Abstract
An abnormal increase in proteolytic enzymes is thought to play a key role in pulmonary emphysema. Alveolar macrophage proteolytic enzymes include cathepsin L, cathepsin S, matrix metalloproteinase 1, 9, and 12, and a number of studies have implicated these proteinases in the alveolar destruction that characterizes emphysema. The aim of this study was to investigate cathepsin L, cathepsin S, matrix metalloproteinase 1, 9, and 12 mRNA expression in alveolar macrophages isolated from patients with varying degrees of emphysema and to correlate their level of expression with measures of emphysema. Alveolar macrophages were isolated from fifty-four patients who underwent surgical resection for lung carcinoma. The level of mRNA expression was determined using real-time PCR. Emphysema was quantified using high-resolution CT scans. Alveolar macrophages were also cultured for 24 h and 48 h; the effect of proinflammatory mediators and promoter polymorphisms on expression was analyzed. There was a significant correlation between matrix metalloproteinase 1 mRNA expression and emphysema. A higher level of matrix metalloproteinase 1 mRNA was associated with more severe emphysema. Matrix metalloproteinase 12 mRNA expression was increased in current smokers as compared with former smokers. Furthermore, there was a negative correlation between matrix metalloproteinase 12 gene expression and carbon monoxide diffusing capacity. The matrix metalloproteinase 9 C-1562T polymorphism significantly influenced matrix metalloproteinase 9 mRNA expression in alveolar macrophages. These results suggest that alveolar macrophage matrix metalloproteinase 1 and 12 may have a role in the lung structural changes leading to the development of emphysema. Furthermore, these data provide evidence to support the concept that multiple proteinases, causing both elastin and collagen degradation, are important in the pathogenesis of pulmonary emphysema.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a debilitating pulmonary condition characterized by “airflow limitation that is not fully reversible; the airflow limitation is usually both progressive and associated with an abnormal inflammatory response of the lungs to noxious particles or gases” (Citation[1]).
According to this NHLBI/WHO Workshop Summary of the Global Initiative for COPD destruction of the lung parenchyma in COPD typically occurs as centrilobular emphysema; an imbalance of endogenous proteinases and antiproteinases in the lung is thought to be a major mechanism leading to emphysema (Citation[1]). In order to develop effective therapies that will halt progression of the disease there needs to be a greater understanding of the cellular and molecular mechanisms involved in its pathogenesis. The discovery that individuals who have severe alpha-1 antitrypsin deficiency develop emphysema at an early age (Citation[2]), in conjunction with the observation that intratracheal instillation of papain, an elastolytic enzyme, produces emphysema in experimental animals (Citation[3]), led to the proteinase:antiproteinase hypothesis in the 1960s (Citation[4]).
The idea that an imbalance in the proteinase:antiproteinase ratio underlies the pathogenesis of emphysema continues to dominate current thinking (Citation[5]). Alpha-1 antitrypsin is the primary inhibitor of neutrophil elastase, which has the capacity to digest lung elastin. Because neutrophil elastase is expressed almost exclusively by neutrophils, these cells, and their release of elastase, were considered to play a major role in pathogenesis. More recently, studies have shown that other cell types, such as alveolar macrophages and epithelial cells, and degradation of other connective tissue proteins, such as collagen, may contribute to the development of emphysema. Neutrophils and macrophages are the two main proteinase-producing cell types in smokers' lungs.
There is increased interest in a potential role for alveolar macrophages in the pathogenesis of emphysema because of the large increase in this cell type in response to cigarette smoke (Citation[6]). In addition, there is a direct relationship between the number of alveolar macrophages in the alveolar parenchyma and lung destruction; an association that is not true for neutrophils except in alpha-1 antitrypsin deficiency (Citation[7]). In 1997, Hautamaki and colleagues (Citation[8]) reported that in contrast to wild-type mice, MMP12 knockout mice did not develop emphysema after chronic exposure to cigarette smoke. Furthermore, they did not accumulate macrophages in their lungs, as did the wild-type mice suggesting that macrophage MMP12 was involved in the development of emphysema.
Human alveolar macrophages express a number of proteolytic enzymes that are capable of degrading elastin including cathepsin L (CTSL) and cathepsin S (CTSS) (Citation[9]), two of the most potent elastases, as well as matrix metalloproteinase 9 (MMP9) and MMP12 (Citation[10]); MMP1, a collagenase, is also produced by human alveolar macrophages (Citation[10]). The finding that mice overexpressing MMP1 develop emphysematous changes in their lungs showed a direct contribution of MMPs to the proteolytic destruction in emphysema (Citation[11]). This finding modified the focus on elastin-degradation to include collagen-degradation as an important process that could lead to the lung destruction characteristic of emphysema. The role of elastases and collagenases in emphysema is supported by both animal and human studies (Citation[12]).
The aim of our study was to investigate the levels of mRNA expression of five proteolytic enzymes (CTSL, CTSS, MMP1, MMP9, and MMP12) produced by alveolar macrophages from patients with varying degrees of airflow limitation in relation to emphysema. Furthermore, factors that may influence the levels of expression of these proteinases were assessed in vitro.
MATERIALS AND METHODS
Experimental design
Alveolar macrophages were obtained by bronchoalveolar lavage (BAL) of resected lung specimens from fifty-four subjects having surgery for lung carcinoma. Expression of CTSL, CTSS, MMP1, MMP9, and MMP12 mRNA was assessed with the use of real-time PCR. Emphysema was quantified on high-resolution computed tomography (CT) scans. The baseline expression levels of the proteinases were correlated with emphysema, smoking history, and lung function data. Alveolar macrophages were also cultured for 24 h and 48 h; the effect of proinflammatory mediators and promoter polymorphisms on expression of these proteolytic enzymes was analyzed.
Study subjects
A total of 54 subjects (48 Caucasian, 6 Asian) who underwent lung resection for a small (< 3 cm), Stage I or II, peripheral tumour at Vancouver General Hospital between April 2001 and March 2003 were recruited for the study. Patients who had chemotherapy or radiation therapy were excluded from the study. The characteristics of the study subjects are described in and the distribution of lung function data is shown in . Genotypic analysis was performed to determine if subjects had alpha-1 antitrypsin deficiency; there were 8 subjects with the MS phenotype and none with the MZ or ZZ phenotypes. This study was approved by the University of British Columbia/Providence Health Care and Vancouver General Hospital Research Ethics Boards. All subjects provided written informed consent for the use of clinical data, pulmonary function test data, CT scans, and for the studies on alveolar macrophages from the surgically resected tissue. All subjects completed a questionnaire reviewing respiratory symptoms and smoking history.
Figure 1 Distribution of lung function data. Panel A shows the distribution of forced expiratory volume in 1 second (FEV1), % predicted (n = 47). Panel B shows the distribution of forced vital capacity (FVC), % predicted (n = 47). Panel C shows the distribution of FEV1/FVC ratio (n = 49). Panel D shows the distribution of carbon monoxide diffusing capacity (DLCO), % predicted (n = 40).
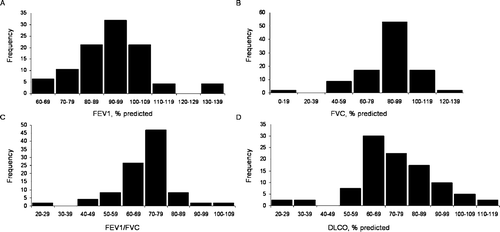
Table 1 Characteristics of subjects, smoking history, lung function data, distribution of emphysema score, and cellular content of bal fluid
Cell preparation and assays
Bronchoalveolar lavage
Lung tissue was obtained from patients undergoing surgical resection for carcinoma. BAL was performed within 1 h after the completion of lung resection, under sterile conditions. After washing the lumen of the bronchus with saline to remove blood and mucus, a Foley catheter (14 gauge) was inserted into an airway supplying a segment or lobe uninvolved by tumour. Sixty ml of cold saline was instilled into the segment or lobe and after a few instillations to expand the lung, BAL fluid was aspirated back. The tissue was gently massaged to help recover the instilled saline. This procedure was repeated several times. Aspirated BAL fluid was collected in sterile plastic bottles. Recovery was between 240 and 480 ml.
The BAL fluid was filtered through sterile gauze to remove debris and mucus, and the filtrate was centrifuged (250 g, 10 min). Cell viability was consistently > 90% as assessed using trypan blue exclusion. Cytospin preparations were made for differential cell counts, using a Shandon II cytocentrifuge, and air dried for 1 h at room temperature. Differential counts were determined on Diff-Quik stained preparations.
Alveolar macrophage culture
Alveolar macrophages were separated by Ficoll-Hypaque density centrifugation. Cells were washed twice with RPMI 1640 (Gibco, Carlsbad, California, USA) and resuspended in RPMI 1640 at a final concentration of 106 alveolar macrophages/ml. An aliquot of freshly isolated alveolar macrophages was used to determine baseline levels of mRNA expression. Cells were plated onto 6-well plates (Costar, Acton, MA, USA) at a density of 4 million cells/well. Following adherence at 37°C in a humidified incubator (5% CO2 in air) for 2 h, cells were washed two times with RPMI 1640 to remove non-adherent cells. Fresh Macrophage Serum-Free Medium (Gibco), supplemented with 2 mM L-glutamine and penicillin-streptomycin (100 U/ml-100 μ g/ml; Gibco), was added to the adhered cells (> 99% alveolar macrophages). Alveolar macrophages were then cultured for a further 24 h and 48 h in the presence of medium alone, medium + lipopolysaccharide (LPS) (2 μ g/ml), medium + interleukin-1beta (IL-1beta) (20 ng/ml), or medium + tumor necrosis factor (TNF-alpha) (20 ng/ml). LPS (Escherichia coli) was purchased from Sigma (St. Louis, MO, USA). Recombinant human IL-1beta and TNF-alpha were purchased from R&D Systems (Minneapolis, MN, USA).
Quantification of mRNA by real-time PCR
Total RNA was isolated from alveolar macrophages using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Reverse transcription was performed with the use of SuperScript RNase h- Reverse Transcriptase (Invitrogen, Carlsbad, California, USA). Quantitative real-time PCR was performed using a LightCycler (Roche Molecular Biochemicals, Indianapolis, IN, USA) to assess CTSL, CTSS, MMP1, MMP9, and MMP12 mRNA levels in alveolar macrophages. Beta-actin (ACTB) was selected as the endogenous control (Citation[13]). Expression was analyzed at baseline (0 h; uncultured cells) and after cell culture for 24 h and 48 h under four culture conditions: (1) control conditions without stimulation (CTRL); (2) LPS stimulation (2 μ g/ml); (3) IL-1beta stimulation (20 ng/ml); and (4) TNF-alpha stimulation (20 ng/ml). Primers, annealing temperatures, and single detection temperatures used to amplify the RT-generated cDNAs are presented in (PCR conditions available upon request). Primer pairs were designed to flank an intron-containing sequence and alignment to other known human genes was excluded by running a BLAST (http://www.ncbi.nih.gov/BLAST/).
Table 2 Primers and conditions for real-time PCR experiments
Genotyping
Genomic DNA was isolated from alveolar macrophages using DNeasy Tissue Kit (Qiagen, Valencia, CA, USA). Study participants were genotyped for alpha-1 antitrypsin S and Z mutations as previously described by our laboratory (Citation[14], Citation[15]) and the following common promoter polymorphisms were assessed: (1) Cathepsin L A-1622C (rs3118869); (2) Cathepsin S G-25A (rs1136774); (3) MMP1 1G-1607/2G (rs1799750); (4) MMP9 C-1562T (rs3918242); (5) MMP9 cytosine-adenine (CA) repeat polymorphism (rs3222264); and (6) MMP12 A-82G (rs2276109) (genotyping protocols available upon request). Template-free controls and known genotype controls were included in each experiment. Genotypes were assigned by two independent investigators who were unaware of the patients' identities and phenotypes. Inconsistencies were resolved by two additional genotyping reactions. Ten samples were selected at random and sequenced to confirm each genotyping protocol.
Computed tomographic morphometry
A semiquantitative emphysema score was assigned to the CT scans by two independent observers for 42 of the subjects. The observers graded the extent of emphysema using a 6-point scale (0: no emphysema and 5: > 75% emphysema). The visual scoring of emphysema was included because only 20 cases had digital CT scan data available for objective quantification but 42 of the subjects had CT films that could be analyzed visually. Therefore, we had an additional 24 cases where emphysema could only be assessed visually. For subjects where their digital CT data were available, a quantitative analysis of the lung parenchyma was performed using custom software (EmphylxJ, Vancouver, BC, Canada) as previously described (Citation[16], Citation[17], Citation[18]).
Briefly, the lung parenchyma was segmented from the chest wall and large central blood vessels using a modified border-tracing algorithm with a prior position-knowledge algorithm (Citation[16]). Lung volume was calculated by summing the segmented pixel area in each slice and multiplying by the slice thickness to acquire the total lung volume. The CT density of the lung (g/ml) was estimated from x-ray attenuation of each of the CT voxels (Citation[19], Citation[20]). Lung weight and air volume were calculated from the lung volume and density measurements (Citation[17], Citation[18]). Emphysema was defined using a density mask cut-off < −855 Hounsfield units (overall emphysema) and < −950 Hounsfield units (severe emphysema), which are appropriate for this CT acquisition technique (Citation[21]). From this analysis, we obtained two quantitative computerized measures of emphysema: 1) a measure of the overall extent of emphysema (density mask cut-off of −855 Hounsfield units; denoted emphysema extent) and 2) a measure of the extent of severe emphysema (density mask cut-off of −950 Hounsfield units; denoted emphysema severity). CT scans from the twenty cases done at our institution were available for quantitative analysis.
Statistical analysis
Results are expressed as means ± SE. Linear regression analysis was used to evaluate the degree of correlation between baseline mRNA expression and computerized measures of emphysema. Age, sex, and smoking history (pack-years) were included as covariates in a multiple regression model. The correlation between mRNA expression at baseline with subjective emphysema score (with ordinal values of 0, 1, 2, 3, 4, and 5) was analyzed using ordinal categorical regression (Citation[22]). Other statistics were performed by analysis of variance and Student's t-test (two-sided). All tests were performed using SAS software version 8.02 (SAS Institute Inc., Cary, NC). P values less than 0.05 were considered to indicate statistical significance.
RESULTS
All target genes were detected in alveolar macrophages (n = 54). At baseline, expression of CTSS mRNA, normalized according to the amount of beta-actin, was significantly higher than expression of the other 4 genes. The expression of CTSS was 2.6-fold higher than CTSL (p = 0.002), 346-fold higher than MMP12 (p < 0.001), 1,520-fold higher than MMP9 (p < 0.001), and 22,800-fold higher than MMP1 (p < 0.001).
Expression levels and emphysema
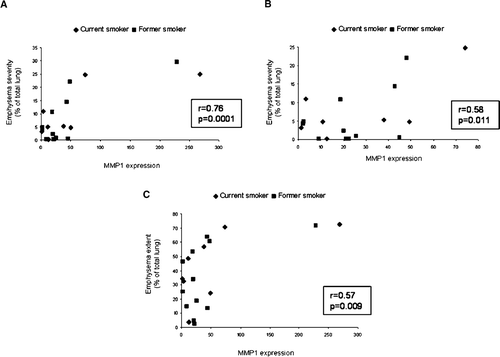
and show that baseline MMP1 mRNA expression was significantly correlated with computerized measures of emphysema extent (cut-off −855 Hounsfield units) (r = 0.57, p = 0.009, n = 20) and emphysema severity (cut-off −950 Hounsfield units) (r = 0.76, p < 0.001, n = 20). Since this measure of MMP1 mRNA expression was compared with two measures of emphysema, we used the Bonferroni method to correct for multiple comparisons and a p < (0.05/2) = 0.025 to indicate statistical significance. Multiple regression modeling to adjust for covariates (age, sex, smoking history (pack-years)) confirmed the findings for severity (r = 0.79, p = 0.002, n = 20) but the findings for extent (r = 0.62, p = 0.034, n = 20) did not reach statistical significance using the Bonferroni correction and p = 0.025 to indicate statistical significance. When the two outliers are removed, the correlation between MMP1 mRNA and computerized emphysema severity (cut-off −950 Hounsfield units) remained significant (r = 0.58, p = 0.011, n = 18), while that with emphysema extent (cut-off −855 Hounsfield units) was not significant (r = 0.40, p = 0.098, n = 18). The correlation between levels of MMP1 expression at baseline with subjective emphysema score () using ordinal categorical regression was borderline significant (p = 0.058). However, the baseline levels of CTSL, CTSS, MMP9, and MMP12 mRNA were not significantly correlated with computerized quantification of emphysema or subjective emphysema score (data not shown).
Figure 2 Comparison of matrix metalloproteinase 1 (MMP1) mRNA expression and emphysema. Panel A shows a plot comparing MMP1 expression with emphysema severity (current smokers, n = 8; former smokers, n = 12). Panel B shows a plot comparing MMP1 expression with emphysema severity, excluding the two outliers (current smokers, n = 7; former smokers, n = 11). The extent of severe emphysema was defined using a density mask cut-off of −950 Hounsfield units. Panel C shows a plot comparing MMP1 expression with emphysema extent (current smokers, n = 8; former smokers, n = 12). The overall extent of emphysema was defined using a density mask cut-off of −855 Hounsfield units.
Expression levels and smoking history
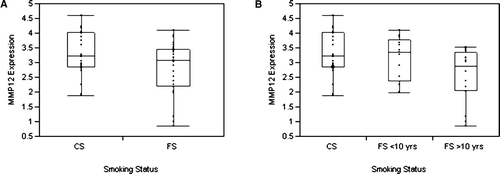
Expression of MMP12 mRNA at baseline was significantly higher in cells from current smokers (CS) compared with cells from former smokers (FS) according to analysis of variance (CS vs. FS [mean ± SE]: 3.3 ± 0.2 vs. 2.8 ± 0.2, p = 0.038) (). Furthermore, when patients were grouped as current smokers or as former smokers according to the length of smoking cessation: (1) less than 10 years (FS < 10 yrs) or (2) greater than or equal to 10 years (FS > 10 yrs), levels of MMP12 mRNA differed significantly according to analysis of variance among these groups (CS vs. FS < 10 yrs vs. FS > 10 yrs [mean ± SE]: 3.3 ± 0.2 vs. 3.1 ± 0.2 vs. 2.6 ± 0.2, p = 0.020) (). There was no significant correlation between MMP12 gene expression and exposure to cigarettes measured in pack-years. Baseline expression levels of CTSL, CTSS, MMP1, and MMP9 were not related to any measures of smoking history.
Figure 3 Matrix metalloproteinase 12 (MMP12) gene expression in current smokers and former smokers. Panel A shows the difference in levels of MMP12 mRNA expression, normalized according to the amount of beta-actin, in study participants who are current smokers (CS) (n = 24) and former smokers (FS) (study participants who had quit smoking for a least 6 months prior to surgery) (n = 29). Panel B shows the difference in levels of MMP12 expression in current smokers and former smokers classified according to the length of smoking cessation: (1) less than 10 years (FS < 10 yrs) (n = 13) or (2) greater than or equal to 10 years (FS > 10 yrs) (n = 16). The ends of the box are the first and third quartiles; the line across the middle of the box identifies the median.
Expression levels and lung function data
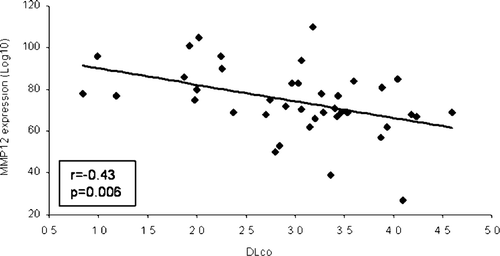
Baseline MMP12 mRNA expression was significantly inversely correlated with carbon monoxide diffusing capacity (DLCO) (r = −0.43, p = 0.006, n = 40); the higher the levels of MMP12 expression, the lower the DLCO (). However, there was no relationship between baseline expression levels of CTSL, CTSS, MMP1, and MMP9 and lung function data (data not shown).
Figure 4 Baseline matrix metalloproteinase 12 (MMP12) mRNA expression and carbon monoxide diffusing capacity (DLCO). MMP12 expression was significantly inversely correlated with DLCO (r = −0.43, p = 0.006, n = 40).
Baseline expression levels and genotype
Baseline levels of CTSL, CTSS, MMP1, MMP9, and MMP12 mRNA did not differ significantly among any of the genotypic groups (data not shown).
Cultured alveolar macrophages
We next determined the response of alveolar macrophages to pro-inflammatory mediators after culture for 24 h and 48 h. Levels of CTSL, CTSS, MMP1, MMP9, and MMP12 mRNA expression under the various culture conditions are shown in .
Figure 5 Relative gene expression in cultured alveolar macrophages. Alveolar macrophages were cultured for 24 h and 48 h in the presence of medium alone (CTRL), medium + lipopolysaccharide (LPS) (2 μ g/ml), medium + interleukin-1beta (IL-1beta) (20 ng/ml), or medium + tumor necrosis factor (TNF-alpha) (20 ng/ml). Panels A through E show the expression of cathepsin L (CTSL), cathepsin S (CTSS), matrix metalloproteinase 1 (MMP1), MMP9, and MMP12, respectively, with the use of real-time PCR. Samples from 54 subjects were run in triplicate. Data are means ± SE. *Statistically different compared with time control, p < 0.05. **Statistically different compared with time control, p < 0.001. † Statistically different between 24 h and 48 h, p < 0.05. ‡ Statistically different between 24 h and 48 h, p < 0.001. Note the log scale for levels of expression of MMP1.
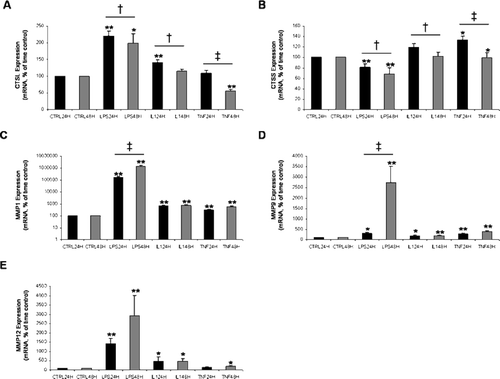
LPS and IL-1beta stimulation caused a significant increase in CTSL expression at 24 h compared with the time control (both, p < 0.001). There was no significant difference between TNF-alpha stimulated expression of CTSL at 24 h and the time control (p = 0.52). LPS stimulation caused a significant increase and TNF-alpha stimulation caused significant decrease in expression of CTSL at 48 h relative to the time control (p = 0.003 and p < 0.001, respectively). There was no significant difference between IL-1beta stimulated expression at 48 h and the time control (p = 0.63). For all 3 conditions (LPS, IL-1beta, and TNF-alpha), expression of CTSL at 24 h was significantly greater than expression at 48 h (p = 0.006, p = 0.004, and p < 0.001, respectively).
LPS stimulation caused a significant decrease in CTSS expression at 24 h and 48 h compared with the time controls (both, p < 0.001). TNF-alpha stimulation caused a significant increase in CTSS expression at 24 h and 48 h compared with the time controls (p = 0.015 and p = 0.025, respectively). There was no significant difference between IL-1beta stimulated expression of CTSS and the time controls (p = 0.13 and p = 0.11, respectively). LPS, IL-1beta, and TNF-alpha stimulated expression levels of CTSS at 24 h were all significantly greater than at 48 h (p = 0.008, p = 0.009, and p < 0.001, respectively).
MMP1 expression showed the largest response to the different treatments. Expression was increased in response to the stimuli (LPS, IL-1beta, and TNF-alpha) at 24 h (all, p < 0.001) and at 48 h (all, p < 0.001). Expression levels were not significantly different between 24 h and 48 h in culture except for MMP1 mRNA expression levels in cells stimulated with LPS; this expression increased from 24 h to 48 h (p < 0.001).
The level of expression of MMP9 in alveolar macrophages stimulated with LPS, IL-1beta, and TNF-alpha was significantly greater than the 24 h time control (p = 0.001, p = 0.003, and p < 0.001, respectively) and the 48 h time control (all, p < 0.001). In LPS treated cells, MMP9 expression increased significantly from 24 h to 48 h (p < 0.001). IL-1beta and TNF-alpha stimulated expression did not differ between 24 h and 48 h (p = 0.77 and p = 0.10, respectively).
LPS treatment caused markedly increased expression of MMP12 compared with the respective time control at 24 h (p < 0.001) and 48 h (p < 0.001). IL-1beta stimulated expression of MMP12 at 24 h and 48 h was greater than the time controls (p = 0.008 and p = 0.028, respectively). MMP12 expression in cells treated with TNF-alpha for 48 h differed significantly from the time control (p = 0.044). Expression levels did not differ significantly between 24 h and 48 h.
Expression levels in culture and genotype.
The MMP9 C-1562T polymorphism significantly influenced MMP9 mRNA expression in culture according to analysis of variance (p = 0.042). Heterozygous individuals (n = 18) had markedly increased expression compared with homozygous CC individuals (n = 35). (Only 1 TT individual was involved in the study; therefore, this individual was not included in this analysis). In contrast, the MMP9 CA repeat polymorphism did not have a significant effect on the level of expression of the gene product (p = 0.99). Polymorphisms examined in the other proteinases did not have a significant effect on the level of expression of the gene product.
DISCUSSION
This study is the first to use CT scans to evaluate the relationship of lung destruction in COPD to message expression of several alveolar macrophage proteinases that could have a potential role in the development of emphysema in COPD. However, Ito and coworkers (Citation[23]) reported the use of CT scanning to show an association between a MMP9 promoter polymorphism and upper lobe emphysema. Our data showed that baseline levels of MMP1 expression in alveolar macrophages were significantly related to CT measures of emphysema. We did not see the same relationship between FEV1 values and expression suggesting that changes in CT may be better linked than FEV1 values to changes in expression of proteolytic enzymes.
For the objective quantification of emphysema, we analyzed the extent of emphysema using a density mask cut-off < −855 Hounsfield units and the severity of emphysema using a density mask cut-off < −950 Hounsfield units. We chose to analyze the data in this way because we hypothesized that an increase in a proteinase could increase emphysema by leading to more widespread emphysema (increased extent), but could also lead to more severe destruction of the lung tissue and hence, an increase in emphysema severity.
The finding that transgenic mice overexpressing human MMP1 in their lungs develop progressive, adult-onset emphysema with physiological changes in lung compliance, consistent with changes noted in humans, first indicated that collagen degradation was an important process in the disease (Citation[11]). Since then, human studies have indicated a potential role of MMP1 in emphysema (Citation[24], Citation[25]). In humans, structural cells such as the lung epithelium have proven to be an important source of this enzyme and it has been shown that cigarette smoke is capable of directly inducing MMP1 expression in normal human small airway epithelial cells in vitro (Citation[26]). Our results provide support for a role of alveolar macrophage MMP1 in association with structural changes associated with pulmonary emphysema in humans. Alternatives to a causal relationship to explain the correlations between elevated MMP1 mRNA expression levels and emphysema include the possibility that proteinase elevations may be secondary to other events that actually trigger emphysema, and that alveolar macrophages may respond to the emphysema microenvironment by increasing proteinase expression.
In the present study, we found higher levels of MMP12 mRNA in alveolar macrophages isolated from current smokers as compared with former smokers. Furthermore, we show that there is a negative correlation between MMP12 gene expression and diffusing capacity for carbon monoxide, suggesting that MMP12 may have a pathogenic role in the destruction of the lung parenchyma. MMP12 is consistently a key player in both cigarette smoke-induced and non-smoke-related mouse models of emphysema (Citation[27]); however, the importance of MMP12 in human emphysema is uncertain. Recent studies indicate that MMP12 is increased in lungs and induced sputum from patients with COPD (Citation[28], Citation[29]). Our study shows, for the first time, increased MMP12 expression in human alveolar macrophages in relation to a decrease in diffusing capacity. The precise role of MMP12 in the disease process is unclear. Recent data has shown that the elastin fragments generated by MMP12 are chemotactic for monocytes, suggesting that MMP12 is critical in disease progression (Citation[30]). MMP12 may not only be involved in emphysema through its direct role in ECM degradation, but also as regulator of inflammation. Churg and colleagues (Citation[31]) showed that in the same mouse model, MMP12 acts as a pro-inflammatory mediator through the activation of TNF-alpha and speculate that this may be its primary role in the pathogenesis of emphysema.
Our data show that the MMP9 C-1562T polymorphism significantly influences MMP9 mRNA expression by alveolar macrophages; individuals with the CT genotype have greater MMP9 mRNA expression levels than individuals with the CC genotype, which is consistent with a previous in vitro study (Citation[32]). In addition, Minematsu et al. (Citation[33]) found an association between the -1562T allele and the development of emphysema in Japanese smokers. The number of subjects we studied may have been too small to show a difference in emphysema extent between the two genotypes.
Alveolar macrophages were treated in vitro with LPS, IL-1beta, and TNF-alpha in order to reflect in vivo stimulation by pro-inflammatory mediators. The bacterial endotoxin, LPS, is one of the most potent inflammatory agents known. LPS may serve as a proxy for bacterial infections and is also present in cigarettes (Citation[34]). In the present study, the most dramatic change in expression was seen with LPS stimulation, which caused significant increases in CTSL, MMP1, MMP9, and MMP12 gene expression and a significant decrease in levels of CTSS mRNA. IL-1beta and TNF-alpha are two ubiquitous and seminal pro-inflammatory cytokines. Studies have shown that IL-1 (Citation[35], Citation[36]) and TNF-alpha (Citation[37], Citation[38]) are increased in patients with COPD. In the present study, we show that IL-1beta and TNF-alpha modulate the expression of alveolar macrophages proteinases.
The proteinase:antiproteinase hypothesis suggests that the proteolytic destruction associated with emphysema is due to an imbalance between proteolytic enzymes and their inhibitors. The imbalance may result from altered production or activity of both proteinases and antiproteinases, which may be caused by a number of different factors such as cigarette smoke, oxidative stress, and genetic polymorphisms that alter the expression or function of these enzymes and their inhibitors. Macrophages also release cathepsin and MMP inhibitors. The measurement of these inhibitors and the ratio between each proteinase and its biological inhibitor was beyond the scope of this study but will be evaluated in further studies.
The use of alveolar macrophages from patients with lung cancer adds a potential bias to our study. This was especially a concern for MMP1 given the fact that the MMP1 1G-1607/2G polymorphism is associated with the development of lung cancer. Zhu et al. (Citation[39]) found that the 2G2G genotype was significantly associated with lung cancer and that these individuals developed lung cancer at an earlier age. If the genotype was associated with lung cancer risk, then we might have expected the sample to be enriched for the risk genotype. Alternatively, if the genotype affected the severity of cancer, thereby making the tumour inoperable, our population could have been enriched for the genotype that decreased severity. To explore this selection bias, we examined the observed genotype frequencies and compared them to those expected under Hardy-Weinberg equilibrium.
The MMP1 polymorphism was in Hardy-Weinberg equilibrium (p = 0.085); however, we observed fewer than the expected number of individuals with the 2G2G genotype. These individuals would be underrepresented in our study if their lung cancer was inoperable. The possibility of genotyping errors at the MMP1 locus was minimized by randomly selecting samples and sequencing them to confirm the genotyping protocol. Furthermore, the population frequency in the present study was consistent with a previous study (p = 0.14) (Citation[40]). The age of lung cancer development in the subjects is not known; the mean age at surgery is not significantly different between genotypic classes (p = 0.23). Since we examined genotype in relation to expression, a relationship between genotype and expression would still be expected, even if there was a bias in the way the samples were collected. Although this is probably a reasonable assumption, scenarios where bias could affect the results are still possible.
Limitations of this study include the relatively small number of subjects and the limited number of subjects with advanced COPD. The latter limitation is a consequence of the experimental design; subjects who have advanced COPD are often ineligible for lung resection. On the other hand, the study of mild disease offers certain advantages since the late stages of disease may be complicated by bacterial infection and other late stage consequences of the disease process. For example, there is often a disconnect between structural damage and physiological changes in COPD patients. The use of a cohort with mild disease is a strength in this regard because it allows for the identification of important factors associated with the structural changes as opposed to physiological changes. Despite these limitations, we detected significant relationships between mRNA level and phenotype and between genotype and mRNA level, implication MMPs in disease pathogenesis.
In conclusion, this study provides evidence that human alveolar macrophage MMP1 and MMP12 have a role in the response of the lung to smoking alveolar damage and therefore, may play an important role in the structural changes associated with emphysema. Our in vitro data indicates that pro-inflammatory mediators modulate the expression of proteinases produced by alveolar macrophages. The findings of this study provide evidence to support the concept that degradation of both elastin and collagen are important in the pathogenesis of emphysema and that multiple proteolytic enzymes and mediators play a role in the disease process.
ACKNOWLEDGMENTS
The authors thank Dr. Ken Evans, Dr. John Yee, Dr. Julia Flint, Dr. Sharyn MacDonald without whom this study could not have been completed, and Dr. David Erle for his insightful comments and suggestions. We are indebted to Ms. Barb Moore, Mr. Steve Kalloger, and Dr. Jian Ruan for assistance in preparing clinical samples, to Ms. Dianna Louie and Mr. Anh-Toan Tran for help analyzing the CT scans, and to Dr. Michael Schultzer for help in the statistical analysis of the ordinal categorical emphysema scores. The authors would also like to thank all of the study participants.
Supported by grants from the Canadian Institutes of Health Research, British Columbia Lung Association, and Glaxo UK Health Clinical Research Award. Alison Wallace is supported by a Doctoral Research Award from the Michael Smith Foundation for Health Research. Andrew Sandford is the recipient of a Canada Research Chair.
REFERENCES
- Pauwels R A, Buist A S, Calverley P M, Jenkins C R, Hurd S S. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Nhlbi/who global initiative for chronic obstructive lung disease (gold) workshop summary. AmJ Respir Crit Care Med 2001; 163: 1256–1276
- Laurell C B, Eriksson S. The electrophoretic alpha1-globulin pattern of serum in alpha1-antitrypsin deficiency. ScandJ Clin Lab Invest 1963; 15: 132–140
- Gross P, Pfitzer E A, Tolker E, Babyak M A, Kaschak M. Experimental emphysema: Its production with papain in normal and silicotic rats. Arch Environ Health 1965; 11: 50–58
- Mittman C. Pulmonary emphysema and proteolysis. Academic Press, New York 1972
- Hogg J C, Senior R M. Chronic obstructive pulmonary disease – part 2: Pathology and biochemistry of emphysema. Thorax 2002; 57: 830–834
- Merchant R K, Schwartz D A, Helmers R A, Dayton C S, Hunninghake G W. Bronchoalveolar lavage cellularity. The distribution in normal volunteers. Am Rev Respir Dis 1992; 146: 448–453
- Finkelstein R, Fraser R S, Ghezzo H, Cosio M G. Alveolar inflammation and its relation to emphysema in smokers. AmJ Respir Crit Care Med 1995; 152: 1666–1672
- Hautamaki R D, Kobayashi D K, Senior R M, Shapiro S D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997; 277: 2002–2004
- Punturieri A, Filippov S, Allen E, Caras I, Murray R, Reddy V, Weiss S J. Regulation of elastinolytic cysteine proteinase activity in normal and cathepsin k-deficient human macrophages. J Exp Med 2000; 192: 789–799
- Shapiro S D. The macrophage in chronic obstructive pulmonary disease. AmJ Respir Crit Care Med 1999; 160: S29–32
- D'Armiento J, Dalal S S, Okada Y, Berg R A, Chada K. Collagenase expression in the lungs of transgenic mice causes pulmonary emphysema. Cell 1992; 71: 955–961
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur RespirJ 2003; 22: 672–688
- Ishii T, Wallace A M, Zhang X, Gosselink J, Abboud R T, English J C, Pare P D, Sandford A J. Stability of housekeeping genes in alveolar macrophages from copd patients. Eur RespirJ 2006; 27: 300–306
- Sandford A J, Chagani T, Spinelli J J, Pare P D. Alpha1-antitrypsin genotypes and the acute-phase response to open heart surgery. AmJ Respir Crit Care Med 1999; 159: 1624–1628
- Sandford A J, Chagani T, Weir T D, Connett J E, Anthonisen N R, Pare P D. Susceptibility genes for rapid decline of lung function in the lung health study. AmJ Respir Crit Care Med 2001; 163: 469–473
- Coxson H O, Chan I H, Mayo J R, Hlynsky J, Nakano Y, Birmingham C L. Early emphysema in patients with anorexia nervosa. AmJ Respir Crit Care Med 2004; 170: 748–752
- Coxson H O, Rogers R M, Whittall K P, D'Yachkova Y, Pare P D, Sciurba F C, Hogg J C. A quantification of the lung surface area in emphysema using computed tomography. AmJ Respir Crit Care Med 1999; 159: 851–856
- Rogers R M, Coxson H O, Sciurba F C, Keenan R J, Whittall K P, Hogg J C. Preoperative severity of emphysema predictive of improvement after lung volume reduction surgery: Use of ct morphometry. Chest 2000; 118: 1240–1247
- Coxson H O, Mayo J R, Behzad H, Moore B J, Verburgt L M, Staples C A, Pare P D, Hogg J C. Measurement of lung expansion with computed tomography and comparison with quantitative histology. J Appl Physiol 1995; 79: 1525–1530
- Hedlund L W, Vock P, Effmann E L. Computed tomography of the lung. Densitometric studies. Radiol Clin North Am 1983; 21: 775–788
- Gevenois P A, de Maertelaer V, De Vuyst P, Zanen J, Yernault J C. Comparison of computed density and macroscopic morphometry in pulmonary emphysema. AmJ Respir Crit Care Med 1995; 152: 653–657
- Agresti A. Analysis of ordinal categorical data. Wiley, New York 1984; 104–119
- Ito I, Nagai S, Handa T, Muro S, Hirai T, Tsukino M, Mishima M. Matrix metalloproteinase-9 promoter polymorphism associated with upper lung dominant emphysema. AmJ Respir Crit Care Med 2005; 172: 1378–1382
- Finlay G A, O'Driscoll L R, Russell K J, D'Arcy E M, Masterson J B, Fitz Gerald M X, O'Connor C M. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. AmJ Respir Crit Care Med 1997; 156: 240–247
- Imai K, Dalal S S, Chen E S, Downey R, Schulman L L, Ginsburg M, D'Armiento J. Human collagenase (matrix metalloproteinase-1) expression in the lungs of patients with emphysema. AmJ Respir Crit Care Med 2001; 163: 786–791
- Mercer B A, Kolesnikova N, Sonett J, D'Armiento J. Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smoke. J Biol Chem 2004; 279: 17690–17696
- Churg A, Wright J L. Proteases and emphysema. Curr Opin Pulm Med 2005; 11: 153–159
- Molet S, Belleguic C, Lena H, Germain N, Bertrand C P, Shapiro S D, Planquois J M, Delaval P, Lagente V. Increase in macrophage elastase (mmp-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res 2005; 54: 31–36
- Demedts I K, Morel-Montero A, Lebecque S, Pacheco Y, Cataldo D, Joos G F, Pauwels R A, Brusselle G G. Elevated mmp-12 protein levels in induced sputum from patients with copd. Thorax 2006; 61: 196–201
- Houghton A M, Quintero P A, Perkins D L, Kobayashi D K, Kelley D G, Marconcini L A, Mecham R P, Senior R M, Shapiro S D. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest 2006; 116: 753–759
- Churg A, Wang R D, Tai H, Wang X, Xie C, Dai J, Shapiro S D, Wright J L. Macrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha release. AmJ Respir Crit Care Med 2003; 167: 1083–1089
- Zhang B, Ye S, Herrmann S M, Eriksson P, de Maat M, Evans A, Arveiler D, Luc G, Cambien F, Hamsten A, Watkins H, Henney A M. Functional polymorphism in the regulatory region of gelatinase b gene in relation to severity of coronary atherosclerosis. Circulation 1999; 99: 1788–1794
- Minematsu N, Nakamura H, Tateno H, Nakajima T, Yamaguchi K. Genetic polymorphism in matrix metalloproteinase-9 and pulmonary emphysema. Biochem Biophys Res Commun 2001; 289: 116–119
- Hasday J D, Bascom R, Costa J J, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest 1999; 115: 829–835
- Bevilacqua M P, Schleef R R, Gimbrone M A, Jr., Loskutoff D J. Regulation of the fibrinolytic system of cultured human vascular endothelium by interleukin 1. J Clin Invest 1986; 78: 587–591
- Nachman R L, Hajjar K A, Silverstein R L, Dinarello C A. Interleukin 1 induces endothelial cell synthesis of plasminogen activator inhibitor. J Exp Med 1986; 163: 1595–1600
- Schleef R R, Bevilacqua M P, Sawdey M, Gimbrone M A, Jr., Loskutoff D J. Cytokine activation of vascular endothelium. Effects on tissue-type plasminogen activator and type 1 plasminogen activator inhibitor. J Biol Chem 1988; 263: 5797–5803
- Keatings V M, Collins P D, Scott D M, Barnes P J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. AmJ Respir Crit Care Med 1996; 153: 530–534
- Zhu Y, Spitz M R, Lei L, Mills G B, Wu X. A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter enhances lung cancer susceptibility. Cancer Res 2001; 61: 7825–7829
- Joos L, He J Q, Shepherdson M B, Connett J E, Anthonisen N R, Pare P D, Sandford A J. The role of matrix metalloproteinase polymorphisms in the rate of decline in lung function. Hum Mol Genet 2002; 11: 569–576