Abstract
COPD is a disease with a multi-component pathophysiology in which inflammation plays a key role. An anti-inflammatory effect of salmeterol (S)/fluticasone propionate (FP) combination (SFC), as demonstrated in a number of biopsy studies, may be the mechanism by which it provides a potential survival benefit in COPD patients in the TORCH study. It is possible that the molecular synergy between S and FP shown in COPD results in enhanced anti-inflammatory in the airways. This may also contribute to the reduction in exacerbations and the increase in lung function seen in the TORCH study. Alternatively, SFC may prolong survival by impacting on systemic inflammation and disease co-morbidities in COPD.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) affects 5–15% of all adults in industrialised countries (Citation[1]). It is estimated to become the third-leading cause of disease-related mortality by 2020 (Citation[2]). Annually, the death rate is greater than lung cancer and breast cancer combined (Citation[3]). The mortality rate in women now exceeds that in men (Citation[4]) and the average age of death has declined markedly (Citation[5]).
A number of non-pharmacological interventions reduce mortality in COPD. In patients with mild disease, a smoking cessation intervention decreased all-cause mortality by 15% (Citation[6]). In more severe patients, long-term oxygen therapy had a greater effect, decreasing mortality from 70% to < 30% at 5 years (Citation[7]). Lung volume reduction surgery changed the probability of COPD-related death from 0.5 to 0.3 compared with medical therapy in a sub-group of patients with predominantly upper-lobe emphysema and reduced exercise tolerance, but not in respiratory failure (Citation[8]). In the whole group, there was no evidence of a survival benefit. TORCH was the first study designed to investigate the effect of pharmacotherapy on survival in COPD patients (Citation[9]). It randomised more than 6000 patients to receive salmeterol/fluticasone propionate combination (SFC), salmeterol (S), fluticasone propionate (FP) or placebo over 3 years. The primary endpoint was all-cause mortality, with other efficacy endpoints being exacerbations, health status (SGRQ) and lung function (post-bronchodilator FEV1). The key finding was a 17.5% reduction in mortality with SFC compared with placebo. However, the study did not achieve its pre-specified objective, since a p-value of 0.052 was obtained after adjustments for the two interim analyses, performed for safety assessment.
Cox's proportional hazards model adjustment for smoking status, age, sex, baseline FEV1 and body mass index resulted in a p-value of 0.031 for the difference in all-cause mortality between SFC and placebo (Citation[9]). The apparent reduction in mortality with SFC after 3 years was similar to that achieved after 14.5 years of a smoking cessation intervention (Citation[6]) and of the order of the impact of statins (16%) on all-cause mortality in cardiovascular disease, interestingly where the p-value was 0.051(Citation[11]). There were no significant effects on mortality with FP and there was a significant difference (p = 0.007) between SFC and FP.
Associated with the decrease in mortality, SFC also reduced exacerbations, significantly slowed the decline in FEV1 over time (p < 0.003) and produced a sustained improvement in health status as assessed by the SGRQ (Citation[9], Citation[10]). Taken together, the TORCH study demonstrated substantial clinical efficacy with SFC in COPD over the individual components.
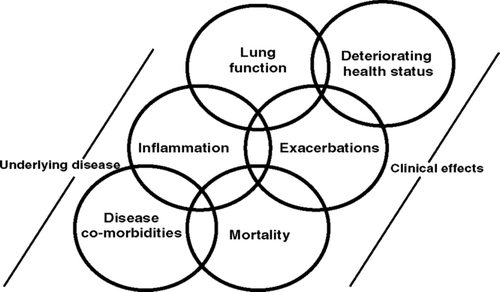
TORCH was not designed to explore potential mechanisms that may be responsible for a change in mortality with pharmacotherapy. The purpose of this paper is to provide a hypothesis to explain the possible survival and other benefits of SFC in COPD patients. To this end, we first highlight and discuss a series of reported associations between elements of the underlying pathophysiology of COPD (inflammation, declining lung function and co-morbidities) and the clinical manifestations of the disease (exacerbations, deteriorating health status and mortality), although it is difficult to prove causality. We then propose a hypothetical model that examines potential mechanisms that could be responsible for the effects seen with SFC, but not salmeterol or FP, in COPD patients in the TORCH study.
REPORTED ASSOCIATIONS
Inflammation and COPD
COPD is a multi-component disease with airway inflammation as one of the key drivers, but which also involves muco-ciliary dysfunction, structural changes and an important systemic component (Citation[12]), all of which contribute to the airflow limitation clinically characteristic of the disease and disease progression. Inflammation in COPD is largely triggered, in the industrialised world, by cigarette smoking and differs in nature from that associated with asthma (Citation[13]). It occurs early in the disease, before airflow limitation is detected and increases with severity of COPD (Citation[14]).
There is evidence in both large and small airway tissue of increased numbers of leukocytes (CD45(+) cells), T-lymphocytes (CD8(+) and CD4(+) T-cells), macrophages and neutrophils (Citation[13], Citation[14]) and elevated levels of inflammatory mediators such as IL-8, TNFα, LTB4 and oxidants in sputum, BALF and blood (Citation[15], Citation[16]). An imbalance between proteases and anti-proteases has also been described (Citation[17]). Inflammation is increased in patients with COPD over that in smokers with normal lung function (Citation[18], Citation[19]) and persists for many years after smoking cessation (Citation[14],Citation[20]). Finally, there is growing recognition that inflammation in COPD is not confined to the airways, but that systemic inflammation, either as a spill-over from the lung or a direct consequence of the disease, may play an important role in co-morbidities (e.g., cardiovascular disease, Type II-diabetes) in moderate-severe COPD patients (Citation[21]). It is possible that some patients have a common inflammatory genotype that manifests itself both systemically and in the lung and that there is more systemic inflammation in patients with more severe COPD.
Inflammation and lung function
There is a weak inverse relationship between large airway CD8(+) T-cells and FEV1 in COPD (Citation[22]). This relationship is much stronger (r = −0.63, p = 0.01) when the numbers of CD8(+)-cells in the small airways is correlated with lung function (Citation[23]). Similarly, patients with rapidly declining FEV1 (> 30 ml/year) exhibited significantly higher levels of sputum neutrophilia, compared with those with an annual decline of < 20 ml (Citation[24]). Sputum levels of IL-8 and TNFα also appear useful indicators of declining lung function. Neutrophil-derived elastase is a potent mucus-secretagogue and COPD patients with the highest decline in lung function also had chronic mucus secretion (Citation[25]). Recent studies (Citation[26], Citation[27]) have suggested that systemic inflammation may also have an effect on lung function. There was an inverse association between pulmonary function and C-reactive protein (CRP) levels (Citation[26]) and increases in serum CRP over time were correlated with steeper FEV1 decline (Citation[27]). Plasma fibrinogen levels are also associated with decline in lung function (Citation[28]).
The number of leukocytes (CD45(+) -cells) in airway tissue was related to lung hyper-inflation, measured as residual volume (Citation[29]). Indeed, it has been suggested that the abnormal inflammatory response that characterises COPD could augment dynamic hyper-inflation through oedema, alveolar destruction and mucus plugging (Citation[30]). Saetta et al. (Citation[31]) have reported a negative correlation between airway inflammation and the number of intact alveolar attachments and the number of macrophages in the alveolar airspaces increases with emphysema score (Citation[32]). Conversely, dynamic hyper-inflation could contribute to increased inflammation in the lung via cellular stretching, damage and hypoxia (Citation[30]).
There is, therefore, evidence that inflammation (airway and systemic) may contribute to abnormal lung function in COPD (). In turn, reduced pulmonary function as assessed by FEV1 and FVC is related to an increase in mortality in COPD patients (Citation[33]). For every 10% decrease in FEV1, all-cause mortality increases by 14%, cardiovascular mortality by 28% and non-fatal coronary events by 20% (Citation[34]).
Figure 1 Hypothetical model: associations between the underlying disease and clinical manifestations in COPD.
Inflammation and COPD exacerbations
A number of studies have shown that airway inflammation in patients with stable COPD increases further during exacerbations of the disease (Citation[35], Citation[36]).This increase in inflammation occurs in some, but not all individuals.The causes of exacerbations of COPD are heterogeneous, but bacterial and viral infection, which are major triggers have been shown to increase airway inflammation, for example, by releasing cytokines and chemokines from the epithelium (Citation[37]). IL-8 and RANTES are increased during exacerbations (Citation[35]) and as chemo-attractants may be responsible for the elevated numbers of neutrophils and eosinophils respectively, which have been detected in the small airways (Citation[36]). Markers of oxidative stress (e.g., H2O2) are also increased in the breath condensates of COPD patients with an exacerbation compared with stable disease (Citation[38]). Similarly, systemic inflammation, as measured by circulating levels of CRP and fibrinogen, is enhanced during an exacerbation of COPD (Citation[39]).
Donaldson et al. (Citation[40]) have reported an increased annual decline in lung function in patients with frequent exacerbations (> 2.92/year), compared with infrequent exacerbations, thereby suggesting an association between inflammation, exacerbations and declining lung function (). Again, this does not prove causality.
Inflammation and disease co-morbidities
It is now clearly recognised that the inflammatory process in COPD extends beyond the lung, resulting in a state of persistent low-grade systemic inflammation (Citation[41]) that has been implicated in various complications of the disease. It is well recognised that COPD is associated with excess risk of cardiovascular morbidity and mortality. For example, approximately 50% of COPD deaths in moderate disease are related to asymptomatic cardiovascular disease (Citation[42]). Recently, arterial stiffness, an independent risk factor for cardiovascular disease, has been reported to be elevated in COPD and there was a strong correlation with circulating IL-6 levels (Citation[43]).
Exacerbations of COPD increase both airway and systemic inflammation and there is a direct correlation between inflammatory indices in the two compartments. Although there is no data showing that inflammation is directly associated with death in COPD, some elevated inflammatory mediators, such as CRP and fibrinogen, have been implicated in risk of cardiovascular disease. Serum CRP levels are in fact a predictor of all-cause and cardiovascular mortality (Citation[44]), with a relative risk of 4.03 for 1 year mortality being reported from the highest to the lowest CRP quintile. High CRP is associated with elevated levels of IL-6 (Citation[45]), which is synthesised and released by a number of both resident and infiltrating inflammatory cells in the lung ().
Figure 2 Potential link between airway inflammation and disease co-morbidities in COPD.
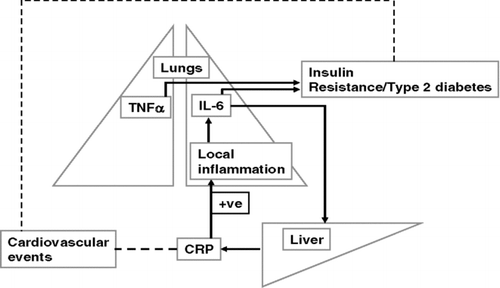
The Nurses Health Study (Citation[46]) showed that the age-adjusted relative risk for Type II-diabetes is 1.8 for COPD, compared with 1.0 for asthma. There is evidence of increased insulin-resistance in COPD, which is significantly correlated with circulating concentrations of IL-6 and TNFα sRI (Citation[47], Citation[48]). Many COPD patients also suffer from cachexia leading to muscle weakness and wasting (Citation[49]) as a result of apoptosis of skeletal muscle cells (Citation[50]). TNFα, which is a cachexic cytokine, is significantly elevated in mild-moderate and severe COPD compared with smokers (without COPD) and healthy controls (Citation[51]). There is, therefore, a possible direct or indirect link between airway inflammation, systemic inflammation and disease co-morbidities such as cardiovascular disease (Citation[42]), diabetes (Citation[46]), and osteoporosis (Citation[43]) in COPD ( and ).
Exacerbations, health status and mortality
There is a weak association between declining lung function and health status (Citation[40]). Annual exacerbation frequency has, however, been shown to have a negative impact on health status. COPD patients having 0-2 exacerbations per year had an SGRQ score of 48.9 compared with 64.1, where the exacerbation frequency was 3–8 (Citation[52]). In another study, following hospitalisation for an exacerbation, health status improved significantly over a period of 26 weeks, provided there was no further exacerbation (Citation[53]). However, if the patient had an additional exacerbation, then the SGRQ score remained > 50 even after 6 months.
Exacerbations of COPD are also a predictor of mortality. A review by Donaldson and Wedizcha (Citation[54]) showed that in-hospital mortality rates were 8% and 11% in the Netherlands and United States. However, these rose to 23% and 43%, respectively, in the year following discharge. A close association is therefore apparent between exacerbations, deteriorating health status and mortality in COPD. Finally, airway inflammation has been shown to contribute to changes in health status (Citation[55]) and progression of disease (Citation[56]) in patients with COPD.
THE MODEL: POTENTIAL MECHANISM (S) FOR THE EFFECT OF SFC ON MORTALITY IN COPD
The model places inflammation at the centre of the disease process in COPD (). This has yet to be confirmed, but evidence is accumulating that inflammation(airway and systemic), either directly or indirectly, contributes to disease severity and disease progression.
Effects of SFC on inflammation
As discussed previously, airway inflammation in COPD is distinct from that in asthma (Citation[13]). A number of studies have found only modest anti-inflammatory activity for inhaled corticosteroids (ICS) in COPD (Citation[57], Citation[58], Citation[59]). Over 3–6 months, FP (500 μ g b.d.) resulted in small changes in CD68 (+)-cells (macrophages) and reduced the CD8/CD4-cell ratio in the epithelium (Citation[59]). However, three biopsy studies have now confirmed a broad-spectrum anti-inflammatory effect for SFC therapy in COPD patients. In the first of these (Citation[60]), three months treatment with SFC (50/500 μ g b.d.) significantly reduced the number of CD45(+), CD4(+), CD8 (+)-cells, mast cells and cells staining positive for IFNγ and TNFα mRNA.
In parallel, there was a time-dependent decrease in the sputum neutrophil differential count, which was significant after 13 weeks compared with placebo (Citation[60]). The degree of change in tissue CD8 (+)-cells and in sputum neutrophils was of an order associated with clinically relevant changes in lung function (Citation[23], Citation[24]). A second study used SFC (50/500μ g b.d.), FP (500 μ g b.d.) and placebo for 3 months. In this study, there was a reduction in CD8(+) and CD68+ cells in the SFC group whereas FP alone had no effect (Citation[61]). In addition, the significant increase in neutrophils observed after FP treatment, was not seen with SFC. Finally, over 6 months, SFC reduced CD8 (+) cells and CD4 (+) cells in the biopsy tissue, whereas FP (500 μ g b.d.) only affected (and to a lesser extent) CD4(+)-cells (Citation[62]). It is interesting, therefore, that SFC, which demonstrates anti-inflammatory in COPD patients, also possibly reduces mortality whilst FP, which does not appear to impact inflammation to the same degree, has no effect.
In support of this hypothesis, in patients with moderate-severe COPD, SFC also decreased arterial stiffness, which is associated with systemic inflammation, after 8 weeks treatment (Citation[63]). In parallel, statins, which also have anti-inflammmatory effects, reducing for example, serum levels of CRP, were associated with a 43% improvement in survival (p = 0.009) after an exacerbation of COPD (Citation[64]).
A possible explanation for the difference between SFC and FP is the synergistic interaction which has been demonstrated between long-acting β2-agonists (LABAs) such as salmeterol and formoterol and corticosteroids (Citation[65], Citation[66]). Fundamental to the anti-inflammatory activity of ICS is binding of the corticosteroid molecule to the intra-cellular glucocorticoid receptor (GR) and subsequent translocation of the active receptor complex from the cytosol to the nucleus of the cell (Citation[67]). Binding to glucocorticoid responsive elements (GRE) on target genes (transactivation) or protein-protein interactions with transcription factors (transrepression) then evoke the anti-inflammatory effects of the corticosteroid (Citation[67]). In a study by Haque et al. (Citation[68]), salmeterol was shown to enhance both the extent and duration of GR translocation following stimulation with FP in airway epithelial cells.
The mechanism of this effect appears to involve phosphorylation of the GR via a MAP kinase pathway (Citation[65], Citation[66]) and activation of the binding enhancer protein CEBP alpha (Citation[69]). The resulting synergy has been demonstrated in a number of cellular systems relevant to COPD, such as suppression of cigarette-smoke induced IL-8 production by macrophages (Citation[70]) and inhibition of rhinovirus-stimulated RANTES release by airway epithelial cells (Citation[37]). In peripheral blood mononuclear cells taken from COPD patients, which exhibit reduced sensitivity to corticosteroids ex vivo, the addition of a LABA restored sensitivity towards the normal range (Citation[71]). Inhalation of SFC (50/100 μ g) by COPD patients significantly enhanced GR translocation and GRE binding in sputum macrophages compared with FP (100 μ g) alone, and equivalent to a five-fold higher dose of the ICS (Citation[72]).
Effects of SFC on exacerbations
Exacerbations have been shown to be a risk factor for increased mortality (Citation[54]). In the TORCH study, SFC reduced the rate of moderate-severe exacerbations of COPD by 25%, which was significantly greater than salmeterol, FP or placebo (Citation[9]). It is possible that the survival benefit of SFC in TORCH is the result of a reduction in exacerbations. However, FP also reduced exacerbations (18%) but had no effect on mortality. Importantly, in the INSPIRE trial (Citation[73]) both SFC and tiotropium had the same effect on exacerbations (1.28/yr, 1.32/yr respectively), but there was a significant (p = 0.031) decrease in all-cause mortality over 2 years in the SFC group (21/641) compared with tiotropium (38/650). Further analysis showed less cardiovascular deaths with SFC (9/641) than tiotropium (19/650). This may suggest that a reduction in exacerbations per se may not provide a survival benefit and that an additional effect is required. Muscarinic receptors are present on inflammatory cells, including lymphocytes and macrophages (Citation[74]), but there is no evidence of any anti-inflammatory activity of tiotropium in vivo or in the COPD patient (Citation[75]). The UPLIFT study (Citation[76]) may provide additional insight on the relative impact of bronchodilatation on exacerbations and mortality.
Effects of SFC on lung function decline
Finally, it is possible that the reduction in mortality in COPD patients with SFC is due to an effect on declining lung function (). Over the first 24 weeks of treatment, there was a improvement in FEV1 of approximately 75 mls over baseline, whereas with placebo, there was no change (Citation[9]). At the end of 3 years, patients in the SFC group, had an increase in post-bronchodilator FEV1 of approximately 30 mls compared with baseline, whereas there was a loss of 62 mls in the placebo group. The effects of SFC were significantly (p < 0.001) greater than salmeterol (−21 mls) or FP (−15 mls) alone (Citation[9]).
After 24 weeks, there was a disease-related decline in lung function in all the groups (Citation[9]). However, the rate of decline in FEV1 was significantly slower (p < 0.003) in the SFC-treated patients compared with those receiving placebo, suggesting a “disease-modifying” effect (Citation[10]). In the INSPIRE study (Citation[73]), there was little difference between the effects of SFC and tiotropium on change in lung function, but a significant mortality benefit with the LABA/ICS combination. Finally, statin use, associated with anti-inflammatory and anti-oxidant effects, has been reported to reduce the decline in lung function in the elderly population (Citation[77]) and mortality in COPD (Citation[64]).
CONCLUSION/PROPOSAL
COPD is a disease with a multi-component pathophysiology in which inflammation plays a key role. Although there are possible alternative explanations, taken together, the TORCH, INSPIRE and biopsy data suggest that an anti-inflammatory effect (airway and/or systemic) of SFC may be the mechanism by which it provides a potential survival benefit in COPD patients. It is possible that the demonstrated synergy between salmeterol and FP in COPD, thereby reversing the sub-sensitivity to ICS characteristic of the disease, results in enhanced anti-inflammatory activity in the airways (as demonstrated in the three biopsy studies) and this is responsible for the decrease in mortality exhibited by SFC in the TORCH study (). It may also contribute to the reduction in exacerbations and the slowing of the decline in lung function (). Alternatively, such an anti-inflammatory effect may prolong survival by impacting on systemic inflammation and disease co-morbidities ().
Figure 3 Possible mechanisms for a survival benefit of salmeterol/fluticasone propionate combination in COPD patients.
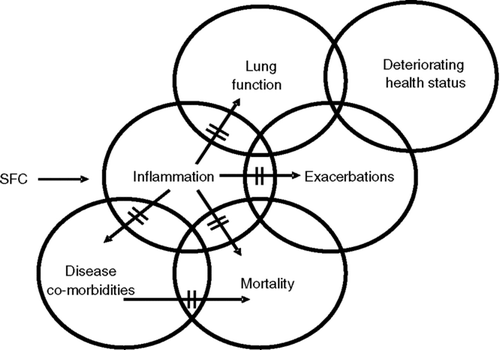
All in all, by reducing inflammation, SFC may affect the underlying disease process to improve lung function, decrease exacerbations and prolong survival. These hypotheses cannot be proven by the current clinical studies, but it is important to recognise that there are potential mechanisms, acting alone or in concert, that could explain the data from the TORCH study. As more studies become available, which compare drugs that are purely bronchodilators, such as anti-cholinergics, and combination therapy with ICS and LABA on a variety of outcome measures, including decline of lung function, exacerbations and mortality, we will be able to determine if the hypothesis that the reduction in mortality is associated with inhibition of inflammation is correct.
REFERENCES
- Pauwels R A, Buist A S, Calverley P M, et al. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD). Am J Respir Crit Care Med 2001; 163: 1256–1276
- Murray C J, Lopez A D. Alternative projections of mortality and disability by cause 1990–2006: Global Burden of Disease Study. Lancet 1997; 349: 1498–1504
- Ferlay J, et al. Globocan 2002, Cancer Incidence, mortality and prevalence worldwide. IARC press, Lyon 2004
- Mannino D, Homa D M, Akinbami L J, et al. Chronic obstructive pulmonary disease surveillance–United States 1971–2000. In Surveillance Summaries MMWR 2002; 51: 1–16
- DeMarco R, Accordini S, Cerveri I, et al. An international survey of Chronic Obstructive Pulmonary Disease in young patients according to GOLD stages. Thorax 2004; 59: 120–125
- Anthonisen N R, et al. The effects of a smoking cessation intervention on 14.5 year mortality: a randomised clinical trial. Ann Intern Med 2005; 142: 233–239
- Nocturnal Oxygen Therapy Trial Group. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease. Ann Intern Med 1980; 93: 391–398
- Fishman A, Martinez F, Naunheim K, et al. A randomised trial comparing lung-volume–reduction surgery with medical therapy for severe emphysema. New Engl J Med 2003; 348: 2059–2073
- Calverley P MA, Anderson J A, Celli B, et al. Salmeterol and fluticasone propionate and survival in Chronic Obstructive Pulmonary Disease. N Engl J Med 2007; 356: 775–789
- Celli B R, Thomas N E, Anderson J A, et al. Effect of pharmacotherapy on rate of decline of lung function in COPD: Results from the TORCH study. Am J Respir Crit Care Med 2008; 178: 332–338, doi:10.1164/rccm.200712-18690C
- Wilt T J, Bloomfield H E, Macdonald R, et al. Effectiveness of statin therapy in adults with coronary heart disease. Arch Intern Med 2004; 164: 1427–1436
- Agusti A GN. COPD, a multi component disease: implications for management. Resp Med 2005; 99: 670–682
- Jeffery P K. Differences and similarities between chronic pulmonary disease and asthma. Clin Exp Allergy 1999; 29: 14–26
- Hogg J C, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2645–2653
- Ronchi M C, Piragino C, Rosi E, et al. Role of sputum differential cell count in detecting airway inflammation in patients with COPD. Thorax 1996; 51: 1001–1004
- Riise G C, Ahlstedt S, Larsson S, et al. Bronchial inflammation in chronic bronchitis assessed by measurement of cell products in bronchial lavage. Thorax 1995; 5: 360–365
- Eriksson S. Pulmonary emphysema and α1-antitrypsin deficiency. AnnN Y Acad Sci 1991; 624: 1–6
- Thompson A B, Daughton D, Robbins R A, et al. Intraluminal airway inflammation in chronic bronchitis. Characterisation and correlation with clinical parameters. Am Rev Respir Dis 1989; 140: 1527–1537
- Saetta M, Turato G, Facchini F M, et al. Inflammatory cells in the bronchial glands of smokers with chronic bronchitis. Am J Respir Crit Care Med 1997; 156: 1633–1639
- Gamble E, Grootendorst D C, Hattotuwa K, et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers:a pooled analysis. Eur RespirJ 2007; 30: 1–5
- Wouters E FM. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2: 26–33
- O'Shaughnessy T C, Ansari T W, Barnes N C, et al. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+T lymphocytes with FEV1. Am J Respir Crit Care Med 1997; 155: 852–857
- Saetta M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157: 822–826
- Stanescu D, Sanna A, Veriter C, et al. Airways obstruction, chronic expectoration and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 1996; 51: 267–271
- Vestbo J, Prescott E, Lange P. Association of chronic mucus hypersecretion with FEV1 decline and chronic obstructive pulmonary disease mortality. Am J Respir Crit Care Med 1996; 153: 1530–1535
- Aronson D, Roterman J, Yigla M, et al. Inverse association between pulmonary function and C-reactive protein in apparently healthy subjects. Am J Repir Crit Care Med 2006; 174: 626–632
- Shaaban R, Kony S, Driss F, et al. Change in C-reactive protein levels and FEV1 decline: a longitudinal population-based study. Resp Med 2006; 100: 2112–2120
- Donaldson G C, Seemungal T A, Patel I S. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 2005; 128: 1995–2004
- Turato G, Zuin R, Miniati M, et al. Airway inflammation in severe chronic obstructive pulmonary disease: relationship with lung function and radiologic emphysema. Am J Respir Crit Care Med 2002; 166: 105–110
- Agusti A, Soriano J B. Dynamic hyperinflation and pulmonary inflammation: a potentially relevant relationship?. Proc Am Thorac Soc 2007; 4: 522–526
- Saetta M, Ghezzo H, Wong Dong K, et al. Loss of alveolar attachments in smokers. A morphometric correlate of lung function impairment. Am Rev Respir Dis 1985; 132: 894–900
- Retamales I, Elliott W M, Meshi B, et al. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am J Respir Crit Care Med 2001; 164: 469–473
- Hole D J, Watt G C, Davey-Smith G, et al. Impaired lung function and mortality risk in men and women: findings from the Renfrew and Paisley prospective population study. Br MedJ 1996; 313: 711–751
- Sin D D, Man S F. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases: The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation 2003; 107: 1514–1519
- Zhu J, Qui Y S, Majumdar S, et al. Exacerbations of bronchitis. Bronchial eosinophilia and gene expression for interleukin-4, interleukin-5 and eosinophil chemoattractants. Am J Respir Crit Care Med 2001; 164: 109–116
- Saetta M, Di Stefano A, Maestrelli P, et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med 1994; 150: 1646–1652
- Edwards M R, Johnson M, Johnston S L. Combination therapy: synergistic suppression of virus-induced chemokines in airway epithelial cells. Am J Respir Cell Mol Biol 2006; 34: 616–624
- Dekhuijzen P NR, Aben K KH, Dekker I, et al. Increased exhalation of hydrogen peroxide in patients with stable and unstable COPD. Am J Respir Crit Care Med 1996; 154: 813–816
- Wedzicha J A, Seemungal T A, MacCallum P K, et al. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost 2000; 84: 210–215
- Donaldson G C, Seemungal T AR, Bhownik A, et al. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57: 847–852
- Gan W Q, Man S F, Senthilselvan A, et al. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax 2004; 59: 574–580
- Mannino D M, Brown C, Giovina G A. Obstructive lung disease deaths in the United States from 1979 through 1993. An analysis using multiple-cause mortality data. Am J Respir Crit Care Med 1997; 156: 814–818
- Sabit R, Bolton C E, Edwards P H, et al. Arterial stiffness and osteoporosis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 175: 1259–1265
- Man S FP, Connett J E, Anthonisen N R, et al. C-reactive protein and mortality in mild to moderate chronic obstructive pulmonary disease. Thorax 2006; 61: 849–853
- Pai J R, Pischon T, Ma J, et al. Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med 2004; 351: 2599–2600
- Rana J S, Mittleman M A, Sheikh J, et al. Chronic obstructive pulmonary disease, asthma and risk of type 2 diabetes in women. Diabetes Care 2004; 27: 2478–2484
- Pradham A D, Manson J E, Rifai N, et al. C-reactive protein, interleukin-6 and risk of developing type 2 diabetes mellitus. JAMA 2001; 286: 327–334
- Hotamisligil G S. The role of TNF ∝ and TNF receptors in obesity and insulin resistance. J Intern Med 1999; 245: 621–625
- Bernard S, Le Blanc P, Whittom F, et al. Peripheral muscle weakness in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 158: 629–634
- Agusti A G, Sauleda J, Miralles C, et al. Skeletal muscle apoptosis and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166: 485–489
- De Godoy I, Donahoe M, Calhoun W J, et al, Elevated T NF. ∝ production by peripheral blood monocytes of weight-losing COPD patients. Am J Respir Crit Care Med 1996; 53: 633–637
- Seemungal T AR, Donaldson G C, Paul E A, et al. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157: 1418–1422
- Spencer S, Jones P W. GLOBE study group. Time course of recovery of health status following an infective exacerbation of chronic bronchitis. Thorax 2003; 58: 589–593
- Donaldson G C, Wedzicha J A. COPD exacerbations: 1. Epidemiology. Thorax 2006; 61: 164–168
- Snoeck-Stroband J B, Postma D S, Lapperre T S, et al. Airway inflammation contributes to health status in COPD: a cross-sectional study. Respir Res 2006; 7: 140
- Parr D G, White A J, Bayley D L, et al. Inflammation in sputum relates to progression of disease in subjects with COPD:a prospective descriptive study. Respir Res 2006; 7: 136
- Keatings V S, Jatakanon A, Worsdell Y M, et al. Effects of inhaled and oral corticosteroids on inflammatory indices in asthma and COPD. Am J Respir Crit Care Med 1997; 155: 542–548
- Verhoeven G T, Hegmans JP JJ, Mulder P GH, et al. Effects of fluticasone in COPD patients with bronchial hyperresponsiveness. Thorax 2002; 57: 694–700
- Hattotuwa K L, Gizycki M, Ansari T W, et al. The effects of inhaled fluticasone on airway inflammation in chronic obstructive pulmonary disease: a double-blind placebo-controlled biopsy study. Am J Respir Crit Care Med 2002; 165: 1592–1596
- Barnes N C, Qui Y S, Pavord I D, et al. Anti-inflammatory effects of salmeterol/fluticasone propionate in chronic obstructive lung disease. Am J Respir Crit Care Med 2006; 173: 736–743
- Bourbeau J, Christodoulopoulos P, Maltais F, et al. Effect of salmeterol/fluticasone combination on airway inflammation in COPD: a randomised trial. Thorax 2007; 62: 938–943
- Gosman M ME, Lappere T S, Snoek-Stroband J B, et al. Effect of 6 months therapy with inhaled fluticasone propionate (FP) with or without salmeterol (S) on bronchial inflammation in COPD. Proc Am Thorac Soc 2006; 3: A111
- Sabit R, Bolton C E, Allanby C, et al. Arterial stiffness is reduced by combination inhaled corticosteroid/long-acting beta -2 agonist therapy in patients with COPD. Thorax 2007; 62: A142
- Soyseth V, Brekke P H, Smith P, et al. Statin use is associated with reduced mortality in COPD. Eur Respir J 2007; 29: 279–283
- Johnson M. Interaction between corticosteroids and β2-agonists in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc 2004; 1: 200–206
- Adcock I M, Maneechotesuwan K, Usmani O. Molecular interactions between glucocorticoids and long-acting β2–agonists. J Allergy Clin Immunol 2002; 110: S261–S268
- Barnes P J. Molecular mechanisms of steroid action in asthma. J Allergy Clin Immunol 1997; 1: 159–168
- Haque R A, Johnson M, Adcock I M, et al. Addition of salmeterol to fluticasone prolongs the retention of glucocorticoid receptors within the nucleus of BEAS-2B cells and enhances downstream glucocorticoid effects. Proc Am Thorac Soc 2006; 3: A78
- Roth M, Johnson P R, Rudiger J J, et al. Interaction between glucocorticoids and β2-agonists on bronchial airway smooth muscle cells through synchronised cellular signalling. Lancet 2002; 360: 1293–1299
- Sarir H, Mortaz E, Karimi K, et al. Combination of fluticasone propionate and salmeterol potentiates the suppression of cigarette smoke-induced IL-8 production by macrophages. Eur J Pharmacol 2007; 571: 55–61
- Tp Y, Ito M, Adcock I M, et al. Formoterol enhances anti-inflammatory effects of corticosteroids in peripheral blood mononuclear cells from COPD patients. Eur Respir J 2006; 28: 215S
- Haque R A, Torego A, Essilfie-Quaye, et al. Effect of salmeterol and fluticasone on glucocorticoid receptor translocation in sputum macrophages and peripheral blood mononuclear cells from patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006; 3: A848
- Wedzicha J A, Calverely P MA, Seemungal T A, et al. The prevention of chronic pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med 2008; 177: 19–26
- Verbout N G, Lorton J K, Jacoby D B, et al. A functional role for muscarinic receptors on eosinophils in the airways. Proc Am Thorac Soc 2006; 3: A587
- Powrie D J, Wilkinson T MA, Donaldson G C, et al. Effect of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur RespirJ 2007; 30: 472–478
- Decramer M, Celli B, Taskin D, et al. Clinical Trial Design Considerations in assessing long-term functional impacts of tiotropium in COPD: The UPLIFT trial. J COPD 2004; 1: 303–312
- Alexeeff S E, Litonjua A A, Sparrow D, et al. Statin use reduces decline in lung function. Am J Respir Crit Care Med 2007; 176: 742–747