Abstract
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are responsible for a large proportion of the health care dollar expenditure, morbidity, and mortality related to COPD. Respiratory infections are the most common cause of acute exacerbations, but recent evidence indicates that the importance of respiratory syncytial virus (RSV) infection in COPD is under-appreciated. Improved detection of RSV using techniques based on the polymerase chain reaction accounts for much of the increased recognition of the importance of this virus in COPD patients. Furthermore, COPD patients may be more susceptible to RSV infection, possibly due to RSV-or immune response-induced pulmonary effects that are altered by age, environmental exposures, genetics, COPD itself, or a combination of these. However, although RSV infection occurs throughout life, viral and host factors that place COPD patients at increased risk are unclear. The complexities of RSV effects in COPD present opportunities for research with the goal of developing approaches to selectively modify damaging viral effects (e.g., altered airway function), while retaining beneficial immunity (e.g., clearance of virus) in COPD patients. This review explores the role RSV plays in acute exacerbations of COPD, the potential for RSV disease in chronic stable COPD, and newer concepts in RSV diagnosis, epidemiology, and host defense.
INTRODUCTION
Chronic Obstructive Pulmonary Disease (COPD) is a major cause of morbidity and mortality in the United States and around the world. It is the 4th-leading cause of death in the U.S., with an age-adjusted mortality rate that is rising (Citation[1]). The U.S. veteran population has one of the highest burdens of COPD (Citation[2]). It is notable that exacerbations of this disease account for a large proportion of the health care dollar expenditure, morbidity, and mortality related to COPD (Citation[3]). Respiratory viral and bacterial infections are the most common cause of acute exacerbations of COPD (AECOPD) (Citation[4]).
Studies that detect viral infection using methods based on the polymerase chain reaction (PCR) have determined the incidence of virus-related AECOPD to be 40–56% (Citation[5], Citation[6], Citation[7], Citation[8], Citation[9]). These observations indicate the importance of developing a better understanding of the interactions between viruses and the human lung in normal individuals and patients with COPD. Viruses that are commonly associated with AECOPD include influenza viruses, picornaviruses, coronaviruses, and paramyxoviruses. Although the paramyxovirus respiratory syncytial virus (RSV) is well known as an important respiratory pathogen in infants and young children, it is increasingly being recognized as a significant cause of morbidity and mortality in older adults and COPD patients (Citation[7]). Furthermore, COPD patients may be more susceptible to RSV infection and are potentially chronic carriers of this virus. This review explores the role RSV plays in AECOPD and chronic stable COPD, with focus on newer concepts in RSV diagnosis, epidemiology, and host defense.
CLINICAL FEATURES AND DIAGNOSIS OF RSV INFECTION
The importance of RSV in the elderly and especially in AECOPD has been poorly recognized. Comprehensive reviews of this virus published as recently as 2001 acknowledge that RSV has primarily been known as a virus that infects children (Citation[10]). Factors relating to RSV being synonymous with pediatric disease are mainly historical and include its first description in infants, the epidemic nature of RSV disease in children, the ability to diagnose RSV infection in infants with nasal antigen testing, and the severity of RSV bronchiolitis in some infants and children (Citation[10], Citation[11]). In contrast, RSV in the adult population has defied precise clinical and epidemiologic correlates that separate it from other respiratory viruses (Citation[10]). Only recently, with the advent of improved detection by PCR has the importance of RSV in adults been recognized.
Clinical symptoms of RSV infection in all age groups include upper or lower respiratory viral infection and/or respiratory distress. Symptoms in RSV-associated AECOPD include nasal congestion, sore throat, hoarseness, cough, increased or altered sputum, dyspnea, wheezing, chest pain, myalgia, and fever. RSV-associated symptoms are generally indistinguishable from other respiratory viral infection, including influenza virus (Citation[5], Citation[7], Citation[8]). RSV infections in young adults are usually self-limited and mild, but infections in COPD patients are often of greater severity and may require hospitalization (Citation[7]). The severity of RSV infection is host-dependent and varies with age, immune status, and location of respiratory system involvement (Citation[10]).
In regard to the diagnosis of RSV infection, older studies that focused on the incidence of RSV infection appear to have largely underestimated the involvement of this virus. However, current PCR-based diagnostics have greatly increased sensitivity and specificity for detection of RSV. Indeed this is true for the majority of respiratory viral infections as studies that included testing for multiple different viruses using PCR (including RSV) demonstrated markedly increased virus detection rates. For example, PCR without and with reverse transcription (RT) doubled the yield for detection of respiratory viruses over culture methods in an ambulatory cohort of young healthy patients presenting with respiratory infection symptoms (Citation[12]). A study of wheezing children ages 9–11 documented virus infection in 80–85% using RT-PCR techniques, which is markedly higher that previous reports using other detection methods (Citation[13]).
Similarly, using a combination of RT-PCR, complement fixation, and enzyme-linked immunosorbent assay (ELISA), at least half of adult asthma exacerbations were found to be associated with infection by respiratory viruses (Citation[14]). For COPD patients, diagnosis using PCR has also detected higher incidences of respiratory viral infection. For example, one study using traditional serology and culture methods detected respiratory virus infection in 19% of patients with AECOPD (Citation[6]). In contrast, PCR-based analysis of nasal secretion sampling or throat swabs from the same cohort increased the detection rate for respiratory viruses to 42%. Similar findings extend to studies of critically ill patients with AECOPD in which non-PCR-based diagnosis resulted in a reported incidence of RSV and all viruses of approximately 2% and 10–16%, respectively (Citation[15], Citation[16]). In contrast, although the cohort was not the same, use of PCR-based detection combined with other techniques for diagnosis in one intensive care unit (ICU) resulted in a detected incidence of RSV and all viruses in AECOPD of 6.5% and 43%, respectively (Citation[17]). Other studies verify that PCR-based testing of samples has significantly increased sensitivity and specificity in diagnosis of RSV and other respiratory viral infections (Citation[5], Citation[18], Citation[19]).
An ideal method for detection of viral infection should be safe, convenient, promote early detection, and test positive during the entire period of active infection. Nasopharyngeal swab or washes and sputum samples are often used in PCR analysis of RSV-associated AECOPD (Citation[5], Citation[7]). However, it is unclear which site is optimal and whether sites should be used alone or in combination for detection of RSV infection. In one study of respiratory viral infection in AECOPD that included RSV, detection rates for respiratory viruses by PCR were 31% from nasopharyngeal samples and 47% from induced sputum. Diagnostic yield correlated with duration of symptoms at initial presentation, with patients with positive nasopharyngeal samples being symptomatic for a mean of 8 days, compared to 13 days for patients with positive sputum samples (Citation[5]).
It is unclear if differences in yield between sites reflected a preferred site (upper versus lower airways) for virus infection in AECOPD or greater severity and/or persistence of infection in laryngeal, tracheal, or bronchial locations. Nevertheless, these results suggest that sputum samples display greater sensitivity in PCR analysis of virus-induced AECOPD. In contrast, there is controversy regarding the safety and utility of induced sputum samples in AECOPD, especially since a positive sputum sample might simply represent a chronic RSV carrier state or viral persistence (Citation[9], Citation[20]). In addition, nasopharyngeal swabs are more convenient and reliably obtained. Thus, a common research practice in detection of RSV infection in AECOPD that combines optimal sensitivity with convenience is to collect nasopharyngeal swab samples for PCR analysis during the symptomatic phase and combine this with serologic testing (Citation[7], Citation[9]).
EPIDEMIOLOGY OF RSV INFECTION IN COPD PATIENTS
AECOPD can be precipitated by multiple factors including infection (viral and bacterial) and certain environmental exposures (e.g., air pollution, ozone, nitrogen dioxide, microfine particulates, and temperature) (Citation[21], Citation[22]). Prior to the use of PCR-based techniques for viral detection in patient samples, approximately 50–70% of exacerbations were considered due to infection, 10% due to environmental agents, and 30% due to unknown etiologies (Citation[23], Citation[24], Citation[25]). Infectious agents isolated were most often bacteria, with one study reporting frequencies of 11% Haemophilus influenzae, 10% Streptococcus pneumoniae, 10% Moraxella catarrhalis, 10% Haemophilus parainfluenzae, and 4% Pseudomonas aeruginosa (Citation[4]). Viruses were detected in less than 20% of AECOPD, even when viral culture, serologic testing, and/or antigen detection methods were used. However, more recent studies using PCR-based detection have found that up to 56% of exacerbations may be attributed to respiratory viruses (Citation[5], Citation[6], Citation[7], Citation[8], Citation[9]). Rhinovirus appears to be the virus most commonly detected in AECOPD, but RSV is also important and may accounts for up to 15% of all exacerbations (Citation[5]). Variation in the incidence of RSV infection among the different studies is likely attributable to differences in study populations, seasonal and regional variation, sample acquisition and type, and PCR assay systems ().
Table 1 Incidence of virus detection using testing that included PCR in adult populations with acute symptomatic respiratory illness
COPD has been identified as an independent and significant risk factor for RSV infection that causes severe illness, hospitalization, and ICU admission (Citation[26]). In two recent, prospective, adult cohort studies, 9–11.4% of patients with RSV-associated AECOPD required hospitalization, 13% of these hospitalized patients required intensive care treatment, and hospital mortality was approximately 8% (Citation[7], Citation[9]). Interestingly, mortality rates for RSV and influenza virus in AECOPD are similar, although the importance of influenza is more commonly acknowledged (Citation[7]).
The importance of RSV in the adult population can be further underscored by longitudinal data obtained in 1990–1999 by the Centers for Disease Control (Citation[27]). In this report, mortality from RSV was greatest in the adult population, with 78% of deaths due to RSV-related respiratory and circulatory exacerbations occurring in individuals over 65 years of age. Indeed, the all-cause death rate from RSV infection for those over 65 (29.6 per 100,000 person-years) was greater than for infants (5.4 per 100,000 person-years). Taken together, these studies confirm RSV as an important cause of morbidity and mortality in elderly and COPD populations.
One interesting and related question is whether the presence of COPD increases susceptibility to RSV infection. In one study, the incidence of symptomatic RSV infection in adults with COPD and cardiac disease was 1.5/100 person-months compared to 0.9/100 person-months in healthy adults (relative risk 1.61) (Citation[7]). This could be similar in other airway inflammatory diseases such as asthma. In a study involving young ambulatory adults, patients with RSV infection had a higher prevalence of asthma compared to other viral infections (Citation[12]). These findings might also be explained by altered disease severity although comparison between studies makes conclusions difficult. For example, in one study 50% of young healthy adults exposed to RSV in an experimental setting developed symptoms (Citation[28]). By comparison, in another report 73% of COPD patients followed closely for natural RSV infection were symptomatic (Citation[9]). Thus, it is unclear if differences in symptom rates between those with COPD or other obstructive lung diseases and healthy adults are a reflection of susceptibility to disease or increased symptom severity, and this question warrants further investigation.
RSV in stable COPD
The seasonality of RSV, with very little activity in summer months, has led to speculation about a natural “hiatal” reservoir for the virus (Citation[29], Citation[27], Citation[28], Citation[29], Citation[30], Citation[31]). To date, no animal reservoir is known to exist (Citation[20]). However, it is possible that after acute infection the reservoir for RSV is the host, and under suitable conditions viral reactivation could result in reinfection and recurrence of the natural cycle. Indeed, experimental RSV infection in animal models may result in acute infection followed by viral persistence in the lung detected at 60–100 days following inoculation (Citation[32], Citation[33]). In a more recent study, RSV infection of mice resulted in acute infection followed by resolution of all signs, symptoms, and detectable virus from BAL within 14 days. However, viral RNA was detected in lung tissue using PCR at 100 days after initial infection (Citation[34]). In addition, depletion of CD4+ and CD8+ T cells allowed infective virus to be recovered from the lungs of experimental mice (Citation[34]).
In archived postmortem lung tissue from infants, RSV was detected even in the summer months suggesting that the virus can persist in lungs after acute infection (Citation[35]). Taken together, these studies suggest the possibility that acute RSV infection in the winter with subsequent symptom resolution could be followed by chronic persistence in lung tissue. Furthermore, viral recovery and infectivity might be triggered through suppression of adaptive immunity resulting in reinfection and recurrence of the RSV season the following winter. This “hit and hide” hypothesis for RSV infection would be particularly relevant in elderly and COPD patients, who display age-related declines in adaptive immunity (Citation[34]).
A related concept is whether RSV or RSV-related viral components could persist in the lung and modulate lung function, as has been proposed for adenovirus (Citation[33], Citation[36]). For example, RSV persistence in the lungs of guinea pigs is associated with increased airway eosinophilia and airway hyper-responsiveness, even 100 days after infection (Citation[33]). In one report, 28.3% of COPD patients hospitalized for reasons other than AECOPD had RSV detected using PCR testing of their nasopharyngeal secretions, although viral load as assessed by RNA copy number was significantly lower when compared to children with acute RSV infection (Citation[19]). Another study in which a cohort of patients with moderate to severe COPD was followed for 16 months, RSV was detected in 23.5% of stable COPD patients using PCR testing of nasal aspirates (Citation[37]).
Furthermore, these patients with RSV infection had higher levels of plasma fibrinogen, serum IL-6, and arterial PCO2, and had more severe and frequent future AECOPD raising the question of whether chronic low-grade viral infection influences COPD severity. Indeed, a followup study of similar patients with stable moderate to severe COPD followed quarterly for two years found that subjects with persistent RSV detected through PCR testing of sputum samples had evidence of more airway inflammation and a greater decline in lung function over time as measured by FEV1compared to patients with RSV detected less frequently (Citation[20]).
However, evidence of RSV latency and persistence has been difficult to find consistently in humans. In one study from North America, sputum, nasal washes, and serum samples were collected every two months and during respiratory illnesses for one year from COPD patients both at baseline and during exacerbations (Citation[9]). In the 95 individuals that completed the study, the possibility of a chronic carrier state or persistent infection was considered in four patients because they had clinically unrecognized infection but were RSV positive by sputum PCR testing or rise in titer on serologic testing. Two of these patients were considered to have had an asymptomatic infection because they had a serologic response. The two other asymptomatic patients tested had no serologic response, and after initially having positive results for RSV by PCR testing of samples, viral RNA cleared on follow-up testing. These individuals were considered to have had an aborted infection, possibly cleared by the innate immune response.
Thus, the issue of RSV persistence versus intermittent subclinical infections remains controversial. Potential reasons for disparity in detection of persistent infection among studies include differences in sample populations such as the use of hospitalized COPD patients versus outpatients, unequal duration of follow-up, inclusion of serologic testing, use of different respiratory sampling and PCR techniques, and different study focus such as on pulmonary function or host response over time. A more definitive multicenter study with a larger patient population is needed to clarify this issue.
COMPARISON OF RSV TO INFLUENZA IN COPD EXACERBATION
The notion of RSV as a childhood respiratory illness and influenza as the more common adult respiratory illness has limited our understanding about the surprising similarities between the two RNA viruses. Elderly COPD and cardiac disease patients with RSV or influenza infection suffered similar symptoms, average 2-week duration of illness, functional impairment, medication use, radiology findings, and hospitalization rates (e.g., 16–20% in high risk ambulatory patients) (Citation[7]). Among hospitalized patients, lengths of stay, ICU admission rates (12–15%), mechanical ventilation need (10–13%), and mortality rate (7–8%) were similar (Citation[7]). Furthermore, RSV tracks with influenza virus in seasonality with a peak incidence around the same time in the winter (Citation[12], Citation[30], Citation[31]).
This temporal relationship appears to be specific for RSV and influenza because other important respiratory viruses, like rhinovirus and parainfluenza virus, are detected throughout the year (Citation[38]). The incidence of influenza in COPD patients is dependent on the dominance and virulence of the circulating strain and vaccination rates (Citation[7]). In contrast, RSV has more stable circulating strains and the incidence in COPD patients is subject to less variation (Citation[7]). Nonetheless, the incidences of influenza and RSV in COPD populations with high influenza vaccination rates when followed over many seasons are similar, as is the morbidity and mortality from the two diseases (Citation[7], Citation[27]).
HOST DEFENSE AGAINST RSV
RSV infection occurs throughout life, but viral and host factors that place COPD patients at increased risk are unclear. Possibilities include RSV-or immune response-induced pulmonary effects that are altered by age, environmental exposures, genetics, COPD itself, or a combination of these. The following section highlights different aspects of this important topic.
Host behavior and susceptibility to RSV
Tobacco use is the most important factor in the development of COPD, and about 15% of cigarette smokers develop clinically significant disease (Citation[39]). Indirect evidence from studies of other respiratory viruses suggests that constituents of cigarette smoke may impact susceptibility to and severity of RSV disease. For example, in mice exposed to chronic cigarette smoke, anti-adenoviral immunity has been shown to be impaired at multiple levels, including fewer antigen processing dendritic cells, impaired humoral immunity with decreased total levels and impaired neutralizing capacity of the serum IgG response, and altered adaptive immunity with both decreased total and activated CD4+ cell numbers and poor T cell effector function (Citation[40]).
In contrast, the increased mortality of cigarette smoke-exposed mice infected with influenza was not associated with an impairment in adaptive immunity, but rather with heightened innate inflammatory responses, including increased expression of tumor necrosis factor (TNF)-α, interferon (IFN)-α, and interleukin (IL)-6 (Citation[41]). These studies suggest an increased risk of respiratory viral disease with smoking, but interestingly also suggest virus-specific effects in the interplay between smoking and antiviral immunity. Whether other smoke-related mechanisms, such as an increased respiratory epithelial permeability, play a role in the increased risk of respiratory viral infection is subject to speculation (Citation[42]). There is a paucity of data linking smoking to increased susceptibility and severity of RSV disease. An improved understanding of cigarette smoke effects on RSV infection may also highlight important immune pathways in RSV disease.
Host genetics and susceptibility to RSV
There is very little information available on the genetic markers that predict either susceptibility to or severity of RSV infection in older individuals and COPD patients, as the majority of human genetic studies on RSV infection have been carried out in pediatric populations. As would be predicted, the majority of associations that have been reported for severe RSV infection are in genes that regulate antiviral defense, inflammation, and immunity. In vitro studies have shown that RSV infection up-regulates toll-like receptor (TLR) 4 expression and converts airway epithelial cells from a LPS-tolerant to LPS-sensitive phenotype (Citation[43]). This finding may correlate with observed associations between polymorphisms in the TLR4 receptor and severity of RSV infection (Citation[44], Citation[45]).
Other genes with polymorphisms that positively predict severity of RSV disease include surfactant proteins A-D, IL-4, −8, −10, and −13, IL-4 receptor, and CC chemokine receptor 5 gene (Citation[46], Citation[47], Citation[48], Citation[49], Citation[50], Citation[51], Citation[52], Citation[53], Citation[54]). In contrast, immune and inflammatory genes that have been shown to contain polymorphisms not associated with severity of RSV infection include intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1, E-selectin, IL-9 and TNF-α (Citation[51], Citation[55]). As the field of prediction genomics continues to evolve, the hope is that COPD patients and elderly individuals with the highest susceptibility to severe RSV infection can be identified. This would help not only in understanding disease pathogenesis, but will likely have profound implications for disease treatment and prevention.
Innate immunity and susceptibility to RSV
Epithelial cells lining the airway surface are targeted for viral infection and replication and are often the first host cell to encounter respiratory viruses (Citation[56]). Pathologic studies of infants who died following unexplained respiratory illnesses between 1925–1959, and more recently from confirmed RSV-induced bronchiolitis, indicate that distal airway and alveolar epithelial cells are the natural host cells for RSV infection (Citation[57], Citation[58]). In fact, in severe RSV bronchiolitis the distal nature of the disease correlates with infection of small bronchiole epithelium and type 1 and 2 pneumocytes. These cells are not passive bystanders during an assault of the epithelial barrier, but instead are active participants in antiviral responses (Citation[59], Citation[60], Citation[61]).
Following RSV infection, airway epithelial cells produce increased levels of potent inflammatory and immunomodulatory molecules important in pulmonary defense including IFN-β, IL-1α, IL-6, IL-8, IL-11, Regulated upon Activation, Normal T cell Expressed, and Secreted (RANTES), macrophage inflammatory protein-1α, TNF-α, granulocyte macrophage colony-stimulating factor, ICAM-1, major histocompatibility (MHC) class I, and inducible nitric oxide synthase (Citation[62], Citation[63], Citation[64], Citation[65], Citation[66], Citation[67], Citation[68], Citation[69], Citation[70], Citation[71]). In this way, epithelial cells contribute to the specific processes of cytokine generation, induction of the host cell antiviral state, and leukocyte recruitment, retention, and activation, that are critical for viral defense in the airway.
Efficient clearance of RSV from the respiratory system requires T cell-dependent cell mediated immunity that is regulated by interferons (Citation[72], Citation[73], Citation[74], Citation[75]). Interferons are a family of related cytokines that mediate diverse functions including antiviral, antiproliferative, antitumor, and immunomodulatory effects (Citation[76]). The two best studied types of interferons activate two different type-specific receptor complexes. Type I interferons, which are produced from 14 different IFN-α genes and one IFN-β gene (as well as the less well studied IFN-ω and IFN-τ genes), are released by most types of cells. IFN-β is generally produced directly by airway epithelial cells themselves (while IFN-α is often induced in leukocytes recruited to the airway) in response to viral infection (Citation[77]). This group of interferons activate many antiviral genes, particularly genes involved in establishment of a host cell antiviral state such as MxA, protein kinase R (PKR), 2′-5′oligoadenylate synthetase, and interferon regulatory factor-7 (Citation[78]). Type II interferon or IFN-γ is produced mainly by T cells and natural killer cells and also activates many antiviral genes, particularly genes that augment immune cell recognition of virus-infected cells. Airway epithelial cells are highly sensitive to IFN-γ, resulting in induction of genes that augment immune cell recognition of virus-infected cells, such as ICAM-1, major histocompatibility complex class I, and transporter associated with antigen processing-1 (Citation[61], Citation[79], Citation[80]). There are also several other antiviral genes induced by both the type I and II interferon pathways (Citation[81]). In total, the interferon system for antiviral defense, coupled with other innate and acquired immune mechanisms, most often results in clearance of pulmonary viral infection. However, the inflammatory and immune response generated to combat viral infections may contribute to lung damage or dysfunction under certain conditions. Therefore, tight regulation of airway defense mechanisms by interferons and other factors is critical for generation of an effective response to viral infection that does not result in uncontrolled cell loss.
Viruses have developed various strategies to overcome antiviral defense in host cells. One example is inhibition of type I interferon-dependent defense gene expression by RSV nonstructural protein 2 (Citation[77], Citation[82]). This RSV protein mediates proteasome-dependent degradation of the transcription factor signal transducer and activator of transcription (Stat) 2 that is required for responses to type I interferon. This early inhibition of type I interferon-dependent signaling is one mechanism through which RSV thwarts interferon-dependent antiviral mechanisms in infected airway epithelial cells. Another mechanism by which RSV potentiates infection is by delaying infection-induced apoptosis and allowing for prolonged infected cell survival (Citation[83]). RSV activates both pro- and anti-apoptotic pathways in airway epithelial cells, but during the first few hours of infection the anti-apoptotic effects that are mediated through phosphatidylinositol 3-kinase and possibly the downstream mediator AKT appear dominant (Citation[84]).
In addition, RSV activation of the epidermal growth factor receptor and subsequent ERK mitogen-activated protein (MAP) activation lead to an alteration in the Bcl2 protein balance, favoring survival and delayed apoptosis (Citation[85]). Furthermore, RSV delays apoptosis by decreasing the amount of the tumor suppressor and pro-apoptotic protein p53 (Citation[86]). RSV also modulates epithelial cell function and enhances inflammatory signals in airway epithelial cells. RSV infection increases the expression of TLR3 and PKR, leading to enhanced inflammatory response upon exposure to the viral replication intermediate, double-stranded RNA (Citation[87], Citation[88]).
Taken together, the published evidence strongly suggests that pulmonary responses to RSV infection are critical in determining the susceptibility and severity of RSV infection. Furthermore, the evolution of this virus has generated several mechanisms that do battle with pulmonary antiviral defenses. However, chronic cigarette smokers and COPD patients display structural and functional dysregulation of lung epithelia that likely impact antiviral defense. For example, cigarette smoke alters epithelial electrolyte transport and induces epithelial cell apoptosis (Citation[89], Citation[90]). Similarly, there is altered mediator expression and greater neutrophil infiltration in the airway epithelium of COPD patients (Citation[91], Citation[92]). Epithelial repair processes after injury are also impaired in COPD patients (Citation[93]). Whether these or other alterations of airway epithelia in COPD patients alter the biology of RSV and other respiratory viral infections is currently unknown. This area is primed for research that clarifies critical mechanisms of virus-induced exacerbations in COPD patients (Citation[94]).
Humoral immunity and susceptibility to RSV
At full-term birth, the risk of RSV infection is low because neonates receive maternal antibodies against RSV through passive placental transfer (Citation[95]). In the first 6 months of life, protection derived from maternal antibodies lasts about 2 months. For the next 4 months, there is a window of increased susceptibility due to waning maternal antibody levels combined with infant inability to mount an adequate humoral response (Citation[96]). Accordingly, the incidence of bronchiolitis is highest during this latter period. RSV infection after 6 months of age is associated with a more robust antibody response that correlates with milder disease. Antibody levels following first infection subsequently wane to undetectable levels after one year, which then places children at risk of RSV reinfection (Citation[96]). However, subsequent infections are usually milder and correlate with an accelerated memory humoral antibody response (Citation[96]). These findings establish a protective role for the humoral antibody response against RSV infection. Data showing that variations in antibody levels at birth correlate with both age of first infection and severity of infection supports this protective role. Accordingly, pre-term infants with lower antibody levels at birth are at a higher risk of severe RSV disease, and administration of monoclonal antibody to high-risk infants protects against severe RSV infection (Citation[97], Citation[98], Citation[99]).
In contrast to children, the correlation between viral immunity and antibody levels in adults is less well studied. Based on limited evidence, it appears that all asymptomatic adults, regardless of age and health status, have detectable antibody levels (Citation[100], Citation[101], Citation[102], Citation[103]). During RSV infection, adults develop a robust antibody response that subsequently wanes to a detectable baseline level in approximately one year (Citation[101], Citation[103]). Antibody levels in adults likely reflect persistent basal levels of antibodies following remote infection(s) in childhood or a cross-sectional “snap-shot” of antibody levels between adult infections. Baseline antibody titers in adults correlate with susceptibility to RSV infection, with lower anti-RSV antibody titers associated with viral infection (Citation[102]). In a cohort study of individuals age 10 month to 89 years, high antibody titers protected against RSV-related hospitalizations (Citation[100]).
Similarly, another report of elderly patients that included individuals with COPD and/or cardiac disease, or in good health, antibody titers determined at onset of illness correlated with severity of RSV infection (Citation[26]). For these patients, 100% of those with antibody titers < 8.01 log2 were hospitalized while no one with titers > 13.0 log2 was hospitalized. Indeed, a multivariate analysis of these patients indicated that titers < 10.0 log2 carried the greatest relative risk for hospitalization. Interestingly, in this analysis the presence of COPD emerged as the second most significant risk factor for hospitalization from RSV infection. Therefore, current information suggests an important role for both antibody titers and COPD in determining severity of RSV infection. Although decreases antibody levels against RSV have not been reported in COPD patients, it is known that antibody production is less efficient and antibody affinity for antigens is reduced with advancing age (Citation[104]). Accordingly, it is reasonable to speculate that decreased humoral immunity may affect defense against RSV in COPD patients.
Adaptive immunity and susceptibility to RSV
Severe and fatal RSV disease in immunocompromised children suggests a critical role for adaptive immunity in the control of viral infections (Citation[105]). In adults, the age-related decline in adaptive immunity likely also contributes to severity of RSV infection. In support of this possibility, efficient clearance of RSV from the respiratory system in murine models requires interferon-regulated T cell-dependent cell mediated immunity, particularly CD8+ T cell responses (Citation[72], Citation[73], Citation[74], Citation[75]). However, control of viral infection through CD8+ T cell function wanes with animal age (Citation[73], Citation[106]). Similarly, infection of senescence-accelerated mice by RSV resulted in greater severity of infection that was likely directly attributable to a lack of clonal expansion of CD4+ and CD8+ T cells together with a Th1/Th2 imbalance in the respiratory tract (Citation[107]). These findings in murine models appear to correlate with observations in humans. Under normal conditions, RSV infection in childhood results in a protective T lymphocyte response followed by creation of a memory-specific CD8+ T cell pool that is capable of re-expansion on re-challenge (Citation[108]). In both elderly adults and COPD patients, the circulating pool of memory-specific T cells against RSV is significantly lower than that of healthy adults (Citation[109]). Peripheral blood mononuclear cells from elderly individuals display a compromised in vitro IFN-γ response to RSV (Citation[110]).
Likewise, in CD4+ T cells isolated from elderly and young adults and subjected to in vitro RSV infection, the ratio of IL-10 (anti-inflammatory) versus IFN-γ (pro-inflammatory) producing cells was significantly lower in the elderly group (Citation[111]). Taken together, the findings suggest an age-related decline in adaptive immunity that predisposes to more severe RSV infection. Indeed, these findings are consistent with the established concept of cellular immune senescence and age-related decline in CD4+ and CD8+ T cell responses (Citation[112]). Another interesting aspect of T cell immunity to RSV is that the memory pool of T cells against RSV in COPD patients is significantly lower than the T cell pool against influenza, suggesting the possibility that virus-specific effects could modulate T cell immunity against RSV (Citation[113]). Therefore, it appears that both virus-specific mechanisms and an age-related decline of memory T cell function can modulate adaptive immunity, predisposing elderly (and COPD) patients to RSV infection.
Cytokine network alterations and susceptibility to RSV
COPD-related susceptibility to viral infection could also be related to alterations in cytokine production. One example may be the allergy-associated cytokine IL-13, which is produced by T helper type 2 lymphocytes, mast cells, eosinophils, and possibly alveolar macrophages (Citation[114], Citation[115]). IL-13 has a beneficial effect in protease/anti-protease imbalance models of COPD, and promotes anti-apoptotic effects on epithelial and endothelial cells, but can promote collagen synthesis and tissue fibrosis (Citation[116], Citation[117], Citation[118]). Moreover, IL-13 appears to play a protective role in modulating viral infections. In a murine model of RSV infection, elevated IL-13 levels decreased RSV disease (Citation[119]).
Further evidence for the role of IL-13 in RSV disease comes from a study of infants with RSV bronchiolitis in which certain IL-13 polymorphisms were associated with the risk of developing late-onset wheezing (Citation[120]). These findings are relevant because IL-13 levels are decreased in the lung tissue of COPD patients (Citation[121]). Taken together, these studies suggest that reduced IL-13 levels could not only contribute to development of emphysema but also enhance susceptibility to severe RSV disease. It is possible that other cytokines altered in COPD could have similar effects on RSV infection.
Viral factors and susceptibility to RSV
Two distinct antigenic groups (A and B) of RSV have been identified, but whether infection by different strains affects COPD differently is not clear (Citation[122]). Mice infected with two different strains of RSV under conditions in which there was little difference in viral titer throughout infection demonstrated strain-specific variations in cytokine expression, goblet cell hyperplasia, gob5 and MUC5AC expression, and airway hyperreactivity (Citation[123]). In contrast, studies in humans have not uncovered a clear difference in virulence or viral effects of different RSV strains. One cohort study of elderly patients and high risk adults found that group A and B contributed 45% and 55% to RSV infections, respectively (Citation[7]). Both groups circulated each year without significant difference in the percentage of each group detected in mildly and severely ill patients. Similarly, disease severity was not associated with virus group detected in infants hospitalized with RSV-induced bronchiolitis (Citation[124]). However, it remains conceivable that differences between strains might affect virulence or host effects of RSV disease. Further studies using molecular typing techniques and correlating disease severity to RSV strains are needed to help resolve this issue.
Viral-bacterial interactions and susceptibility to RSV
Interactions between RSV and bacteria in the airway could place COPD patients at an increased risk for AECOPD. COPD patients with acute RSV infections could be predisposed to bacterial super-infection and/or increased severity of AECOPD. For example, infection under in vitro conditions by RSV or certain other respiratory viruses increased adherence of both H. influenzae and S. pneumoniae to airway epithelial cells, possibly by viral induction of host cell bacterial receptor expression (Citation[125], Citation[126]). It has also been observed that the RSV G protein may function as a cell surface receptor for H. influenzae and S. pneumoniae, thereby facilitating increased bacterial binding to virus-infected cells (Citation[127]). Similarly, infection of calves with bovine RSV six days before Haemophilus somnus infection resulted in enhanced severity of infection compared to infection with bacteria alone (Citation[128]).
Conversely, bacterial colonization or infection of the airway in COPD patients could create a permissive environment for subsequent viral-induced AECOPD. Up to one-half of COPD patients have airway colonization by bacteria, often H. influenzae, M. catarrhalis, S. pneumoniae, and P. aeruginosa (Citation[129], Citation[130], Citation[131], Citation[132], Citation[133]). In these patients, AECOPD is often associated with an acquisition of a new bacterial strain that may be more pro-inflammatory (Citation[21], Citation[134]). Studies with other respiratory viruses support the possibility that bacteria may affect a later viral infection. Airway epithelial cells infected in vitro with H. influenzae and subsequently with rhinovirus display increased virus binding and epithelial cell release of neutrophil chemoattractant cytokines (Citation[135]). Furthermore, AECOPD patients with coinfected with H. influenzae and rhinovirus had greater symptom counts, airway IL-6 levels, and fall in lung function when compared to patients with AECOPD due to bacteria alone (Citation[136]). Taken together, the current body of evidence suggests that RSV and other respiratory viruses interact with bacteria in ways that enhance the severity of illness during AECOPD.
Other host factors and susceptibility to RSV
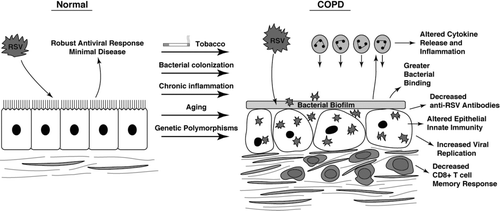
There are multiple potential mechanisms for increased risk of AECOPD after RSV infection in addition to effects on inflammation or immunity (). Respiratory viruses may compromise mucociliary clearance, causing retention of secretions and bacterial overgrowth (Citation[57], Citation[137]). Additionally, recurrent exacerbations predispose to an increased risk of future, more symptomatic exacerbations, and it is reasonable to propose that persistent inflammation in the airway could affect subsequent responses to viral infection (Citation[138]).
Figure 1 Potential differences in pulmonary defense against RSV in COPD patients. Factors that include age, environmental exposures, genetics, COPD itself (e.g., bacterial colonization, chronic inflammation), or a combination of these may alter the host response to RSV thereby increasing susceptibility to or severity of infection. Altered defense responses include antiviral innate and adaptive immunity, as well as inflammation that may lead to AECOPD.
SUMMARY
Although RSV is an important cause of AECOPD and contributes to significant morbidity, health-care utilization, and mortality, the role of RSV in AECOPD is under-recognized. The mechanisms that alter the risk and severity of RSV infection in COPD patients are unclear, but likely include host genetic factors, aging-related immune decline, COPD-specific factors, virus-specific modulation of innate, humoral and adaptive immunity, and viral-bacterial interactions. Furthermore, the possibility that RSV exists in a chronic carrier state and contributes to the decline in lung function observed in COPD patients needs to be clarified. The complexities of RSV effects in COPD present opportunities for research with the goal of developing approaches to selectively modify damaging viral effects (e.g., altered airway function), while retaining beneficial immunity (e.g., clearance of virus) in COPD patients.
ACKNOWLEDGMENTS
The authors gratefully acknowledge M. McCormick for review of the manuscript and helpful discussion. The research programs of the authors are supported by grants from the National Institutes of Health and the Veterans Medical Research Foundation of San Diego. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
REFERENCES
- Jemal A, Ward E, Hao Y, Thun M. Trends in the leading causes of death in the United States, 1970–2002. JAMA 2005; 294: 1255–1259
- Roman J, Perez R L. COPD in VA hospitals. Clinical Cornerstone 2003; 5: 37–44
- Donaldson G C, Wedzicha J A. COPD exacerbations: 1. Epidemiology. Thorax 2006; 61: 164–168
- Sapey E, Stockley R A. COPD exacerbations: 2. Aetiology. Thorax 2006; 61: 250–258
- Rohde G, Wiethege A, Borg I, Kauth M, Bauer T T, Gillissen A, Bufe A, Schultze-Werninghaus G. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax 2003; 58: 37–42
- Beckham J D, Cadena A, Lin J, Piedra P A, Glezen W P, Greenberg S B, Atmar R L. Respiratory viral infections in patients with chronic obstructive pulmonary disease. J Infect 2005; 50: 322–330
- Falsey A R, Hennessey P A, Formica M A, Cox C, Walsh E E. Respiratory syncytial virus infection in elderly and high-risk adults. N Eng J Med 2005; 352: 1749–1759
- Falsey A R, Criddle M C, Walsh E E. Detection of respiratory syncytial virus and human metapneumovirus by reverse transcription polymerase chain reaction in adults with and without respiratory illness. J Clin Virol 2006; 35: 46–50
- Falsey A R, Formica M A, Hennessey P A, Criddle M M, Sullender W M, Walsh E E. Detection of respiratory syncytial virus in adults with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 173: 639–643
- Hall C B. Respiratory syncytial virus and parainfluenza virus. N Eng J Med 2001; 344: 1917–1928
- Chanock R, Roizman B, Myers R. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). I. Isolation, properties and characterization. Am J Hyg 1957; 66: 281–290
- Louie J K, Hacker J K R. G., Mark J, Maselli J H, Yagi S, Drew W L. Characterization of viral agents causing acute respiratory infection in a San Francisco university medical center clinic during the influenza season. Clin Infect Dis 2005; 41: 822–828
- Johnston S L, Pattemore P K, Sanderson G, Smith S, Lampe F, Josephs L, Symington P, O'Toole S, Myint S H, Tyrrell D AJ, Holgate S T. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. Br Med J 1995; 310: 1225–1228
- Nicholson K G, Kent J, Ireland D C. Respiratory viruses and exacerbations of asthma in adults. Br Med J 1993; 307: 982–986
- Soler N, Torres A, Ewig S, Gonzalez J, Celis R, El-Ebiary M, Hernandez C, Rodriguez-Roisin R. Bronchial microbial patterns in severe exacerbations of chronic obstructive pulmonary disease (COPD) requiring mechanical ventilation. Am J Respir Crit Care Med 1998; 157: 1498–1505
- Ko F W, Ip M, Chan P K, Fok J P, Chan M C, Ngai J C, Chan D P, Hui D S. A 1-year prospective study of the infectious etiology in patients hospitalized with acute exacerbations of COPD. Chest 2007; 131: 44–52
- Cameron R J, de Wit D, Welsh T N, Ferguson J, Grissell T V, Rye P J. Virus infection in exacerbations of chronic obstructive pulmonary disease requiring ventilation. Intensive Care Med 2006; 32: 968–970
- Falsey A R, Formica M A, Treanor J J, Walsh E E. Comparison of quantitative reverse transcription-PCR to viral culture for assessment of respiratory syncytial virus shedding. J Clin Microbiol 2003; 41: 4160–4165
- Borg I, Rohde G, Loseke S, Bittscheidt J, Schultze-Werninghaus G, Stephan V, Bufe A. Evaluation of a quantitative real-time PCR for the detection of respiratory syncytial virus in pulmonary diseases. Eur Resp J 2003; 21: 944–951
- Wilkinson T M, Donaldson G C, Johnston S L, Openshaw P J, Wedzicha J A. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 173: 871–876
- Sethi S, Evans N, Grant B J, Murphy T F. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Eng J Med 2002; 347: 465–471
- Burge S, Wedzicha J A. COPD exacerbations: definitions and classifications. Eur Resp J 2003; 21(Suppl 41)46s–53s
- Sunyer J, Sáez M, Murillo C, Castellsague J, Martínez F, Antó J M. Air pollution and emergency room admissions for chronic obstructive pulmonary disease: a 5-year study. Am J Epidemiol 1993; 137: 701–705
- Ball P. Epidemiology and treatment of chronic bronchitis and its exacerbations. Chest 1995; 108: 43S–52S
- Connors A FJ, Dawson N V, Thomas C, Harrell F EJ, Desbiens N, Fulkerson W J, Kussin P, Bellamy P, Goldman L, Knaus W A. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (study to understand prognoses and preferences for outcomes and risks of treatments). Am J Respir Crit Care Med 1996; 154: 959–967
- Walsh E E, Peterson D R, Falsey A R. Risk factors for severe respiratory syncytial virus infection in elderly persons. J Infect Dis 2004; 189: 233–238
- Thompson W W, Shay D K, Weintraub E, Brammer L, Cox N, Anderson L J, Fukuda K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003; 289: 179–186
- Lee F EH, Walsh E E, Falsey A R, Betts R F, Treanor J J. Experimental infection of humans with A2 respiratory syncytial virus. Antiviral Res 2004; 63: 191–196
- Glezen P, Denny F W. Epidemiology of acute lower respiratory disease in children. N Engl J Med 1973; 288: 498–505
- Upshur R EG, Moineddin R, Crighton E J, Mamdani M. Interactions of viral pathogens on hospital admissions for pneumonia, croup and chronic obstructive pulmonary diseases: Results of a multivariate time-series analysis. Epidemiol Infect 2006; 134: 1174–1178
- Mullooly J P, Bridges C B, Thompson W W, Chen J, Weintraub E, Jackson L A, Black S, Shay D K, Vaccine Safety Datalink Adult Working Group. Influenza- and RSV-associated hospitalizations among adults. Vaccine 2007; 25: 846–855
- Dakhama A, Vitalis T Z, Hegele R G. Persistence of respiratory syncytial virus (RSV) infection and development of RSV-specific IgG1 response in a guinea-pig model of acute bronchiolitis. Eur Respir J 1997; 10: 20–26
- Bramley A M, Vitalis T Z, Wiggs B R, Hegele R G. Effects of respiratory syncytial virus persistence on airway responsiveness and inflammation in guinea-pigs. Eur Resp J 1999; 14: 1061–1067
- Schwarze J, O'Donnell D R, Rohwedder A, Openshaw P J. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am J Respir Crit Care Med 2004; 169: 801–805
- Cubie H A, Duncan L A, Marshall L A, Smith N M. Detection of respiratory syncytial virus nucleic acid in archival postmortem tissue from infants. Pediatr Pathol Lab Med 1997; 17: 927–938
- Hogg J C. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med 2001; 164: S71–S75
- Seemungal T, Harper-Owen R, Bhowmik A, Moric I, Sanderson G, Message S, Maccallum P, Meade T W, Jeffries D J, Johnston S L, Wedzicha J A. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164: 1618–1623
- Greenberg S B, Allen M, Wilson J, Atmar R L. Respiratory viral infections in adults with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 162: 167–173
- Croxton T L, Weinmann G G, Senior R M, Wise R A, Crapo J D, Buist A S. Clinical research in chronic obstructive pulmonary disease-needs and opportunities. Am J Respir Crit Care Med 2003; 17: 1142–1149
- Robbins C S, Dawe D E, Goncharova S I, Pouladi M A, Drannik A G, Swirski F K, Cox G, Stämpfli M R. Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsiveness. Am J Respir Cell Mol Biol 2004; 30: 202–211
- Robbins C S, Bauer C M, Vujicic N, Gaschler G J, Lichty B D, Brown E G, Stämpfli M R. Cigarette smoke impacts immune inflammatory responses to influenza in mice. Am J Respir Crit Care Med 2006; 174: 1342–1351
- Huchon G J, Russell J A, Barritault L G, Lipavsky A, Murray J F. Chronic air-flow limitation does not increase respiratory epithelial permeability assessed by aerosolized solute, but smoking does. Am Rev Respir Dis 1984; 130: 457–460
- Monick M M, Yarovinsky T O, Powers L S, Butler N S, Carter A B, Gudmundsson G, Hunninghake G W. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem 2003; 278: 53035–53044
- Tal G, Mandelberg A, Dalal I, Cesar K, Somekh E, Tal A, Oron A, Itskovich S, Ballin A, Houri S, Beigelman A, Lider O, Rechavi G, Amariglio N. Association between common toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J Infect Dis 2004; 189: 2057–2063
- Puthothu B, Forster J, Heinzmann A, Krueger M. TLR-4 and CD14 polymorphisms in respiratory syncytial virus associated disease. Dis Markers 2006; 22: 303–308
- Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax 2000; 55: 1023–1027
- Löfgren J, Rämet M, Renko M, Marttila R, Hallman M. Association between surfactant protein A gene locus and severe respiratory syncytial virus infection in infants. J Infect Dis 2002; 185: 283–289
- Lahti M, Lofgren J, Marttila R, Renko M, Klaavuniemi T, Haataja R, Ramet M, Hallman M. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res 2002; 51: 696–699
- Hoebee B, Rietveld E, Bont L, Oosten M, Hodemaekers H M, Nagelkerke N J, Neijens H J, Kimpen J L, Kimman T G. Association of severe respiratory syncytial virus bronchiolitis with interleukin-4 and interleukin-4 receptor alpha polymorphisms. J Infect Dis 2003; 187: 2–11
- Hull J, Rowlands K, Lockhart E, Moore C, Sharland M, Kwiatkowski D. Variants of the chemokine receptor CCR5 are associated with severe bronchiolitis caused by respiratory syncytial virus. J Infect Dis 2003; 188: 904–907
- Hoebee B, Bont L, Rietveld E, van Oosten M, Hodemaekers H M, Nagelkerke N J, Neijens H J, Kimpen J L, Kimman T G. Influence of promoter variants of interleukin-10, interleukin-9, and tumor necrosis factor-alpha genes on respiratory syncytial virus bronchiolitis. J Infect Dis 2004; 189: 239–247
- Puthothu B, Krueger M, Forster J, Heinzmann A. Association between severe respiratory syncytial virus infection and IL13/IL4 haplotypes. J Infect Dis 2006; 193: 438–441
- Puthothu B, Krueger M, Heinze J, Forster J, Heinzmann A. Haplotypes of surfactant protein C are associated with common paediatric lung diseases. Pediatr Allergy Immunol 2006; 17: 572–577
- Puthothu B, Forster J, Heinze J, Heinzmann A, Krueger M. Surfactant protein B polymorphisms are associated with severe respiratory syncytial virus infection, but not with asthma. BMC Pulm Med 2007; 7: 6
- Krueger M, Puthothu B, Heinze J, Forster J, Heinzmann A. Genetic polymorphisms of adhesion molecules in children with severe RSV-associated diseases. Int J Immunogenet 2006; 33: 233–235
- Becker S, Soukup J, Yankaskas J R. Respiratory syncytial virus infection of human primary nasal and bronchial epithelial cell cultures and bronchoalveolar macrophages. Am J Respir Cell Mol Biol 1992; 6: 369–374
- Look D C, Walter M J, Williamson M R, Pang L, You Y, Sreshta J N, Johnson J E, Zander D S, Brody S L. Effects of paramyxoviral infection on airway epithelial cell Foxj1 expression, ciliogenesis, and mucociliary function. Am J Pathol 2001; 159: 2055–2069
- Johnson J E, Gonzales R A, Olson S J, Wright P F, Graham B S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol 2007; 20: 108–119
- Levin S J. Bronchial epithelial cell-cytokine interactions in airway inflammation. J Invest Med 1995; 43: 241–249
- Holtzman M J, Look D C, Sampath D, Castro M, Koga T, Walter M J. Control of epithelial immune-response genes and implications for airway immunity and inflammation. Proc Assoc Am Phys 1998; 110: 1–11
- Look D C, Brody S L. Engineering viral vectors to subvert the airway defense response. Am J Respir Cell Mol Biol 1999; 20: 1103–1106
- Noah T L, Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am J Physiol (Lung Cell Mol Physiol) 1993; 265: L472–L478
- Elias J A, Zheng T, Einarsson O, Landry M, Trow T, Rebert N, Panuska J. Epithelial interleukin-11 -regulation by cytokines, respiratory syncytial virus, and retinoic acid. J Biol Chem 1994; 269: 22261–22268
- Stark J M, Godding V, Sedgwick J B, Busse W W. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells—roles of CD18 and intercellular adhesion molecule-1. J Immunol 1996; 156: 4774–4782
- Garofalo R, Mei F, Espejo R, Ye G, Haeberle H, Baron S, Ogra P L, Reyes V E. Respiratory syncytial virus infection of human respiratory epithelial cells up-regulates class I MHC expression through the induction of IFN-β and IL-1α. J Immunol 1996; 157: 2506–2513
- Saito T, Deskin R W, Casola A, Haeberle H, Olszewska B, Ernst P B, Alam R, Ogra P L, Garofalo R. Respiratory syncytial virus induces selective production of the chemokine rantes by upper airway epithelial cells. J Infect Dis 1997; 175: 497–504
- Becker S, Reed W, Henderson F W, Noah T L. RSV infection of human airway epithelial cells causes production of the β -chemokine RANTES. Am J Physiol (Lung Cell Mol Physiol) 1997; 272: L512–L520
- Olszewska-Pazdrak B, Casola A, Saito T, Alam R, Crowe S E, Mei F, Ogra P L, Garofalo R P. Cell-specific expression of RANTES, MCP-1, and MIP-1α by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol 1998; 72: 4756–4764
- Koga T, Sardina E, Tidwell R M, Pelletier M, Look D C, Holtzman M J. Virus-inducible expression of a host chemokine gene relies on replication-linked mRNA stabilization. Proc Natl Acad Sci USA 1999; 9(6)5680–5685
- Garofalo R P, Haeberle H. Epithelial regulation of innate immunity to respiratory syncytial virus. Am J Respir Cell Mol Biol 2000; 23: 581–585
- Kao Y J, Piedra P A, Larsen G L, Colasurdo G N. Induction and regulation of nitric oxide synthase in airway epithelial cells by respiratory syncytial virus. Am J Respir Crit Care Med 2001; 163: 532–539
- Milner M E, Monte S DL, Hutchins G M. Fatal respiratory syncytial virus infection in severe combined immunodeficiency syndrome. Am J Dis Child 1985; 139: 1111–1114
- Cannon M J, Openshaw P JM, Askonas B A. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med 1988; 168: 1163–1168
- Tang Y W, Graham B S. T cell source of type I cytokines determines illness patterns in respiratory syncytial virus-infected mice. J Clin Invest 1997; 99: 2183–2191
- Durbin J E, Johnson T R, Durbin R K, Mertz S E, Morotti R A, Peebles R S, Graham B S. The role of IFN in respiratory syncytial virus pathogenesis. J Immunol 2002; 168: 2944–2951
- Stark G R, Kerr I M, Williams B R, Silverman R H, Schreiber R D. How cells respond to interferons. Annu Rev Biochem 1998; 67: 227–264
- Ramaswamy M, Shi L, Monick M M, Hunninghake G W, Look D C. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am J Respir Cell Mol Biol 2004; 30: 893–900
- Katze M G, He Y, Gale M J. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2002; 2: 675–687
- Look D C, Roswit W T, Frick A G, Gris-Alevy Y, Dickhaus D M, Walter M J, Holtzman M J. Direct suppression of Stat1 function during adenoviral infection. Immunity 1998; 9: 871–880
- Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-α /β revisited. Nature Rev 2001; 2: 378–386
- Der S D, Zhou A, Williams B RG, Silverman R H. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci USA 1998; 95: 15623–15628
- Ramaswamy M, Shi L, Varga S M, Barik S, Behlke M A, Look D C. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 2006; 344: 328–339
- Bitko V, Shulyayeva O, Mazumder B, Musiyenko A, Ramaswamy M, Look D C, Barik S. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J Virol 2007; 81: 1786–1795
- Thomas K W, Monick M M, Staber J M, Yarovinsky T, Carter A B, Hunninghake G W. Respiratory syncytial virus inhibits apoptosis and induces NF-kappa B activity through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem 2002; 277: 492–501
- Monick M M, Cameron K, Staber J, Powers L S, Yarovinsky T O, Koland J G, Hunninghake G W. Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J Biol Chem 2005; 280: 2147–2158
- Groskreutz D J, Monick M M, Yarovinsky T O, Powers L S, Quelle D E, Varga S M, Look D C, Hunninghake G W. Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J Immunol 2007; 179: 2741–2747
- Rudd B D, Burstein E, Duckett C S, Li X, Lukacs N W. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J Virol 2005; 79: 3350–3357
- Groskreutz D J, Monick M M, Powers L S, Yarovinsky T O, Look D C, Hunninghake G W. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J Immunol 2006; 176: 1733–1740
- Kreindler J L, Jackson A D, Kemp P A, Bridges R J, Danahay H. Inhibition of chloride secretion in human bronchial epithelial cells by cigarette smoke extract. Am J Physiol (Lung Cell Mol Physiol) 2005; 288: L894–L902
- Gelbman B D, Heguy A, O'Connor T P, Zabner J, Crystal R G. Upregulation of pirin expression by chronic cigarette smoking is associated with bronchial epithelial cell apoptosis. Respir Res 2007; 8: 10
- Pesci A, Majori M, Cuomo A, Borciani N, Bertacco S, Cacciani G, Gabrielli M. Neutrophils infiltrating bronchial epithelium in chronic obstructive pulmonary disease. Respir Med 1998; 92: 863–870
- Takizawa H TM, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Nakahara K, Umeda A. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med 2001; 163: 1476–1483
- Puchelle E, Zahm J M, Tournier J M, Coraux C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2006; 3: 726–733
- Caramori G, Ito K, Contoli M, Di Stefano A, Johnston S L, Adcock I M, Papi A. Molecular mechanisms of respiratory virus-induced asthma and COPD exacerbations and pneumonia. Curr Med Chem 2006; 13: 2267–2290
- Beem M, Egerer R, Anderson J. Respiratory syncytial virus neturalizing antibodies in persons residing in Chicago, Illinois. Pediatrics 1964; 34: 761–770
- Welliver R C, Kaul T N, Putnam T I, Sun M, Riddlesberger K, Ogra P L. The antibody response to primary and secondary infection with respiratory syncytial virus: kinetics of class-specific responses. J Pediatr 1980; 96: 808–813
- Glezen W P, Paredes A, Allison J E, Taber L H, Frank A L. Risk of respiratory syncytial virus infection for infants from low-income families in relationship to age, sex, ethnic group, and maternal antibody level. J Pediatr 1981; 98: 708–715
- de Sierra T M, Kumar M L, Wasser T E, Murphy B R, Subbarao E K. Respiratory syncytial virus-specific immunoglobulins in preterm infants. J Pediatr 1993; 122: 787–791
- The IMpact-RSV Study Group. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 1998; 102: 531–537
- Piedra P A, Jewell A M, Cron S G, Atmar R L, Glezen W P. Correlates of immunity to respiratory syncytial virus (RSV) associated-hospitalization: establishment of minimum protective threshold levels of serum neutralizing antibodies. Vaccine 2003; 21: 3479–3482
- Walsh E E, Falsey A R. Age related differences in humoral immune response to respiratory syncytial virus infection in adults. J Med Virol 2004; 73: 295–299
- Walsh E E, Falsey A R. Humoral and mucosal immunity in protection from natural respiratory syncytial virus infection in adults. J Infect Dis 2004; 190: 373–378
- Falsey A R, Singh H K, Walsh E E. Serum antibody decay in adults following natural respiratory syncytial virus infection. J Med Virol 2006; 78: 1493–1497
- Meyer K C. Aging. Proc Am Thorac Soc 2005; 2: 433–439
- Hall C B, Powell K R, MacDonald N E, Gala C L, Menegus M E, Suffin S C, Cohen H J. Respiratory syncytial viral infection in children with compromised immune function. N Eng J Med 1986; 315: 77–81
- Zhang Y, Wang Y, Gilmore X, Xu K, Wyde P R, Mbawuike I N. An aged mouse model for RSV infection and diminished CD8(+) CTL responses. Exp Biol Med 2002; 227: 133–140
- Liu B, Kimura Y. Local immune response to respiratory syncytial virus infection is diminished in senescence-accelerated mice. J Gen Virol 2007; 88: 2552–2558
- Mbawuike I N, Wells J, Byrd R, Cron S G, Glezen W P, Piedra P A. HLA-restricted CD8+ cytotoxic T lymphocyte, interferon-gamma, and interleukin-4 responses to respiratory syncytial virus infection in infants and children. J Infect Dis 2001; 183: 687–696
- de Bree G J, Heidema J, van Leeuwen E M, van Bleek G M, Jonkers R E, Jansen H M, van Lier R A, Out T A. Respiratory syncytial virus-specific CD8+ memory T cell responses in elderly persons. J Infect Dis 2005; 191: 1710–1718
- Looney R J, Falsey A R, Walsh E, Campbell D. Effect of aging on cytokine production in response to respiratory syncytial virus infection. J Infect Dis 2002; 185: 682–685
- Lee F E, Walsh E E, Falsey A R, Liu N, Liu D, Divekar A, Snyder-Cappione J E, Mosmann T R. The balance between influenza- and RSV-specific CD4 t cells secreting IL-10 or IFNgamma in young and healthy-elderly subjects. Mech Ageing Dev 2005; 126: 1223–1229
- Linton P J, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol 2004; 5: 133–139
- de Bree G J, Daniels H, Schilfgaarde M, Jansen H M, Out T A, van Lier R A, Jonkers R E. Characterization of CD4+ memory T cell responses directed against common respiratory pathogens in peripheral blood and lung. J Infect Dis 2007; 195: 1718–1725
- Hancock A, Armstrong L, Gama R, Millar A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol 1998; 18: 60–65
- Wills-Karp M. Interleukin-13 in asthma pathogenesis. Immunol Rev 2004; 202: 175–190
- Lee C G, Homer R J, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley J M, Gotwals P, Noble P, Chen Q, Senior R M, Elias J A. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta (1). J Exp Med 2001; 194: 809–821
- Ke B, Shen X D, Zhai Y, Gao F, Busuttil R W, Volk H D, Kupiec-Weglinski J W. Heme oxygenase 1 mediates the immunomodulatory and antiapoptotic effects of interleukin 13 gene therapy in vivo and in vitro. Hum Gene Ther 2002; 13: 1845–1857
- Wynn T A. IL-13 effector functions. Annu Rev Immunol 2003; 21: 425–456
- Zhou W, Hashimoto K, Moore M L, Elias J A, Zhu Z, Durbin J, Colasurdo G, Rutigliano J A, Chiappetta C L, Goleniewska K, O'Neal J F, Graham B S, Peebles R S. IL-13 is associated with reduced illness and replication in primary respiratory syncytial virus infection in the mouse. Microbes Infect 2006; 8: 2880–2889
- Ermers M J, Hoebee B, Hodemaekers H M, Kimman T G, Kimpen J L, Bont L. IL-13 genetic polymorphism identifies children with late wheezing after respiratory syncytial virus infection. J Allergy Clin Immunol 2007; 119: 1086–1091
- Boutten A, Bonay M, Laribe S, Leseche G, Castier Y, Leçon-Malas V, Fournier M, Durand G, Aubier M, Dehoux M, Crestani B. Decreased expression of interleukin 13 in human lung emphysema. Thorax 2004; 59: 850–854
- Mufson M A, Örvell C, Rafnar B, Norrby E. Two distinct subtypes of human respiratory syncytial virus. J Gen Virol 1985; 66: 2111–2124
- Lukacs N W, Moore M L, Rudd B D, Berlin A A, Collins R D, Olson S J, Ho S B, Peebles R S. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am J Pathol 2006; 169: 977–986
- Fodha I, Vabret A, Ghedira L, Seboui H, Chouchane S, Dewar J, Gueddiche N, Trabelsi A, Boujaafar N, Freymuth F. Respiratory syncytial virus infections in hospitalized infants: association between viral load, virus subgroup, and disease severity. J Med Virol 2007; 79: 1951–1958
- Plotkowski M C, Puchelle E, Beck G, Jacquot J, Hannoun C. Adherence of type I Streptococcus pneumoniae to tracheal epithelium of mice infected with influenza A/PR8 virus. Am Rev Respir Dis 1986; 134: 1040–1044
- Avadhanula V, Rodriguez C A, Devincenzo J P, Wang Y, Webby R J, Ulett G C, Adderson E E. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. J Virol 2006; 80: 1629–1636
- Avadhanula V, Wang Y, Portner A, Adderson E. Nontypeable Haemophilus influenzae and Streptococcus pneumoniae bind respiratory syncytial virus glycoprotein. J Med Microbiol 2007; 56: 1133–1137
- Corbeil L B, Arnold K F, Kimball R, Berghaus L, Gershwin L J. Specificity of IgG and IgE antibody responses to Haemophilus somnus infection of calves. Vet Immunol Immunopathol 2006; 113: 191–199
- Monso E, Ruiz J, Rosell A, Manterola J, Fiz J, Morera J, Ausina V. Bacterial infection in chronic obstructive pulmonary disease. A study of stable and exacerbated outpatients using the protected specimen brush. Am J Respir Crit Care Med 1995; 152: 1316–1320
- Cabello H, Torres A, Celis R, El-Ebiary M, Puig de la Bellacasa J, Xaubet A, Gonzalez J, Agusti C, Soler N. Bacterial colonization of distal airways in healthy subjects and chronic lung disease: A bronchoscopic study. Eur Respir J 1997; 10: 1137–1144
- Zalacain R, Sobradillo V, Amilibia J, Barron J, Achotegui V, Pijoan J I, Llorente J L. Predisposing factors to bacterial colonization in chronic obstructive pulmonary disease. Eur Respir J 1999; 13: 343–348
- Soler N, Ewig S, Torres A, Filella X, Gonzalez J, Zaubet A. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J 1999; 14: 1015–1022
- Hill A T, Campbell E J, Hill S L, Bayley D L, Stockley R A. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med 2000; 109: 288–295
- Chin C L, Manzel L J, Lehman E E, Humlicek A L, Shi L, Starner T D, Denning G M, Murphy T F, Sethi S, Look D C. Haemophilus influenzae from patients with chronic obstructive pulmonary disease exacerbation induce more inflammation than colonizers. Am J Respir Crit Care Med 2005; 172: 85–91
- Sajjan U S, Jia Y, Newcomb D C, Bentley J K, Lukacs N W, LiPuma J J, Hershenson M B. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J 2006; 20: 2121–2123
- Wilkinson T M, Hurst J R, Perera W R, Wilks M, Donaldson G C, Wedzicha J A. Effect of interactions between lower airway bacterial and rhinoviral infection in exacerbations of COPD. Chest 2006; 129: 317–324
- Sethi S. Coinfection in exacerbations of COPD: a new frontier. Chest 2006; 129: 223–224
- Donaldson G C, Seemungal T A, Patel I S, Lloyd-Owen S J, Wilkinson T M, Wedzicha J A. Longitudinal changes in the nature, severity and frequency of COPD exacerbations. Eur Respir J 2003; 22: 931–936