and perpetuating the inflammatory response in COPD. Tissue inflammation in COPD is characterized by a predominant neutrophil, CD8+ lymphocyte, and macrophage infiltration (Citation[6], Citation[7], Citation[8], Citation[9]). It has been proposed that the mechanism of tissue damage involves the recruitment and activation of neutrophils, macrophages, and CD8+ T cells with concomitant upregulation of several cellular proteases and inflammatory cytokines. While smoking can stimulate inflammation in the lungs, smokers with COPD have an increased inflammatory response compared to smokers without COPD, and numbers of inflammatory cells, particularly neutrophils and CD8+ lymphocytes, correlate directly with degree of airflow obstruction (Citation[6], Citation[7], Citation[8], Citation[9]). In addition, such inflammation can persist despite smoking cessation and has been found to involve increases in cell types including B cells, CD4+ lymphocytes and eosinophils (Citation[10], Citation[11]). Furthermore, recent studies have documented the presence of inflammatory mucous in peripheral airways and lymphoid follicles associated with peripheral airways and in the parenchyma of patients with advanced COPD (Citation[12], Citation[13]). These observations suggest that an ongoing adaptive immune response exists, even in former smokers with advanced disease, and that microbial pathogens are agents that perpetuate the chronic inflammatory response in COPD.
Many pathogens such as adenovirus and Haemophilus influenzae have historically been associated with development and progression of COPD (Citation[10], Citation[14], Citation[15], Citation[16]). Over the past several years, increasing evidence has linked the pathogen Pneumocystis jirovecii (formerly Pneumocystis carinii) with COPD. This pathogen has not traditionally been associated with COPD, but several studies in humans and in a non-human primate model suggest that Pneumocystis is another pathogen that is responsible for differential progression of COPD in smokers. This finding is potentially important as Pneumocystis is treatable with inexpensive, common antibiotics. This review will briefly discuss the role of infections in general in COPD and will present the data supporting the role of Pneumocystis specifically.
ROLE OF INFECTIONS IN COPD
The vicious circle hypothesis
While overt bacterial infection has long been associated with symptomatic chronic bronchitis and acute bronchitis, it has recently been emphasized that inflammatory processes may occur in many patients with or without classic symptoms of sputum production. Cigarette smoke is commonly the initial inciting factor in the inflammatory cascade seen in COPD; however, smokers have varying degrees of inflammation, and ex-smokers can have persistence of inflammation and progression of airflow obstruction. These observations suggest that other agents, such as low level infections, likely act to amplify the smoking-induced inflammatory response. The hypothesis that micro-organisms perpetuate inflammation and lung destruction in COPD has been termed the vicious circle hypothesis (Citation[16]). The theory was originally proposed with bacteria, but could apply to any pathogen. In the vicious circle, cigarette smoke results in impaired mucociliary clearance and defective local immunity and leads to low level infection or colonization with micro-organisms.
The organisms in turn have several effects in the lung. They can further impair the ability of the respiratory system to clear infection, thus resulting in their persistence. The organisms also stimulate chronic inflammation that accelerates pulmonary decline with or without the persistence of tobacco exposure. Accentuation of the complex cellular inflammatory response leads to the release of a variety of cytokines and proteases. This inflammatory response in turn stimulates a variety of tissue responses including fibroblast proliferation with peribronchiolar fibrosis, mucosal hyperplasia, matrix destruction, and cell apoptosis leading to alveolar septal loss.
The role of bacteria in COPD
Several microbiologic agents including bacteria, viruses, and fungi have been proposed to play a role in progression of COPD. Bacteria have been associated with COPD progression, and the most commonly implicated bacteria are Haemophilus influenzae, Moraxella catarrhalis, Streptococcus pneumoniae, and Pseudomonas aeruginosa (Citation[17]). These pathogens can be found in patients with COPD in the stable state and have been detected in as many as 50% of exacerbations (Citation[17]). Pathogens have typically been identified by routine cultures, but polymerase chain reaction (PCR) is now commonly used. As bacterial load increases, forced expiratory volume in one second (FEV1) falls (Citation[18]). Bacterial colonization has been associated with greater sputum purulence, increased sputum neutrophils, and increased levels of interleukin (IL)-8, tumor necrosis factor (TNF)-α, and neutrophil elastase (Citation[18], Citation[19], Citation[20], Citation[21]). Colonization is also associated with a greater degree of airway obstruction, and colonized patients have more exacerbations (Citation[21], Citation[22]).
The role of viruses in COPD
Viruses might also be important in the pathogenesis of COPD. Retamales and colleagues reported a higher rate of detection of adenovirus E1A protein in lung tissue of smokers with emphysema compared to age- and smoking-matched controls without evidence of airway obstruction (Citation[10]). Based on elevated levels of CD8+ lymphocytes and interferon (IFN)-γ in lung samples, the authors speculated that the presence of latent adenovirus in the lungs provokes a heightened inflammatory response that leads to worsening of airway obstruction. Data in animals support this hypothesis, as guinea pigs with adenoviral infections have a more pronounced inflammatory response and accelerated emphysema development compared to non-infected controls (Citation[23]).
Several other viruses seem to be important in COPD, particularly in exacerbations, half of which are associated with PCR-detected viruses (Citation[24]). Exacerbations associated with viruses are more severe and last longer than those without a viral trigger (Citation[25], Citation[26]). Because exacerbations are associated with more rapid progression of COPD over time, these viruses might be important in overall disease course as well. Rhinovirus is the most frequent virus identified during exacerbations and has been detected in one quarter to over half of subjects with COPD exacerbations (Citation[25]). Experimental rhinovirus inoculation of subjects with COPD results in increased respiratory symptoms and airway obstruction (Citation[27]). Respiratory syncytial virus (RSV) might also play a role in COPD as it has been detected in patients both during stable disease and exacerbations (Citation[25]).
Using PCR, Influenza A and B have been found in 16% of patients' nasal aspirates during exacerbation (Citation[25]). Human metapneumovirus is a recently described virus that was found by real time-PCR in nasopharyngeal specimens of 12% of patients hospitalized with COPD exacerbations (Citation[28]). These viruses likely worsen symptoms and progression of COPD via upregulation of pulmonary inflammation. Influenza leads to increases in IL-6, IL-8, and RANTES in bronchial epithelial cells (Citation[29]). RSV also results in increased cytokine expression in airway epithelial cells (Citation[30]).
The role of Pneumocystis in COPD
In the past several years, new data have emerged implicating Pneumocystis as another pathogen with a potentially important role in COPD progression in certain patients. The evidence linking Pneumocystis and COPD includes an increase in Pneumocystis colonization in COPD that corresponds to disease severity and is independent of smoking history, the high rates of both emphysema and Pneumocystis colonization in HIV-infected smokers, COPD-like changes that occur after Pneumocystis pneumonia (PCP), the similarity between the inflammatory response in Pneumocystis colonization and COPD, and the development of airway obstruction and emphysema in a non-human primate model of Pneumocystis colonization (). We will first review the epidemiology of Pneumocystis colonization and then outline the specific data implicating Pneumocystis in COPD.
Table 1 Evidence linking Pneumocystis colonization and COPD
Pneumocystis colonization
Pneumocystis is a eukaryotic pathogen that causes pneumonia in immunocompromised hosts. Despite the fact that PCP is responsible for a large degree of morbidity and mortality in immunosuppressed populations, little is understood about the organism's epidemiology. The historic inability to culture Pneumocystis reliably has severely limited study of its life cycle and transmission. Originally classified as a protozoan, genetic analysis has found greater homology between Pneumocystis and certain fungi (Citation[31], Citation[32], Citation[33]). The infectious form of the organism has not been identified, and routes of transmission are not well understood. Pneumocystis appears to be ubiquitous in the environment as nearly all children develop anti-Pneumocystis antibodies early in life (Citation[34], Citation[35], Citation[36]). Traditionally, PCP has been thought to result from reactivation of latent infection acquired during childhood. This theory has never been definitively proven, and active or de novo infection from person-to-person exposure or from exposure to environmental sources of Pneumocystis may also be responsible for infections. In addition, studies in laboratory animals have shown direct transmission among animals (Citation[37], Citation[38], Citation[39], Citation[40]).
The development of molecular techniques to identify and genotype Pneumocystis has provided an important tool with which to explore its epidemiology. PCR detects low numbers of organisms, particularly in cases where routine histochemical staining methods are negative (Citation[41], Citation[42]). Use of PCR, particularly nested PCR, for detection of Pneumocystis has led to discovery of the organism among asymptomatic subjects. Presence of Pneumocystis in respiratory specimens from subjects without signs or symptoms of clinical infection and who do not progress to infection has been defined as colonization. In healthy subjects, Pneumocystis colonization is uncommon. Several studies have failed to detect any evidence of Pneumocystis colonization in autopsy studies of lungs from non-immunocompromised subjects without lung disease (Citation[42], Citation[43], Citation[44], Citation[45]).
Pneumocystis colonization may occur in patients who are immunocompromised for reasons other than HIV infection. Nevez et al. examined bronchoscopic alveolar lavage (BAL) fluid from 82 patients whose immunosuppression was caused either by use of corticosteroids or by an underlying malignancy (Citation[46]). Thirteen (16%) of these samples were positive for Pneumocystis by non-nested PCR. The prevalence of colonization in asymptomatic HIV-infected subjects appears to be higher than that seen in the general population with estimates ranging from 12 to 46% depending on the subject population and respiratory samples studied (Citation[42], Citation[44], Citation[47], Citation[48], Citation[49], Citation[50]). Colonization appears to be more frequent in symptomatic HIV-infected persons. A recent study found that 69% of HIV-infected subjects who presented with respiratory symptoms, but were found not to have PCP by traditional staining methods, actually had BAL or induced sputum that contained Pneumocystis DNA by nested PCR (Citation[51]).
Pneumocystis colonization is increased in COPD and correlates with disease severity
The first link of Pneumocystis to COPD came from epidemiologic studies demonstrating that Pneumocystis colonization is higher in patients with COPD than healthy subjects or those with other lung disorders (). Calderon and colleagues initially reported that 10% of patients with chronic bronchial disease were colonized with Pneumocystis (Citation[52]). They later studied 37 patients with chronic bronchitis and found that 41% of them were colonized (Citation[53]). The increased colonization seen in COPD is not just a function of lung disease, but seems specific to COPD. For example, two studies have reported about 7% colonization in cystic fibrosis patients, whereas patients with COPD had a colonization frequency around 40% (Citation[54], Citation[55]). A recent study by Helweg-Larsen and colleagues examined HIV-negative patients admitted to the hospital with suspected bacterial pneumonia (Citation[56]). Of these subjects, 16 of 367 (4%) were colonized with Pneumocystis, and 63% of these patients had COPD compared to 20% of non-colonized patients. Morris and colleagues examined the prevalence of Pneumocystis colonization in patients undergoing lung transplantation for severe COPD or other end-stage lung diseases (Citation[57]). They performed PCR on the native lung tissue removed at time of transplant and found that 37% of those with severe COPD were Pneumocystis-colonized compared to only 9% of those with other severe lung diseases. In multivariate analysis, a diagnosis of COPD was the only important predictor of colonization (odds ratio (OR) = 7.3, 95% confidence interval (CI) = 2.4–22.4, p < 0.001). Other clinical characteristics such as age, use of oral corticosteroids, and use of trimethoprim-sulfamethoxazole were not related to colonization risk. Not all studies have found the same association with colonization and COPD, however. A study by Maskell and colleagues found that colonization was associated with use of oral corticosteroids and not with COPD, but this study examined subjects with very mild airway obstruction and these subjects may have been less likely to be colonized (Citation[58]).
Table 2 Summary of studies reporting prevalence of Pneumocystis colonization in COPD
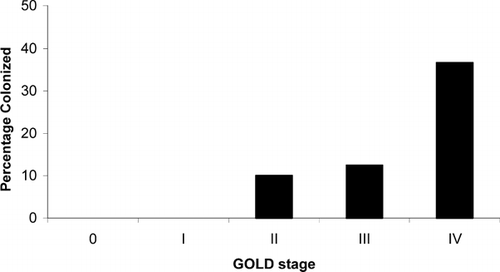
One of the strongest pieces of evidence linking Pneumocystis colonization and progression of COPD is the finding that colonization correlates with degree of airway obstruction independent of smoking history. Morris and colleagues examined Pneumocystis colonization in lung samples of patients with varying degrees of airway obstruction as characterized by Global Health Initiative on Obstructive Lung Disease (GOLD) stage, but who had similar smoking histories (Citation[57]). Colonized subjects had significantly lower FEV1 and FEV1/forced vital capacity (FVC) percent predicted () and colonization was more common as GOLD stage increased (). Over a third (36.7%) of those with severe COPD (GOLD stage IV) were colonized compared to 5.3% of those with less severe COPD (stages 0 to III) (Citation[57]). Although this study does not prove causality, it strongly suggests that Pneumocystis colonization affects COPD progression.
Figure 1 Comparison of FEV1 and FEV1/FVC in Pneumocystis-colonized subjects and Pneumocystis negative. p = 0.006 for both comparisons. Abbreviations: FVC, forced vital capacity; FEV1, forced expiratory volume in 1 second, percent predicted.
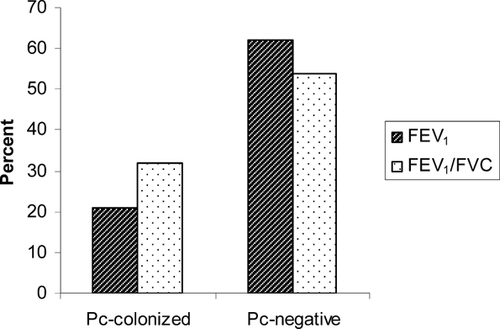
Figure 2 Association of GOLD stage and prevalence of Pneumocystis colonization. Reproduced with permission from American Journal of Respiratory and Critical Care Medicine. Abbreviations: GOLD, Global Health Initiative on Obstructive Lung Disease.
Increased emphysema and Pneumocystis colonization in HIV-infected smokers
Another line of evidence linking Pneumocystis colonization and COPD is the tendency for HIV-infected subjects to develop both emphysema and colonization. HIV-infected subjects are at increased risk for development of emphysema, particularly if they smoke (Citation[59], Citation[60]). Diaz reported that almost 40% of HIV-infected smokers without a history of pulmonary infections had emphysema by pulmonary function and/or chest computed tomography (CT)-scanning (Citation[60]). No HIV-negative controls matched by smoking history had emphysema by either pulmonary function measures or chest CT. CD8+ T lymphocyte levels in BAL were also significantly increased in those HIV-infected smokers with emphysema compared to HIV-positive and HIV-negative smokers without emphysema.
Another study found that a significantly higher number of HIV-infected subjects than HIV-negative controls had evidence of focal air-trapping on CT scan (Citation[59]). Subjects with air-trapping also had worse obstructive changes in pulmonary function including lower FEV1 and diffusing capacity for carbon monoxide (Dlco). Even HIV-infected subjects who do not smoke appear to have an increased risk for developing emphysema. Recent data from a multicenter study of autopsy lungs in HIV infection has shown that 16% of patients with HIV infection who were non-smokers had pathological emphysema (Morris, unpublished data). Although there was no HIV-negative control group, this prevalence of emphysema in a non-smoking population is much higher than would be expected. It has been postulated that latent or subclinical infections might play a role in the development of HIV-associated emphysema, but there have been no studies to date that have explored this hypothesis.
It is possible that Pneumocystis is one such subclinical infection that might play a role in the accelerated emphysema seen in HIV infection. Morris and colleagues have investigated the epidemiology of Pneumocystis colonization in HIV-infected subjects (Citation[50]). They examined lung specimens from subjects who died from causes other than PCP and found that 46% of HIV-infected subjects had Pneumocystis detectable in their lungs. Interestingly, clinical factors such as degree of immunosuppression based on CD4 cell count, history of previous PCP, or use of PCP prophylaxis did not influence colonization risk. One of the strongest independent risk factor for Pneumocystis colonization was having a history of cigarette smoking (OR = 4.5, 95% CI = 1.27–15.6, p = 0.02). Although lung function data were not available in this population, this observation raises the possibility that the accelerated emphysema seen in HIV-infected smokers may in fact be related to Pneumocystis colonization.
PCP produces permanent COPD-like changes
Pulmonary function changes have not been studied in the setting of Pneumocystis colonization, but acute PCP has been associated with the development of obstructive pulmonary changes. A case series of 10 patients with PCP reported a high incidence of small airways dysfunction as demonstrated by a reduced forced expiratory flow (FEF) 25–75% (Citation[61]). Another study of 169 patients with HIV found that DLco, FEV1, and peak flow were decreased in those with acute PCP (Citation[62]). These obstructive changes appear to be permanent and not just a function of acute infection. The Pulmonary Complications of HIV Infection Study, a prospective cohort of over 1,100 HIV-infected subjects followed for a median of four years, performed serial pulmonary function studies in subjects at pre-defined intervals and after an episode of pneumonia. In a study of pulmonary function after PCP in this cohort, HIV-infected subjects with PCP had accelerated declines in FEV1, FEV1/FVC, and DLco beyond that expected from age and smoking history (Citation[63]). These changes in pulmonary function were indistinguishable from those seen clinically with COPD and persisted for years after the acute infection resolved.
Similarity of the inflammatory response in COPD and Pneumocystis colonization
Airflow limitation in COPD is progressive and associated with an abnormal inflammatory response in the lungs. While the inflammatory response in COPD is complex and heterogeneous, it most often includes neutrophil, CD8+ lymphocyte and macrophage infiltration (Citation[7], Citation[9], Citation[64], Citation[65]). It has long been proposed that the mechanism of tissue damage involves the recruitment and activation of these inflammatory cells with concomitant upregulation of several cellular proteases and inflammatory cytokines. These cells seem to play an important role in development of disease as the numbers of CD8+ lymphocytes and neutrophils are correlated with disease severity (Citation[7], Citation[8], Citation[9], Citation[10], Citation[64], Citation[66]). Macrophages produce chemokines that lead to an influx of neutrophils and CD8+ T-lymphocytes. The neutrophils are potent producers of proteases that may contribute to tissue destruction. The CD8+ T-lymphocytes in turn produce cytokines such as IFN-γ and IL-8 which perpetuate recruitment and activation of macrophage and neutrophils (Citation[67], Citation[68], Citation[69]). In fact, the lymphocyte in the context of lymphoid follicles may be responsible for the primary events initiating the interaction with microbial antigens, thus precipitating the inflammatory cascade (Citation[70]). TNF-α production by macrophages and CD8+ T-lymphocytes is also elevated and contributes to the inflammatory process by activating nuclear factor (NF)-κ β. NF-κ β subsequently upregulates a number of inflammatory molecules, including IL-8 and proteases.
One hypothesis of COPD pathogenesis is the protease/antiprotease theory (Citation[71]). Several proteases, particularly neutrophil elastase and matrix metalloproteases likely play a role in the degradation of extracellular matrix proteins, particularly elastin and collagen, leading to the tissue damage observed. In addition, it has been proposed, based on the increased incidence of emphysema in patients deficient in alpha-one antitrypsin, that an imbalance between proteases and their inhibitors results in tissue injury. Increased levels of cytokines and chemokines, particularly the neutrophil chemoattractants, IL-8 and leukotriene (LT) B4, as well as TNF-α and IFN-γ, have been reported to contribute to emphysematous changes both in humans and in animal models of COPD (Citation[72]).
Acute PCP elicits a pronounced pulmonary inflammatory response characterized by increases in neutrophils and lymphocytes, primarily CD8+ T cells. There is an enhanced production of pro-inflammatory cytokines such as IFN-γ, TNF-α and IL-8 (Citation[73]). The immune response in colonized humans has not been studied, but preliminary studies indicate that IFN-γ and TNF-α are increased in BAL samples of non-HIV-infected subjects colonized with Pneumocystis (Morris, unpublished data). In a model of Pneumocystis-colonized rhesus macaques, a cascade of cellular infiltrates and mediator release that is similar to that seen in acute PCP occurs. Board and coworkers examined the immune response to Pneumocystis colonization in a simian model of AIDS in which simian immunodeficiency (SIV)-infected monkeys were inoculated with macaque-derived Pneumocystis (Citation[74]). Some animals developed PCP while others remained in a protracted state of asymptomatic colonization.
The early period after inoculation was marked by an influx of CD8+ T cells and neutrophils regardless of whether fulminant PCP or asymptomatic colonization resulted. Interestingly, CD8+ cell and neutrophil infiltration persisted throughout the course of infection in those animals with prolonged colonization and those who progressed to acute PCP. IFN-γ TNF-α and IL-8 also increased during colonization (Citation[75]). The intensity and persistence of the inflammatory response seen in this model raises the possibility that Pneumocystis-induced lung damage results from colonization. Such inflammation may play either a causative or adjunctive role in the evolution of lung diseases in which Pneumocystis colonization is common.
In humans, the systemic inflammatory response to Pneumocystis colonization has been examined. A study of non-immunosuppressed individuals with chronic bronchial disease found that those patients colonized with Pneumocystis had a significantly higher peripheral lymphocyte count than those not colonized. In addition, the mean CD4+ count also appeared higher in the colonized individuals. While the exact clinical significance of this finding was unclear, it was postulated that the increase in the CD4+ cells and overall lymphocyte counts seen in those colonized by Pneumocystis might be related to exacerbations of the underlying chronic bronchitis.
Pneumocystis colonization may also contribute to the development of emphysema through release of endogenous proteases and stimulation of protease release in the host lung. Increased protease activity occurs in the lungs during acute PCP and may be responsible for some of the parenchymal destruction seen in the disease. Host-derived matrix metalloproteases (MMP)-2 and 9 are increased during acute PCP (Citation[77], Citation[78]). Pneumocystis-derived proteases may also contribute to lung destruction. Rat Pneumocystis has protease, elastase, and chymase activity that can degrade the alveolar-capillary interface (Citation[77], Citation[79]). A novel collagenase has also been identified in the lungs of rats with PCP, and it is thought that the enzyme is Pneumocystis-derived (Citation[77]). Pneumocystis also has kexins, a family of serine proteases, on its cell surface (Citation[80], Citation[81], Citation[82]). Although the function of kexin is not completely understood, it may also contribute to lung damage from Pneumocystis.
Pneumocystis colonization and airway obstruction in a simian model of AIDS
Few studies have addressed the clinical significance of prolonged Pneumocystis colonization and whether colonization results in pulmonary damage. The SIV/Pneumocystis primate model offers the opportunity to investigate the consequences of persistent colonization. As discussed above, Pneumocystis-induced inflammatory response in the lungs of SIV-infected macaques is evident very early after Pneumocystis inoculation, persists for weeks to months before any respiratory symptoms associated with acute PCP are observed and might result in airflow obstruction (Citation[74]).
To assess airflow obstruction as a consequence of SIV infection or SIV infection with Pneumocystis colonization, pulmonary physiologic maneuvers using whole body plethysmography and a forced deflation technique were performed longitudinally in SIV-infected and SIV-infected/Pneumocystis-colonized animals (Citation[85]). SIV-infected, Pneumocystis-colonized monkeys had significant airflow obstruction compared to normal or SIV-infected monkeys, supporting the hypothesis that colonization with Pneumocystis in the context of SIV-induced immunosuppression leads to pulmonary impairment.
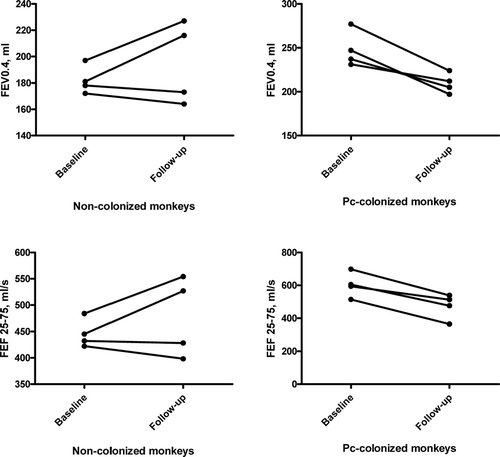
This model was modified using simian immunodeficiency/human immunodeficiency chimeric virus-infected cynomolgus macaques and spontaneous development of Pneumocystis colonization produced by exposure to infected monkeys (Citation[85]). To further test the link between Pneumocystis colonization and lung disease, longitudinal studies of pulmonary function in these animals were performed comparing changes in SHIV-infected alone to SHIV-infected/Pneumocystis-colonized animals (Citation[85]). Pneumocystis-colonized animals developed airway obstruction as determined by decreases in most pulmonary function parameters while SHIV-infected animals did not change significantly (). These changes demonstrated that Pneumocystis colonization leads to the development of airflow obstruction.
Figure 3 Change in forced expiratory volumes and forced expiratory flow values for non-colonized monkeys from baseline (uninfected) to follow-up (SHIV-infected, Pneumocystis-negative) and for Pneumocystis-colonized monkeys from baseline (uninfected) to follow-up (SHIV-infected, Pneumocystis-colonized). Average follow-up of 8 months. * p = 0.02 for comparison of change in FEV0.4 and FEF25 − 75 in non-colonized versus Pneumocystis-colonized. Abbreviations: FEV0.4, forced expiratory volume during first 0.4 seconds; FEF25 − 75, forced expiratory flow over mid 50% of forced vital capacity; Pc, Pneumocystis.
CONCLUSION
Despite the enormous public health burden of COPD, we still do not understand the factors that determine differential development and progression of disease in smokers. Various microbes may be involved in the pathogenesis of COPD by perpetuating the “vicious circle” of airway colonization, inflammation, and destruction. Pneumocystis is an agent recently proposed to play a role in COPD by inducing inflammatory changes that contribute to development and progression of disease. The presence of Pneumocystis in the lung may lead to chronic lung injury and worsening of pulmonary function. Additional studies are needed to better define the role of Pneumocystis in COPD and to determine the exact mechanism of its effects. Understanding the role of Pneumocystis colonization may help us identify a novel, treatable risk factor for progression of COPD.
Supported by NIH HL072837, HL083461 (AM), HL077095 (KN) HL083462 (KN), and HL084948 (FCS).
REFERENCES
- Petty T L. Scope of the COPD problem in North America: early studies of prevalence and NHANES III data: basis for early identification and intervention. Chest 2000; 117: 326S–331S
- Lopez A D, Murray C C. The global burden of disease, 1990–2020. Nat Med 1998; 4: 1241–1243
- Pauwels R A, Buist A S, Ma P, Jenkins C R, Hurd S S. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: National Heart, Lung, and Blood Institute and World Health Organization Global Initiative for Chronic Obstructive Lung Disease (GOLD): executive summary. Respir Care 2001; 46: 798–825
- Mannino D M, Homa D M, Akinbami L J, Ford E S, Redd S C. Chronic obstructive pulmonary disease surveillance–United States, 1971–2000. MMWR Surveill Summ 2002; 51: 1–16
- Buist A S, Connett J E. The Lung Health Study. Baseline characteristics of randomized participants. Chest 1993; 103: 1644
- Lacoste J Y, Bousquet J, Chanez P, Van Vyve T, Simony-Lafontaine J, Lequeu N, Vic P, Enander I, Godard P, Michel F B. Eosinophilic and neutrophilic inflammation in asthma, chronic bronchitis, and chronic obstructive pulmonary disease. J Allergy Clin Immunol 1993; 92: 537–548
- Keatings V M, Collins P D, Scott D M, Barnes P J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. AmJ Respir Crit Care Med 1996; 153: 530–534
- O'Shaughnessy T C, Ansari T W, Barnes N C, Jeffery P K. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. AmJ Respir Crit Care Med 1997; 155: 852–857
- Saetta M, Di Stefano A, Turato G, Facchini F M, Corbino L, Mapp C E, Maestrelli P, Ciaccia A, Fabbri L M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. AmJ Respir Crit Care Med 1998; 157: 822–826
- Retamales I, Elliott W M, Meshi B, Coxson H O, Pare P D, Sciurba F C, Rogers R M, Hayashi S, Hogg J C. Amplification of inflammation in emphysema and its association with latent adenoviral infection. AmJ Respir Crit Care Med 2001; 164: 469–473
- Rutgers S R, Timens W, Kauffman H F, Postma D S. Markers of active airway inflammation and remodelling in chronic obstructive pulmonary disease. Clin Exp Allergy 2001; 31: 193–205
- Hogg J C, Chu F, Utokaparch S, Woods R, Elliott W M, Buzatu L, Cherniack R M, Rogers R M, Sciurba F C, Coxson H O, Pare P D. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N EnglJ Med 2004; 350: 2645–2653
- Kerstjens H, Postma D, ten Hacken N. Chronic obstructive pulmonary disease. Clin Evid 2006; 2077–2100
- Matsuse T, Hayashi S, Kuwano K, Keunecke H, Jefferies W A, Hogg J C. Latent adenoviral infection in the pathogenesis of chronic airways obstruction. Am Rev Respir Dis 1992; 146: 177–184
- Vitalis T Z, Kern I, Croome A, Behzad H, Hayashi S, Hogg J C. The effect of latent adenovirus 5 infection on cigarette smoke-induced lung inflammation. Eur RespirJ 1998; 11: 664–669
- Sethi S. Bacterial infection and the pathogenesis of COPD. Chest 2000; 117: 286S–291S
- Sethi S. Bacteria in exacerbations of chronic obstructive pulmonary disease: phenomenon or epiphenomenon?. Proc Am Thorac Soc 2004; 1: 109–114
- Wilkinson T M, Patel I S, Wilks M, Donaldson G C, Wedzicha J A. Airway bacterial load and FEV1 decline in patients with chronic obstructive pulmonary disease. AmJ Respir Crit Care Med 2003; 167: 1090–1095
- Sethi S. Infectious etiology of acute exacerbations of chronic bronchitis. Chest 2000; 117: 380S–385S
- Stockley R A, Hill A T, Hill S L, Campbell E J. Bronchial inflammation: its relationship to colonizing microbial load and alpha(1)-antitrypsin deficiency. Chest 2000; 117: 291S–293S
- Patel I S, Seemungal T A, Wilks M, Lloyd-Owen S J, Donaldson G C, Wedzicha J A. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax 2002; 57: 759–764
- Zalacain R, Sobradillo V, Amilibia J, Barron J, Achotegui V, Pijoan J I, Llorente J L. Predisposing factors to bacterial colonization in chronic obstructive pulmonary disease. Eur RespirJ 1999; 13: 343–348
- Meshi B, Vitalis T Z, Ionescu D, Elliott W M, Liu C, Wang X D, Hayashi S, Hogg J C. Emphysematous lung destruction by cigarette smoke. The effects of latent adenoviral infection on the lung inflammatory response. AmJ Respir Cell Mol Biol 2002; 26: 52–57
- Rohde G, Wiethege A, Borg I, Kauth M, Bauer T T, Gillissen A, Bufe A, Schultze-Werninghaus G. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax 2003; 58: 37–42
- Seemungal T, Harper-Owen R, Bhowmik A, Moric I, Sanderson G, Message S, Maccallum P, Meade T W, Jeffries D J, Johnston S L, Wedzicha J A. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. AmJ Respir Crit Care Med 2001; 164: 1618–1623
- Papi A, Bellettato C M, Braccioni F, Romagnoli M, Casolari P, Caramori G, Fabbri L M, Johnston S L. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. AmJ Respir Crit Care Med 2006; 173: 1114–1121
- Mallia P, Message S D, Kebadze T, Parker H L, Kon O M, Johnston S L. An experimental model of rhinovirus induced chronic obstructive pulmonary disease exacerbations: a pilot study. Respir Res 2006; 7: 116
- Martinello R A, Esper F, Weibel C, Ferguson D, Landry M L, Kahn J S. Human metapneumovirus and exacerbations of chronic obstructive pulmonary disease. J Infect 2006; 53: 248–254
- Matsukura S, Kokubu F, Noda H, Tokunaga H, Adachi M. Expression of IL-6, IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced by influenza virus A. J Allergy Clin Immunol 1996; 98: 1080–1087
- Zhang Y, Luxon B A, Casola A, Garofalo R P, Jamaluddin M, Brasier A R. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J Virol 2001; 75: 9044–9058
- Edman J C, Kovacs J A, Masur H, Santi D V, Elwood H J, Sogin M L. Ribosomal RNA sequence shows Pneumocystis carinii to be a member of the fungi. Nature 1988; 334: 519–522
- Stringer J R, Edman J C, Cushion M T, Richards F F, Watanabe J. The fungal nature of Pneumocystis. J Med Vet Mycol 1992; 30: 271–278, Suppl 1
- Stringer J R. Pneumocystis carinii: what is it, exactly?. Clin Microbiol Rev 1996; 9: 489–498
- Meuwissen J H, Tauber I, Leeuwenberg A D, Beckers P J, Sieben M. Parasitologic and serologic observations of infection with Pneumocystis in humans. J Inf Dis 1977; 136: 43–49
- Pifer L L, Hughes W T, Stagno S, Woods D. Pneumocystis carinii infection: evidence for high prevalence in normal and immunosuppressed children. Pediatrics 1978; 61: 35–41
- Wakefield A E, Stewart T J, Moxon E R, Marsh K, Hopkin J M. Infection with Pneumocystis carinii is prevalent in healthy Gambian children. Trans Royal Soc Trop Med Hygiene 1990; 84: 800–802
- Hughes W T. Natural mode of acquisition for de novo infection with Pneumocystis carinii. J Infect Dis 1982; 145: 842–848
- Powles M A, McFadden D C, Pittarelli L A, Schmatz D M. Mouse model for Pneumocystis carinii pneumonia that uses natural transmission to initiate infection. Infect Immun 1992; 60: 1397–1400
- Ceré N, Polack B, Chanteloup N K, Coudert P. Natural transmission of Pneumocystis carinii in nonimmunosuppressed animals: early contagiousness of experimentally infected rabbits (Oryctolagus cuniculus). J Clin Micro 1997; 35: 2670–2672
- Gigliotti F, Harmsen A G, Wright T W. Characterization of transmission of Pneumocystis carinii f. sp. muris through immunocompetent BALB/c mice. Infect Immun 2003; 71: 3852–3856
- Wakefield A E, Pixley F J, Banerji S, Sinclair K, Miller R F, Moxon E R, Hopkin J M. Detection of Pneumocystis carinii with DNA amplification. Lancet 1990; 336: 451–453
- Wakefield A E, Pixley F J, Banerji S, Sinclair K, Miller R F, Moxon E R, Hopkin J M. Amplification of mitochondrial ribosomal RNA sequences from Pneumocystis carinii DNA of rat and human origin. Mol Biochem Parasitol 1990; 43: 69–76
- Peters S E, Wakefield A E, Sinclair K, Millard P R, Hopkin J M. A search for Pneumocystis carinii in post-mortem lungs by DNA amplification. J Pathol 1992; 166: 195–198
- Leigh T R, Kangro H O, Gazzard B G, Jeffries D J, Collins J V. DNA amplification by the polymerase chain reaction to detect sub-clinical Pneumocystis carinii colonization in HIV-positive and HIV-negative male homosexuals with and without respiratory symptoms. Respir Med 1993; 87: 525–529
- Tamburrini E, Ortona E, Visconti E, Margutti P, Mencarini P, Zolfo M, Marinaci S, Siracusano A. Detection of Pneumocystis carinii in oropharyngeal washings by PCR-SHELA and nested PCR. J Eukaryot Microbiol 1997; 44: 48S
- Nevez G, Raccurt C, Vincent P, Jounieaux V, Dei-Cas E. Pulmonary colonization with Pneumocystis carinii in human immunodeficiency virus-negative patients: assessing risk with blood CD4+ T cell counts. Clin Infect Dis 1999; 29: 1331–1332
- Olsson M, Elvin K, Lofdahl S, Linder E. Detection of Pneumocystis carinii DNA in sputum and bronchoalveolar lavage samples by polymerase chain reaction. J Clin Microbiol 1993; 31: 221–226
- Leibovitz E, Pollack H, Moore T, Papellas J, Gallo L, Krasinski K, Borkowsky W. Comparison of PCR and standard cytological staining for detection of Pneumocystis carinii from respiratory specimens from patients with or at high risk for infection by human immunodeficiency virus. J Clin Microbiol 1995; 33: 3004–3007
- Matos O, Costa M C, Lundgren B, Caldeira L, Aguiar P, Antunes F. Effect of oral washes on the diagnosis of Pneumocystis carinii pneumonia with a low parasite burden and on detection of organisms in subclinical infections. Eur J Clin Microbiol Infect Dis 2001; 20: 573–575
- Morris A, Kingsley L A, Groner G, Lebedeva I P, Beard C B, Norris K A. Prevalence and clinical predictors of Pneumocystis colonization among HIV-infected men. AIDS 2004; 18: 793–798
- Huang L, Crothers K, Morris A, Groner G, Fox M, Turner J R, Merrifield C, Eiser S, Zucchi P, Beard C B. Pneumocystis colonization in HIV-infected patients. J Eukaryot Microbiol 2003; 50: 616–617, Suppl
- Calderon E J, Regordan C, Medrano F J, Ollero M, Varela J M. Pneumocystis carinii infection in patients with chronic bronchial disease. Lancet 1996; 347: 977
- Calderon E, de la Horra C, Medrano F J, Lopez-Suarez A, Montes-Cano M A, Respaldiza N, Elvira-Gonzalez J, Martin-Juan J, Bascunana A, Varela J M. Pneumocystis jiroveci isolates with dihydropteroate synthase mutations in patients with chronic bronchitis. Eur J Clin Microbiol Infect Dis 2004; 23: 545–549
- Sing A, Roggenkamp A, Autenrieth I B, Heesemann J. Pneumocystis carinii carriage in immunocompetent patients with primary pulmonary disorders as detected by single or nested PCR. J Clin Microbiol 1999; 37: 3409–3410
- Probst M, Ries H, Schmidt-Wieland T, Serr A. Detection of Pneumocystis carinii DNA in patients with chronic lung diseases. Eur J Clin Microbiol Infect Dis 2000; 19: 644–645
- Helweg-Larsen J, Jensen J S, Dohn B, Benfield T L, Lundgren B. Detection of Pneumocystis DNA in samples from patients suspected of bacterial pneumonia–a case-control study. BMC Infect Dis 2002; 2: 28
- Morris A, Sciurba F C, Lebedeva I P, Githaiga A, Elliott W M, Hogg J C, Huang L, Norris K A. Association of chronic obstructive pulmonary disease severity and Pneumocystis colonization. Am J Respir Crit Care Med 2004; 170: 408–413
- Maskell N A, Waine D J, Lindley A, Pepperell J C, Wakefield A E, Miller R F, Davies R J. Asymptomatic carriage of Pneumocystis jiroveci in subjects undergoing bronchoscopy: a prospective study. Thorax 2003; 58: 594–597
- Diaz P T, Clanton T L, Pacht E R. Emphysema-like pulmonary disease associated with human immunodeficiency virus infection. Ann Intern Med 1992; 116: 124–128
- Diaz P T, King M A, Pacht E R, Wewers M D, Gadek J E, Nagaraja H N, Drake J, Clanton T L. Increased susceptibility to pulmonary emphysema among HIV-seropositive smokers. Ann Intern Med 2000; 132: 369–372
- Fleischman J K, Greenberg H, Web A. Small airways dysfunction in patients with AIDS and Pneumocystis carinii pneumonia. AIDS Patient Care STDS 1996; 10: 16–20
- Shaw R J, Roussak C, Forster S M, Harris J R, Pinching A J, Mitchell D M. Lung function abnormalities in patients infected with the human immunodeficiency virus with and without overt pneumonitis. Thorax 1988; 43: 436–440
- Morris A M, Huang L, Bacchetti P, Turner J, Hopewell P C, Wallace J M, Kvale P A, Rosen M J, Glassroth J, Reichman L B, Stansell J D. Permanent declines in pulmonary function following pneumonia in human immunodeficiency virus-infected persons. The Pulmonary Complications of HIV Infection Study Group. AmJ Respir Crit Care Med 2000; 162: 612–616
- Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, Mapp C E, Fabbri L M, Donner C F, Saetta M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. AmJ Respir Crit Care Med 1998; 158: 1277–1285
- Pesci A, Balbi B, Majori M, Cacciani G, Bertacco S, Alciato P, Donner C F. Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur RespirJ 1998; 12: 380–386
- Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, Cavallesco G, Tropeano G, Mapp C E, Maestrelli P, Ciaccia A, Fabbri L M. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. AmJ Respir Crit Care Med 1999; 160: 711–717
- Barnes P J, Cosio M G. Characterization of T lymphocytes in chronic obstructive pulmonary disease. PLoS Med 2004; 1: e20
- Grumelli S, Corry D B, Song L Z, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis D E, Kheradmand F. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med 2004; 1: e8
- Barcelo B, Pons J, Fuster A, Sauleda J, Noguera A, Ferrer J M, Agusti A G. Intracellular cytokine profile of T lymphocytes in patients with chronic obstructive pulmonary disease. Clin Exp Immunol 2006; 145: 474–479
- Gadgil A, Zhu X, Sciurba F C, Duncan S R. Altered T-cell phenotypes in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006; 3: 487–488
- Tetley T D. New perspectives on basic mechanisms in lung disease. 6. Proteinase imbalance: its role in lung disease. Thorax 1993; 48: 560–565
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur RespirJ 2003; 22: 672–688
- Wright T W, Johnston C J, Harmsen A G, Finkelstein J N. Analysis of cytokine mRNA profiles in the lungs of Pneumocystis carinii-infected mice. Am J Respir Cell Mol Biol 1997; 17: 491–500
- Board K F, Patil S, Lebedeva I, Capuano S, 3rd, Trichel A M, Murphey-Corb M, Rajakumar P A, Flynn J L, Haidaris C G, Norris K A. Experimental Pneumocystis carinii pneumonia in simian immunodeficiency virus-infected rhesus macaques. J Infect Dis 2003; 187: 576–588
- Patil S P, Board K F, Lebedeva I P, Norris K A. Immune responses to Pneumocystis colonization and infection in a simian model of AIDS. J Eukaryot Microbiol 2003; 661–662, 50 Suppl
- Varela J M, Respaldiza N, Sanchez B, de la Horra C, Montes-Cano M, Rincon M, Dapena J, Gonzalez-Becerra C, Medrano F J, Calderon E. Lymphocyte response in subjects with chronic pulmonary disease colonized by Pneumocystis jirovecii. J Eukaryot Microbiol 2003; 672–673, 50 Suppl
- Sukura A, Konttinen Y T, Sepper R, Kaartinen L, Sorsa T, Lindberg L A. Collagenases and the serine proteinases elastase and cathepsin G in steroid-induced Pneumocystis carinii pneumonia. J Clin Microbiol 1995; 33: 829–834
- Qu J, Rong Z, He L, Pan J, Chen X. Relationship between the burden of Pneumocystis carinii, the inflammatory reaction and lung injury in Pneumocystis carinii pneumonia. Chin Med J (Engl) 2000; 113: 1071–1074
- Atzori C, Mainini A, Agostoni F, Angeli E, Bartlett M, Bruno A, Scaglia M, Cargnel A. Detection of rat Pneumocystis carinii proteinases and elastase and anti Pneumocystis activity of proteinase inhibitors in vitro. Parasite 1999; 6: 9–16
- Lugli E B, Allen A G, Wakefield A E. A Pneumocystis carinii multi-gene family with homology to subtilisin-like serine proteases. Microbiology 1997; 143: 2223–2236
- Lee L H, Gigliotti F, Wright T W, Simpson-Haidaris P J, Weinberg G A, Haidaris C G. Molecular characterization of KEX1, a kexin-like protease in mouse Pneumocystis carinii. Gene 2000; 242: 141–150
- Kutty G, Kovacs J A. A single-copy gene encodes Kex1, a serine endoprotease of Pneumocystis jiroveci. Infect Immun 2003; 71: 571–574
- Hanrahan J P, Tager I B, Segal M R, Tosteson T D, Castile R G, Van Vunakis H, Weiss S T, Speizer F E. The effect of maternal smoking during pregnancy on early infant lung function. Am Rev Respir Dis 1992; 145: 1129–1135
- Gaultier C. Cardiorespiratory adaptation during sleep in infants and children. Pediatr Pulmonol 1995; 19: 105–117
- Norris K A, Morris A, Patil S, Fernandes E. Pneumocystis colonization, airway inflammation, and pulmonary function decline in acquired immunodeficiency syndrome. Immunol Res 2006; 36: 175–187