Abstract
Background: Deficiency of the antiprotease alpha-1-antitrypsin (AAT) and exposure to cigarette smoke (CS) contribute to the development of early onset emphysema. CS-induced apoptosis of alveolar cells including endothelial cells plays critical role in the lung destruction. AAT deficiency is associated with increased lung tissue destruction as well. We hypothesize that AAT protects lung alveoli from noxious environmental stimuli such as CS-induced apoptosis. Methods: Porcine pulmonary artery endothelial cells (PAEC) were exposed to CS in the presence or absence of AAT (20μ M). AAT internalization and markers for apoptosis were assessed by confocal microscopy. Flow cytometry was performed in parallel to quantify the number of AAT-loaded and apoptotic cells. Results: We demonstrated that exogenous AAT accumulated in PAEC and protected cells from CS-induced apoptosis. AAT-loaded CS-exposed cells exhibited increased amounts of chaperone HSP-70 in their cytosol and less apoptosis inducing factor in their nuclei compared to AAT-untreated, CS-exposed cells. Conclusions: Our results suggest that AAT is taken up by endothelial cells via two mechanisms and that intracellular AAT may have a protective role in CS-induced endothelial apoptosis. This may open new insights into the field of endothelial serpins as agents capable of protecting the vasculature from environment-derived noxious substances.
| Abbreviations | ||
| AAT, | = | alpha-1-antitrypsin, serpins serine protease inhibitors |
| NE | = | neutrophil elastase |
| HSP, | = | heat shock protein |
| AIF, | = | apoptosis inducing factor |
| PAEC, | = | porcine pulmonary artery endothelial cells |
| CS, | = | cigarette smoke |
| WST | = | 4-[3-(4-iodopheny)-2-(4-nitrop henyl)-2H-5-tetrazolio]-1,3-benzene disulfonate |
| MES, | = | 2-(N-morpholino) ethanesulfonic acid) |
| GPCR, | = | G-protein-coupled receptor |
| Rho, | = | Rhodamine |
INTRODUCTION
Alpha-1-antitrypsin (AAT), an abundant 52 kDa serum glycoprotein, is the archetypal member in a family of serpins, inhibitors of serine proteases. Major targets of AAT include neutrophil derived elastase and protease-3. Neutrophil elastase (NE), a proteolytic component of the primary granule of the human granulocyte, is known to degrade elastin in lungs (Citation[1]). In the absence or insufficiency of its major inhibitor AAT, NE can degrade lung parenchyma with resultant panacinar emphysema (Citation[2]). Under normal circumstances, AAT binds to and neutralizes NE via a reactive loop, which contains a methionine residue. Cleaving the high-energy methionine-serine bond causes the AAT molecule to change its structure, locking NE in a mousetrap-like mechanism (Citation[3]).
The inhibitory function of AAT has expanded, and novel cell protective and stimulatory activities have been reported (Citation[4], Citation[5], Citation[6], Citation[7], Citation[8], Citation[9], Citation[10]). A body of evidence suggests that AAT is an anti-apoptotic factor (Citation[4]). For example, elastase-mediated apoptosis can be prevented by AAT (Citation[11]), and AAT has been reported to interact with the enzymes of the apoptotic cascade (Citation[12]).
Apoptosis of lung structural cells is implicated in COPD development as one of the major mechanisms (Citation[13]). Interestingly, in the lungs of patients with emphysema the majority of apoptotic cells were endothelial cells (Citation[14]). AAT has been reported on the vascular endothelial surface (Citation[15]), and the anti-apoptotic effect of intra-cellular AAT in mouse endothelium in vivo has been reported recently (16). The uptake of AAT by endothelial cells and it's direct inhibitory effect on caspase-3 has also been demonstrated (Citation[17]). However, our understanding of the antiprotease-endothelium interaction is still limited.
Endothelium has been always regarded as a semi-permeable barrier through which AAT passively enters tissues from the circulation. It is believed that AAT diffuses from the circulation through intercellular gaps in the lung capillary endothelial surface and reaches the lung epithelial lining fluid and other tissues with tissue specific distribution (Citation[18], Citation[19], Citation[20]).
We hypothesize that AAT is not only passively moving through the gaps in the vascular wall but is also actively taken up and transported into endothelial cells. The uptake of AAT, similar to that of serum albumin, might be based on a caveolin-related uptake mechanism. Caveolin, as previously reported, forms chaperone complexes with heat shock proteins (HSPs) and some of the HSPs are chaperones for AAT (Citation[21], Citation[22]). HSP-70 can act as anti-apoptotic agent in parallel with its chaperoning activities by preventing apoptosis inducing factor (AIF) translocation into the cell nucleus (Citation[23]). We will investigate if this process is influenced by AAT supplementation into the cell environment.
In the present study CS is used to induce apoptosis of endothelial cells because, in parallel with AAT deficiency, it is recognized as one of the major risk factors for lung emphysema development. Cultured pulmonary artery endothelial cells incubated with exogenous AAT in physiological concentrations will be exposed to CS after which cell viability and apoptosis, transport of AAT into endothelial cells, intracellular localization of AAT, and the interaction of AAT with caveolin and HSP-70 will be assessed.
MATERIALS AND METHODS
Cell Culture and CS Exposure
Primary porcine pulmonary artery endothelial cells (PAEC) were obtained from the main pulmonary arteries of 6-month-old pigs from a local slaughterhouse and were grown in monolayer culture as described by Zhang et al (Citation[24]). Fifth to seventh passage cells in confluent monolayers maintained in RPMI 1640 medium (Life Technologies, USA) with 5% fetal bovine serum (HyClone Laboratories, USA), and antibiotics were used for all experiments. Cells were incubated in serum free medium 12 hours prior to and during experiments.
Cultured cells were exposed to mainstream CS derived directly from burning 2R4F reference cigarettes developed by the NIH-NCI, the USDA and the Tobacco and Health Research Institute of the University of Kentucky. Briefly, the inlet of a modular incubator chamber (Billups-Rothenberg, USA) was connected to a cigarette holder in a fume hood, and the outlet was connected to a vacuum line through a valve for adjustment of flow rates and an absorption unit (). PAEC monolayers in 100-mm dishes with a lid were covered by 5 ml of medium to mimic alveolar lining fluid (∼ 4 mm in depth). Cigarettes were burned at a rate of one cigarette per five minutes for 15 min (3 cigarettes).
Figure 1 CS exposure apparatus. The inlet of a modular incubator chamber was connected to a cigarette holder, and the outlet was connected to a vacuum line through a valve for adjustment of flow rates and an absorption unit. Culture dishes in the smoking chamber were exposed to whole CS generated by burning 2R4F cigarettes at a rate of 1 cigarette per 5 minutes. Cells in the control chamber were treated under identical conditions with an unlit cigarette.
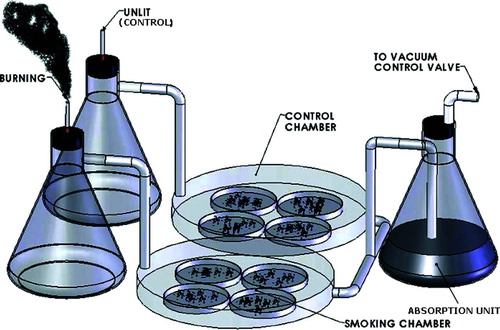
The exposed cells were then returned to normal conditions, i.e., room air-5% CO2 at 37°C, for a 45 min recovery to mimic the intervals between smoking episodes in vivo. These exposure cycles (15 min/3 cigarettes plus 45 min recovery) were repeated 6 times and all studies were conducted after the completion of 6 cycles. PAEC in a parallel chamber were treated under identical conditions with an unlit cigarette and were used as controls in all experiments. Morphology of the monolayers was assessed by phase contrast microscopy.
Specific Reagents
Human plasma AAT (purity > 95%, inhibitory activity > 75%) was obtained from Sigma, USA and an AAT preparation, used for augmentation therapy, Aralast was also used. These two AAT preparations were compared for the effects on cell viability, and no differences were observed (data not presented). Thus, Aralast™ was used for all later experiments. AAT was diluted in PBS, pH 7.4, and tested to ensure the absence of endotoxin using a colorimetric endotoxin detection kit (Cambrex, USA). In all AAT preparations used, endotoxin levels were less than 0.1 EU/mg of protein. Polymerised and oxidized AAT were prepared as described previously (Citation[15]). Human serum albumin was purchased from Sigma (USA). Anti-AAT antibody was obtained from DAKO (Denmark), anti-caveolin antibody from BD Biosciences (USA), anti-HSP-70 antibodies from Abcam and Stressgene (USA), and anti-AIF antibody from Chemicon (USA).
Cell Viability Assay
4-[3-(4-iodopheny)-2-(4-nitrophen yl)-2H-5-tetrazolio]-1,3-benzene disulfonate (WST), Cell Proliferation Reagent (Roche Diagnostics), was used to determine the effects of AAT on cell viability following the manufacturer's instructions. Viability was measured at the same exposure times and dosages of AAT that were used for other experiments.
Cell Lysates and Nuclear Extracts
Cell lysis and nuclear extraction kit, N-XTRACT, from Sigma (Sigma-Aldrich, USA) was used according to the manufacturer's recommendations. This method for nuclei isolation from cultured cells utilizes a hypotonic Igepal CA-360 detergent lysis buffer. Nuclei are separated from cytosol by low speed centrifugation and further purified by repeated washes in the same lysis buffer. The nuclear proteins are then extracted into extraction buffer under agitation and removed from the DNA pellet after centrifugation. Finally, Bradford assay is performed as suggested by manufacturer (Pierce, USA) to assess protein concentration in the samples.
Immunoprecipitation/Western Blotting
Lysates (300 μ g of protein in each) were immunoprecipitated using a mouse monoclonal anti-HSP-70 antibody and protein A/G-Agarose (Santa Cruz). After incubation for 2 h at 4°C, the antigen-antibody complexes were washed 5 times in cold PBS. The immunoprecipitates, lysates (for HSP-70 and β -actin) and nuclear extracts (for AIF) were boiled in Laemmli sample buffer and loaded on a 10% SDS-PAGE gels in a Mini-PROTEAN II electrophoresis cell (Bio-Rad). Separated proteins were then transferred to a nitrocellulose membrane (Bio-Rad, USA) in transfer buffer (25 mM Tris, 190 mM glycine, 20% methanol) using a BioRad blot transfer system (Bio-Rad, USA). The membranes were blocked overnight with Casein/PBS blocking buffer. Blots were developed using primary polyclonal rabbit antibodies against AAT (1:5000), HSP-70 (1:1000), β -actin (1:1000) and AIF (1:750). All membranes were visualized by incubation with horseradish peroxidase-conjugated secondary antibody (1:2500) and chemiluminescence substrate.
Flow Cytometry
AAT was labeled with Rhodamine following the manufacturer's protocol (Pierce, USA) and applied to cell cultures (5 μ M, 18 hours). Accumulation of AAT was monitored using confocal microscopy and then quantified by FACSort II® and Cell Quest® software. For competition assays unlabeled AAT (25 and 50 μ M) was added together with fluorescent AAT.
For apoptosis assays control and AAT-treated (20 μ M, 18 hours) PAEC were exposed to CS, harvested (trypsinised), and incubated with FITC-conjugated Annexin-V and Propidium iodide following the manufacturer's recommendations (Molecular Probes, USA). Stained cells were analyzed using FACSort II® and Cell Quest® software. Percentage of Annexin V positive/Propidium iodine negative cells is presented as a measure of apoptosis.
Immunocytochemistry
Cell monolayers cultured in the presence and absence of AAT (5 μ M, 18 hours) were washed, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton, and stained for caveolin and AAT or HSP-70 and AAT simultaneously using specific primary antibodies. Secondary fluorescent agent conjugated-antibodies were applied following exclusive washes. After a two-hour incubation, cells were washed again, and mounted using Vectashield (Vector Laboratories, USA) mounting medium containing DAPI, which is specialized to preserve fluorescence and counter stain DNA. Stained cells were examined by confocal microscopy.
Confocal Microscopy
Cells were examined for fluorescent AAT uptake and Annexin and Propidium Iodine binding using a Zeiss LSM 510 laser scanning confocal microscope. AAT was labeled with Rhodamine following the manufacturer's protocol (Pierce) or stained using specific antibodies as described in Immunocytochemistry section. Annexin and Propidium Iodine binding were performed using a protocol provided by Molecular Probes. In parallel, real-time monitoring of caspase activity was also performed using CaspACE FITC-VAD-FMK from Promega (USA). It is a cell permeable caspase inhibitor conjugated with FITC which binds active caspase and serves as an in situ marker for apoptosis. The images shown are representative of the results observed in at least 3 experiments. In each sample, 10 fields were monitored.
Preparation of Caveolae Enriched Membrane Fractions
Membrane fractions were prepared by a detergent-free method as previously described (Citation[25]) with modifications that allow maintenance of the functional integrity of caveolar components. In brief, PAEC monolayers were washed twice with ice-cold MES [25 mmol 2-(N-morpholino) ethanesulfonic acid), pH 7.0, 150 mmol NaCl] buffered saline, scraped in MES containing protease-inhibitor cocktails, and homogenized using a glass pestle Dounce homogenizer at 4°C.
The homogenized cell suspensions were subjected to nitrogen cavitation using a Parr Bomb (Parr Instrument) at 650 psi for 15 min as previously described (Citation[26]). The lysed cells were then mixed with an equal volume of 80% sucrose in MES and transferred to ultracentrifuge tubes. Five ml of 30% sucrose/MBS (25 mM MES, pH 6.5, 0.15 M NaCl) were layered on the top of the lysate followed by 4 ml of 5% sucrose/MBS. The sucrose gradients were centrifuged at 38,000 rpm in SW-40 rotor (Beckman Coulter Optima, L-70K ultracentrifuge) at 4°C for 16 h. Twelve 1 ml fractions from the top to the bottom of the centrifuge tube were collected. These 12 fractions were diluted with 3 volumes of MES and centrifuged at 100,000xg in Ti-70 rotor (Beckman Coulter Optima, L-70K ultracentrifuge) at 4°C for 1 hour. The resulting pellets were used in Western Blots to detect AAT and caveolin.
Statistics
In each experiment, control and experimental PAEC were matched for cell line, age, density, number of passages, and confluence state. Results are shown as mean ± SD. The differences in the means of experimental results were analyzed for their statistical significance by two-sided t-test and one-way analysis of variance (ANOVA) combined with a multiple comparisons test (Bonferroni t-test), and p < 0.05 was taken as significant. Sigma Stat software was used for the calculations.
RESULTS
Internalization of AAT by PAEC
To determine whether exogenous AAT enters endothelial cells, PAEC were incubated with physiological levels of Rhodamine-labeled AAT (5 μ M) for 2–18 hours (). Aralast, the comercially available AAT preparation, was used. Uptake of AAT by PAEC started as early as 2 hours after introduction of protein into the cell culture medium and peaked at 18 hours. Unlabeled AAT competed with the intracellular accumulation of labeled AAT in PAEC as assessed by flow cytometry () suggesting that there is a receptor/transporter mediated uptake involved. However, the possibility of a second uptake mechanism still exists since unlabeled competitor doesn't completely block the uptake of labeled AAT. Accumulation of Rhodamine labeled-AAT was identical to that of unlabeled AAT detected by means of antibodies and presented later in this section ( and ).
Figure 2 Visualization and quantification of Rhodamine (Rho) labeled AAT in PAEC. (A) Cells were incubated with Rho-labeled AAT (5 μ mol, 18 hours), fixed and mounted in the mounting medium with DAPI. Confocal microscopy micrographs demonstrate nuclei stained with DAPI (blue) and Rho (red) fluorescence of accumulated AAT. Magnification – 1000×. (B) Graph represents percentage of fluorescent cells (Rho-AAT positive) detected by flow cytometry. The initial bar represents Rho-AAT-(5 μ M, 18 hours) treated cells. The second and third bars are cells treated with Rho-AAT together with increasing concentrations of unlabeled AAT (25 and 50 μ M, respectively). Significance of the difference between cells treated with Rho-AAT plus 10-fold excess of cold AAT (50 μ M) versus Rho-AAT-only treated cells is indicated (p value of < 0.05 was taken as significant, n = 3).
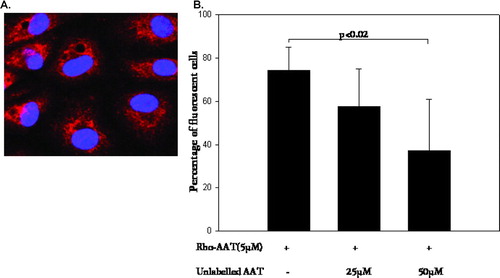
In some experiments native AAT was replaced with modified, i.e. oxidized or polymerized AAT, and the uptake of these modified AAT was identical to that of native AAT (results not shown). However, AAT uptake was not detected when the experiments were performed at +4°C, which suggests that the mechanism of uptake is not passive diffusion but rather an active process.
CS induces endothelial apoptosis
Exposure to CS for 6 hours induces an early apoptotic event, i.e., Annexin V binding to the cell surface (, upper panel), as well as activation of caspases (, lower panel) as detected by the in situ apoptosis marker CaspACE FITC-VAD-FMK. The numbers of apoptotic cells were quantified by flow cytometry () as the percentage of Annexin V positive/Propidium iodine negative cells in the total cell population. These results indicate that CS causes apoptosis in PAEC, as assessed by 2 markers of apoptosis, and that AAT significantly diminishes the apoptotic effects of CS.
Figure 3 Apoptotic PAEC detected by confocal microscopy. Upper panel represents control cells, CS (18 cigarettes/6 hours) exposed cells, and AAT (20 μ M, 18 hours) pretreated/CS exposed cells stained with Annexin-FITC. The bottom panel represents control cells, CS-exposed cells and CS-exposed cells pretreated with AAT stained with an in situ marker for apoptosis, CaspACE FITC-VAD-FMK. Photomicrographs are representatives of at least 3 experiments. Magnification—400×.
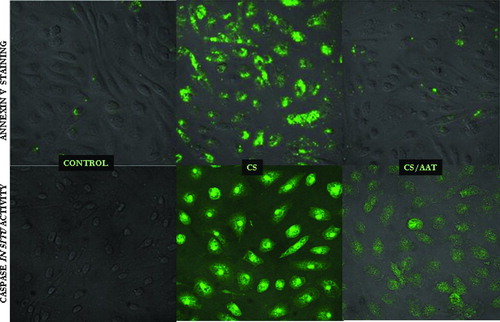
Figure 4 Annexin V-FITC positive/Propidium Iodine negative, i.e., apoptotic, cells detected by flow cytometry. Control cells are represented (first bar) and compared to CS-exposed (second bar) cells, AAT pretreated (20 μ M, 18 hours) and CS-exposed cells (third bar), and, cells treated with AAT but not exposed to CS (fourth bar). The flow cytometry insets above each column show cell distribution according to Annexin V and Propidium Iodine staining, where the lower right quadrant represents Propidium Iodine negative/Annexin V-positive, i.e., apoptotic cells. Significance of differences between the control (first bar) and CS-exposed cells (second bar) and between the CS-exposed (second bar) vs. AAT-treated/CS exposed cells (third bar) are indicated (n = 9).
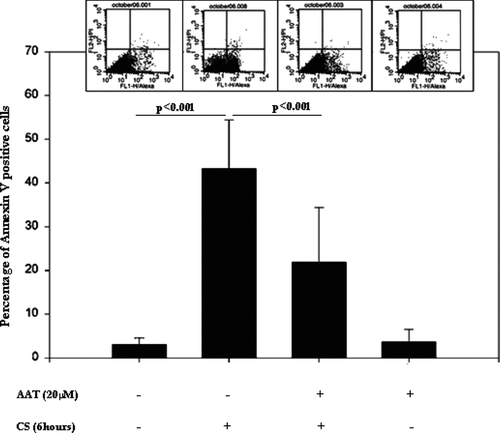
CS induces apoptosis in approximately 50% of cells exposed as quantified by flow cytometry (). AAT pretreatment (for 18 hours prior to exposure) does not change Annexin V binding compared to control cells. When cells were pretreated with polymerized and oxidized AAT instead of native AAT, protection against apoptosis was also observed (results not shown). In contrast, human serum albumin, which is also taken up by PAEC, did not exhibit any anti-apoptotic activity in our model.
Caveolin-enriched membrane fractions of endothelial cells contain AAT
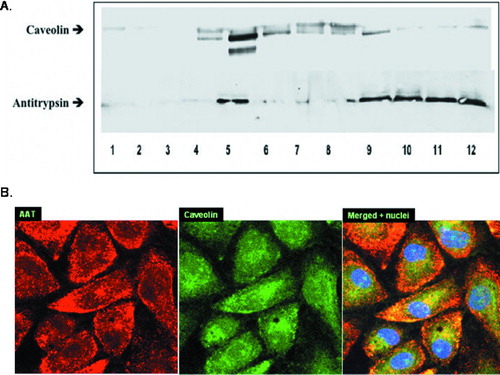
Membrane-associated proteins can be separated into 12 different membrane fractions according to their lipid content (25). The caveolin-enriched fraction, designated the buoyant fraction, is the 5th, while G-protein-coupled receptor (GPCR) signaling components appear in the 9th–12th fractions, or heavy/non-buoyant fractions (Citation[27]). Proteins of the GPCR family, such as muscarinic acetylcholine receptors and mu-opioid receptors, have been shown to localize in both, buoyant and heavy fractions. We were interested to see if AAT co-localizes with caveolin, a protein marker for caveolae, in PAEC membrane fractions.
As shown in , both AAT and caveolin are detected in the 5th fraction. AAT is also detected in fractions 9–12. This result suggests that AAT may target GPCR and caveolae for uptake and intracellular signaling. To confirm the association of AAT and caveolin in our cells, AAT treated (5 μ M, 18 hours) PAEC were fixed and stained for AAT and caveolin using specific primary and fluorescent secondary antibodies. The labeled cells were examined under a confocal microscope (). Yellow areas within the cells suggest that AAT (red) is co-localized with caveolin (green). This observation supports the notion that AAT is associated with caveolin.
Figure 5 Co-localization of caveolin and AAT. (A) Western Blot of 12 membrane fractions derived from endothelial cells incubated with AAT (5 μ M, 18 hours) and probed with anti-Caveolin (upper panel) and anti-AAT (lower panel) antibodies is presented. Both AAT and caveolin are abundant in Lane 5 (5th fraction). AAT is also detected in fractions 9–12 where G-protein-coupled receptor (GPCR) binding components usually appear. Shown is a representative blot from 3 experiments. (B) Confocal microscopy images of AAT treated (5 μ M, 18 hours) PAEC were taken at 1000× magnification. Binding of primary antibodies to AAT and caveolin was visualized using Rhodamine- and FITC-conjugated secondary antibodies for AAT and caveolin, respectively. Yellow areas within the cells indicate co-localization of AAT and caveolin. Three identical experiments were performed.
AT internalization is associated with elevation of HSP-70 and decreased nuclear AIF
Induction of HSP-70 is associated with its chaperone function and protective effects within the endothelium. The top panel in depicts a Western Blot in which PAEC whole cell lysates were analyzed for the presence of inducible HSP-70 using a specific (recognizing inducible form of HSP-70 only) anti-HSP-70 antibody. Control cells have low but detectable amounts of inducible HSP-70 which are up-regulated by cell cultivation in the presence of AAT (20 μ M, 18 hours). CS abolished any detectable HSP-70 in control cell lysates, but not in AAT pretreated cells, where the level of HSP-70 remained unchanged even after CS exposure. Equal protein loading was assessed by monitoring β -actin in the Western blot (the second panel).
Figure 6 Internalized AAT modulates HSP-70 and AIF. (A) Western Blots of cytoplasmic HSP-70 (top panel), β-actin (second panel), precipitated HSP-70/AAT complexes (third panel) and nuclear AIF (fourth or bottom panel) are shown. Control, AAT-treated (20 μ M, 18 hours), CS-exposed (18 cigarettes/6 hours), and AAT-treated/CS-exposed cells are represented. Protein loading is comparable in all four lines as assessed by uniform β -actin staining (panel below). Co-immunoprecipitation with a specific HSP-70 antibody followed by Western Blot analysis was performed to determine the association of AAT and HSP-70 (the third panel from the top). Finally, AIF detected in the nuclear extracts of PAEC is presented in the bottom panel. Nuclear fractions were loaded by protein concentration (30 ng/well). Representative blots of at least 3 experiments are shown. (B) Confocal microscopic images of AAT-treated (5 μ M, 18 hours) PAEC (1000× magnification) are presented. Binding of primary antibodies to AAT and HSP-70 was visualized using Rhodamine- and FITC-conjugated secondary antibodies for AAT and HSP-70, respectively. Yellow areas within the cells indicate co-localization of proteins. Three identical experiments were performed.
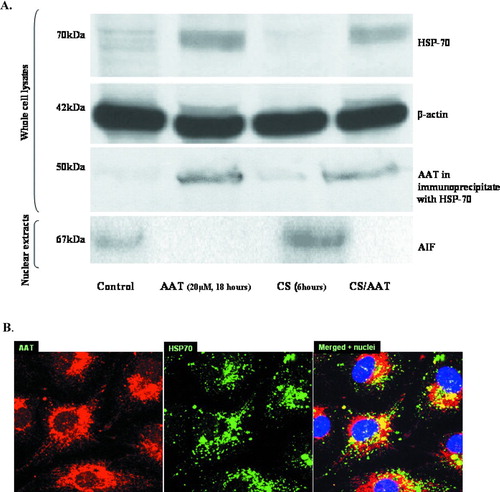
The third panel in represents AAT immunoprecipitated from cell lysates by the HSP-70 antibody suggesting direct protein/protein interaction between AAT and HSP-70 which was not affected by exposure of cells to CS. Immunostaining of PAECs for AAT and HSP-70 presented in shows that they co-localize and accumulate in the perinuclear area of AAT treated-PAEC. These observations support the conclusion that AAT and HSP-70 form a complex within the PAEC.
A possible target protein for the anti-apoptotic action of HSP-70 is AIF, a pro-apoptotic protein known to translocate into the cell nucleus upon apoptosis and to be down-regulated by HSP-70. Levels of AIF in the nuclear extracts of PAEC are presented in the bottom panel of . Control cells have some nuclear AIF while CS-exposed cells exhibit much higher amount of AIF. In contrast, nuclear AIF is not detected in AAT-pretreated cells or in AAT-pretreated cells exposed to CS. Therefore, these results suggest that AAT may protect against apoptosis in PAEC by interfering with the translocation of AIF into the nucleus of CS-exposed cells.
DISCUSSION
The mechanism of AAT and vascular endothelium interaction is not fully understood. Still, there is a body of clinical evidence suggesting involvement of AAT in clinical situations with a vascular disease component, such as vasculitis, autoimmune diseases, cervical artery dissection, aneurysms, and atherosclerosis (Citation[28], Citation[29]). In support, in vitro studies show that AAT might be utilized for endothelial protection from proteolytic attack (Citation[30]). Moreover, in an animal model of emphysema AAT has been shown to play a novel anti-apoptotic role (Citation[16]). Our report provides evidence in support of the protective anti-apoptotic effects of AAT in endothelial cells in vitro.
Recently documented active endothelial transport of several serum proteins via the vascular endothelium (Citation[31]) supports the notion that AAT may also be actively transported. Moreover, the endothelial localization of AAT has already been reported (Citation[15]). There are several possible AAT uptake mechanisms, particularly, caveolae and clathrin-mediated transcytosis and receptor or transporter mediated uptake (Citation[32]). We report here that uptake of AAT, similar to that of serum albumin, appears to be a caveolin-related uptake. This notion comes from our finding that AAT taken up by PAEC is found primarily in a caveolin-enriched cell membrane fraction ().
Co-localization of AAT and caveolin seen with confocal microscopy supports this observation further (). Moreover, caveolin was previously reported to form chaperone complexes with HSPs and to transport lipoproteins (Citation[21]). Thus, AAT co-localization with caveolin and the subsequent induction of HSP-70 in PAEC () may represent a complex mechanism of molecular trafficking. On the other hand, the inhibition of labeled AAT uptake by cold AAT (), which would not be expected if uptake was exclusively due to a caveolae-related mechanism, points to the fact that there is a second operational uptake mechanism for AAT, possibly a receptor and/or transporter-mediated uptake mechanism requiring energy.
The presence of AAT in the membrane fractions where GPCR are predominantly located () suggests the possibility that receptors for AAT are from this family. The existence of specific AAT receptors, although widely discussed, has never been proven, and the putative receptors have never been identified. Our data suggest that AAT can enter cells by 2 distinct mechanisms, via caveolae-mediated transcytosis and/or via receptors, possibly belonging to the GPCR family. Passive diffusion of protein can be excluded since intracellular AAT was not observed in PAEC after incubation at +4°C.
Our knowledge of intracellular AAT is limited to a study in which endothelial cells were transfected with the AAT gene (Citation[16]). The results of our study demonstrate a protective action of exogenous AAT accumulated within the cells. We have looked at the role of exogenous AAT in CS-induced endothelial damage since deficiency of AAT and smoking are two crucial factors for development of emphysema in vivo. Indeed, this situation is particularly relevant in individuals with AAT deficiency who smoke. We have found that intracellular AAT plays a protective role in CS-induced apoptosis ( and ). Non-inhibitory forms of AAT, i.e. oxidized and polymerized AAT, exhibited the same effect on PAEC, suggesting that the inhibitory activity of AAT is not crucial for its anti-apoptotic properties.
The possible downstream events by which AAT prevents apoptosis might be its interaction with inducible cytosolic HSP-70. Expression of HSP-70 is an adaptive response to various stimuli, and its primary function is molecular chaperoning, contributing to the folding and transport of cytosol proteins (Citation[33]). As we hypothesized, supplemental AAT induced cytosolic HSP-70 levels in our PAEC (). Such direct HSP-70 induction was shown previously when novel proteins were introduced in the endothelial cells (Citation[34]).
Co-immunoprecipitation of HSP-70 and AAT proteins from cell lysates by an anti-HSP70 antibody suggests the presence of their interaction in PAEC (). This is further supported by dual labeling studies demonstrating co-localization of AAT and HSP-70 in the perinuclear area of PAEC (). These results are consistent with a report by Finotti and Pagetta (Citation[22]) demonstrating interaction of HSP-70 with AAT.
The protective effects HSP-70 have been documented (Citation[35]). HSP-70 prevents DNA breaks and lipid peroxidation, protects mitochondrial integrity and function (Citation[36]), and prevents nuclear translocation of apoptosis-inducing factor (Citation[37]). AIF involvement in our apoptotic model is suggested by data that CS-exposed cells exhibit high levels of AIF in their nuclear fraction (). However, PAEC treated with AAT and exhibiting inducible HSP-70 had no detectable AIF in their nuclei and also were less apoptotic when exposed to CS (, and ).
Thus, AAT-stimulated synthesis of HSP-70 and subsequent inhibition of AIF nuclear translocation introduce a possible mechanism by which CS-induced damage in endothelial cells can be prevented or reduced. Whether chaperoning of AAT by HSP-70 directly leads to resistance to apoptosis or whether induction of HSP-70 alone is enough to protect cells in our model remains unclear.
In summary, our data suggest that pulmonary endothelial cells are protected from tobacco smoke by an antiprotease shield which is beneficial not only to the lung elastin network but also to the lung vasculature. Vascular cells transport AAT and perhaps other antiproteases from the blood stream to the tissues employing them for self-protection at the same time. Internalized AAT plays a protective role in CS-induced endothelial cell apoptosis. Impaired AAT internalization and/or insufficient amounts of AAT may lead to increased cell susceptibility to damage induced by CS. Therefore, endothelium-bound/internalized AAT may protect the vascular endothelium from a variety of environment-derived noxious species.
The authors would like to acknowledge the Interdisciplinary Center for Biotechnology Research at the University of Florida for assistance with the flow cytometry experiments and the Scanning Confocal Microscopy Unit at the Malcom Randall VA Medical Center for help with confocal imaging.
REFERENCES
- Yasutake A, Powers J. Reactivity of human leukocyte elastase and porcine pancreatic elastase toward peptide 4-nitroanilides containing model desmosine residues. Evidence that human leukocyte elastase is selective for cross-linked regions of elastin. Biochemistry 1981; 20: 3675–3679
- Wiedemann H P, Stoller J K. Lung disease due to alpha 1-antitrypsin deficiency. Curr Opin Pulm Med 1996; 2: 155–160
- Carrell R W, Lomas D A. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346: 45–53
- Daemen M, Heemskerk V, van't Veer C, Denecker G, Wolfs T, Vandenabeele P, Buurman W. Functional protection by acute phase proteins {alpha}1-acid glycoprotein and {alpha}1-antitrypsin against ischemia/reperfusion injury by preventing apoptosis and inflammation. Circulation 2000; 102: 1420–1426
- Dabbagh K, Lauren G J, Shock A, Leoni P, Papakrivopoulou J, Chambers R. Alpha-1-antitrypsin stimulates fibroblast proliferation and procollagen production and activates classical MAP kinase signalling pathways. J Cell Physiol 2001; 186: 73–81
- Jeannin P, Lecoanet-Henchoz S, Delneste Y, Gauchat J, Bonnefoy J. Alpha-1 antitrypsin up-regulates human B cell differentiation selectively into IgE- and IgG4-secreting cells. Euro J Immunol 1998; 28: 1815–1822
- Graziadei I, Weiss G, Egger C, Niederwieser D, Patsch J, Vogel W. Modulation of iron metabolism in monocytic THP-1 cells and cultured human monocytes by the acute-phase protein alpha1-antitrypsin. Exp Hematol 1998; 26: 1053–1060
- Bucurenci N. Inhibition of neutrophil superoxide production by human plasma alpha 1-antitrypsin. FEBS Lett 1992; 300: 21–24
- Tilg H, Vannier E, Vachino G, Dinarello C, Mier J. Antiinflammatory properties of hepatic acute phase proteins: preferential induction of interleukin 1 (IL-1) receptor antagonist over IL-1 beta synthesis by human peripheral blood mononuclear cells. J Exp Med 1993; 178: 1629–1636
- Torras J, Bordalba J R, Seron D, Moliner R, Carrera M, Valles J, Martinez-Castelao A, Alsina J, Grino J M. Protective effect of the PAF antagonist BN 52021 in an experimental renal warm ischemia model. Transpl Int 1993; 6: 236–238
- Wakasugi K, Schimmel P. Two distinct cytokines released from a human aminoacyl-trna synthetase. Science 1999; 284: 147–151
- Van Molle W, Denecker G, Rodriguez I, Brouckaert P, Vandenabeele P, Libert C. Activation of caspases in lethal experimental hepatitis and prevention by acute phase proteins. J Immunol 1999; 163: 5235–5241
- Tuder R M, Petrache I, Elias J A, Voelkel N F, Henson P M. Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol 2003; 28: 551–554
- Voelkel N F, Cool C D. Pulmonary vascular involvement in chronic obstructive pulmonary disease. Eur Respir J 2003; 22: 28S–32
- Aldonyte R, Jansson L, Ljungberg O, Larsson S, Janciauskiene S. Polymerized alpha-1-antitrypsin is present on lung vascular endothelium. New insights into the biological significance of alpha-1-antitrypsin polymerization. Histopathology 2004; 45: 587–592
- Petrache I, Fijalkowska I, Zhen L, Medler T R, Brown E, Cruz P, Choe K-H, Taraseviciene-Stewart L, Scerbavicius R, Shapiro L, Zhang B, Song S, Hicklin D, Voelkel N F, Flotte T, Tuder R M. A novel antiapoptotic role for {alpha}1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med 2006; 173: 1222–1228
- Petrache I, Fijalkowska I, Medler T R, Skirball J, Cruz P, Zhen L, Petrache H I, Flotte T R, Tuder R M. {Alpha}-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol 2006; 169: 1155–1166
- Barker A F, Iwata-Morgan I, Oveson L, Roussel R. Pharmacokinetic study of alpha1-antitrypsin infusion in alpha-1 antitrypsin deficiency. Chest 1997; 112: 607–613
- Laurell C, Nosslin B, Jeppsson J. Catabolic rate of alpha1-antitrypsin of Pi type M and Z in man. Clin Sci Mol Med 1977; 52: 457–461
- Kalsheker N, Morley S, Morgan K. Gene regulation of the serine proteinase inhibitors alpha1-antitrypsin and alpha1-antichymotrypsin. Biochem Soc Trans 2002; 30: 93–98
- Uittenbogaard A, Ying Y-s, Smart E J. Characterization of a cytosolic heat-shock protein-caveolin chaperone complex. Involvment in cholesterol trafficiking. J Biol Chem 1998; 273: 6525–6532
- Finotti P, Pagetta A. A heat shock protein70 fusion protein with [alpha]1-antitrypsin in plasma of Type 1 diabetic subjects. Biochem Biophys Res Commun 2004; 315: 297–305
- Ravagnan L, Gurbuxani S, Susin S A, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger J M, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 2001; 3: 839–843
- Zhang J, Jin B, Li L, Block E R, Patel J M. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol 2005; 288: C840–849
- Song K S, Li S, Okamoto T, Quilliam L A, Sargiacomo M, Lisanti M P. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. J Biol Chem 1996; 271: 9690–9697
- Patel J M, Edwards D A, Block E R, Raizada M K. Effect of nitrogen dioxide on surface membrane fluidity and insulin receptor binding of pulmonary endothelial cells. Biochem Pharmacol 1988; 37: 1497–1507
- Head B P, Patel H H, Roth D M, Lai N C, Niesman I R, Farquhar M G, Insel P A. G-protein-coupled receptor signaling components localize in both sarcolemmal and intracellular caveolin-3-associated microdomains in adult cardiac myocytes. J Biol Chem 2005; 280: 31036–31044
- Patterson C C, Jr, Patrick R, Pope-Harman A L, Knight D A, Magro C M. Alpha-1 anti-trypsin deficiency and Henoch-Schonlein purpura associated with anti-neutrophil cytoplasmic and anti-endothelial cell antibodies of immunoglobulin-A isotype. J Cutan Pathol 2005; 32: 300–306
- Talmud P J, Martin S, Steiner G, Flavell D M, Whitehouse D B, Nagl S, Jackson R, Taskinen M-R, Frick M H, Nieminen M S, Kesaniemi Y A, Pasternack A, Humphries S E, Syvanne M, and the Diabetes Atherosclerosis Intervention Study I. Progression of atherosclerosis is associated with variation in the {alpha}1-antitrypsin gene. Arterioscler Thromb Vasc Biol 2003; 23: 644–649
- Forsyth K, Talbot V, Beckman I. Endothelial serpins—protectors of the vasculature?. Clin Exp Immunol 1994; 95: 277–282
- Rowe D J, Bobilya D J. Albumin facilitates zinc acquisition by endothelial cells. Proc Soc Exp Biol Med 2000; 224: 178–186
- Minshall R, Tiruppathi C, Vogel S, Malik A. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem Cell Biol 2002; 117: 105–112
- Pinot F, el Yaagoubi A, Christie P, Dinh-Xuan A, Polla B. Induction of stress proteins by tobacco smoke in human monocytes. Cell Stress Chaperones 1997; 2: 156–161
- Zhang F, Hackett N R, Lam G, Cheng J, Pergolizzi R, Luo L, Shmelkov S V, Edelberg J, Crystal R G, Rafii S. Green fluorescent protein selectively induces HSP70-mediated up-regulation of COX-2 expression in endothelial cells. Blood 2003; 102: 2115–2121
- Sun Y, Ouyang Y-B, Xu L, Chow AM-Y, Anderson R, Hecker J G, Giffard R G. The carboxyl-terminal domain of inducible Hsp70 protects from ischemic injury in vivo and in vitro. J Cereb Blood Flow Metab 2005; 26: 937–950
- Kim H P, Wang X, Zhang J, Suh G Y, Benjamin I J, Ryter S W, Choi A MK. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38β MAPK and heat shock factor-1. J Immunol 2005; 175: 2622–2629
- Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie 2002; 84: 215–222