Abstract
Chronic obstructive pulmonary disease patients are at increased risk for mortality, particularly from cardiovascular conditions. Acute exacerbation increases heart burden and may lead to release of troponin I. This study investigates the long-term prognostic value of elevated troponin I detected during acute exacerbation of chronic obstructive pulmonary disease. The records of 182 patients with acute exacerbation in whom troponin I levels were sampled during their hospitalization were reviewed retrospectively. Receiver operator curve was used to determine the cut-off level for troponin I that discriminated survivors and non-survivors, and predictors for all-cause mortality were tested in a multivariate analysis. The results showed that, during a mean observation time of 50.1 ± 45.6 months, 66 (36.3%) patients died, providing 1, and 3-year survival rates of 84%, and 54%, respectively. Troponin I levels were significantly higher in non-survivors compared with survivors, mean troponin I ± SD in μ g· L−1: 1.35 ± 3.17 vs. 0.53 ± 2.08, respectively, p = 0.0033. ROC curve analysis identified troponin I > 0.03 μ g· L−1 as the optimal cut-off level for prediction of mortality. Kaplan-Meier survival analysis revealed that the probability of survival was significantly lower in patients with troponin I > 0.03 μ g· L−1 (log-rank test p = 0.0058). On multivariate analysis, only ischemic heart disease (HR = 2.335, p = 0.0017) and troponin I level (HR = 1.31541, p = 0.2513) were independent predictors of mortality. In conclusion, it was found that a mildly elevated troponin I level measured in patients with chronic obstructive pulmonary disease during acute exacerbation is a strong independent predictor of mortality following discharge.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a major public health problem and a major cause of death worldwide (Citation[1], Citation[2]). The natural course of COPD is characterized by a progressive decline in pulmonary function and recurrent exacerbations requiring hospitalization. Acute exacerbation of COPD (AECOPD) is associated with both excessive short (in-hospital) and long-term (following discharge) mortality rates (Citation[3], Citation[4], Citation[5]). There is increasing awareness that ischemic heart disease (IHD) and cardiac co-morbidities are major contributors to mortality in patients with mild to moderate COPD, many of whom are current or former smokers with increased risk for atherosclerosis and IHD (Citation[6], Citation[7], Citation[8]).
During AECOPD, hypoxia and elevation of pulmonary vascular resistance occurs, presenting an increased burden on the myocardium and resulting in myocardial injury and possible release of cardiac-specific biomarkers, namely, troponin. Although cardiac troponins are highly specific markers of myocardial necrosis (Citation[9], Citation[10]), elevated troponin levels have been found in several conditions with non-ischemic mechanisms of cardiac injury (Citation[11], Citation[12], Citation[13], Citation[14], Citation[15], Citation[16], Citation[17]). Mild myocardial damage during AECOPD detected by measuring cardiac troponins may identify a subgroup of vulnerable patients who are at increased risk of death from cardiac conditions. The literature on troponin levels detected in patients during AECOPD is limited (Citation[18], Citation[19], Citation[20]) and, therefore, we sought to investigate the predictive value of elevated troponin levels on long-term survival rates following AECOPD.
PATIENTS AND METHODS
Study population
Approval for this study was obtained from the Rambam Medical Center Review Board. All consecutive patients who were admitted to general medicine wards at Rambam Health Care Campus in Haifa, Israel, over a 7-year period between January 1, 2001, and December 31, 2007, for the primary diagnosis of AECOPD were retrospectively studied. COPD was further verified from the patient's pre morbid records from the Out-Patient Pulmonary Clinic. Patients were included if the following criterion was met: diagnosis of COPD according to the criteria set by The Global Initiative for Chronic Obstructive Lung Disease (GOLD) (Citation[21]). AECOPD was defined by the presence of an increase in at least two of three symptoms—dyspnea, cough, and sputum purulence-severe enough to warrant hospital admission without concomitant evidence of pneumonia. According to hospital guidelines cardiac-specific troponin I (cTnI) levels were obtained in the emergency department in patients with COPD who presented with dyspnea as chief complaint accompanied with chest pain suggestive of myocardial ischemia. The decision to perform cTnI determination was left to the discretion of the attending physician Since cTnI may be elevated in patients with impaired renal function (Citation[22]) patients with chronic renal failure, defined as calculated serum creatinine level of more than 1.5 mg% (normal < 1.1 mg%) for 3 months or more, were excluded. Patients with other conditions known to affect troponin levels (Citation[9]) such as sepsis, pulmonary embolism, myocarditis, cardiomyopathy, and chest contusion were also excluded.
EPIDEMIOLOGICAL AND BASELINE LABORATORY DATA
Data were collected retrospectively. The age, gender, and smoking status of the patients were documented, in addition to medical history and co morbid conditions (diabetes mellitus, IHD, congestive heart failure, IHD was defined as patients with prior history of acute myocardial infarction, coronary artery bypass surgery, or percutaneous coronary intervention. An index case of CHF was based on the following: one inpatient hospitalization under diagnosis-related group 127 or 124 and one of the above ICD-9-CM codes for CHF in the principal position. Active smoking status was defined as having smoked within the last 6 months.
Results of laboratory analyses performed within 24 hours of hospital admission were retrieved from the hospital laboratory database. If multiple results were available, average levels were used. cTnI assay used by the hospital laboratory was AxSYM troponin-I ADV (Abbott Diagnostics, Redwood City CA, USA). In accordance with the manufacturer data, (http://www.abbottdiagnostics.com), the laboratory reported cTnI levels as measurable if the level exceeded the manufacturer limit of detection -0.02 μ g· L−1 and the diagnostic cutoff for acute myocardial injury was 0.4 μ g· L−1.
Outcome and follow-up
The hospital discharge date was used as the starting point regarding the follow-up period. End of observation date was December 31, 2007. In-hospital mortality and length of hospital stay were determined for each patient. The maximal observation time period was 6 years. Information on the current status (living, dead), survival time, was collected by review of the national death registration record for patients who died out-ofhospital.
Statistical analysis
Descriptive data are presented as mean (± SD) or median (range). Comparisons between groups were made by using the Mann–Whitney test (for continuous variables), or Fisher exact test (for categorical variables), where appropriate. The two-sided p-value of < 0.05 was considered as statistically significant. Receiver operator curve (ROC) analysis was used to determine the optimal cut-off level for cTnI that predicted mortality. Survival curves were estimated by the Kaplan-Meier product limit method and compared using the log-rank test.
Multivariate Cox regression analysis was used to determine the correlation between independent parameters and all-cause mortality. The entry criterion was p < 0.20, and the permanence criterion was p < 0.05. Statistical analyses were performed using a statistical software package (MedCalc Version 9.3.0.0, USA).
RESULTS
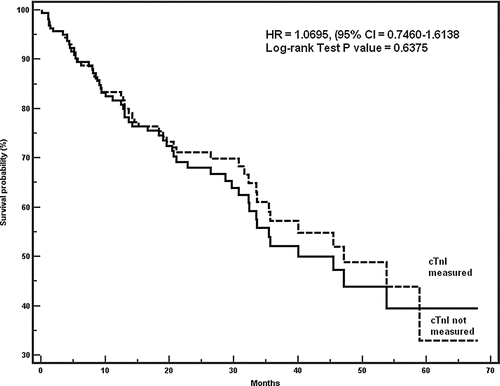
During the study period, 1135 patients were admitted to one of the five General Medicine wards with a primary admission diagnosis of AECOPD. Troponin levels were obtained in 182 patients who comprised the cohort of the study. There were no significant differences in baseline demographic and clinical characteristics between patients in whom cTnI was obtained and those in whom it was not obtained (). Similarly, the probability of survival was not different in COPD patients in whom cTnI levels were obtained compared with those in whom it was not measured (). Log-rank test p = 0.6375, HR = 1.0965 (95% CI = 0.7460-1.6138).
Table 1 Demographic and clinical characteristics of sub-groups of patients with and without cardiac troponin I (cTnI) measurement
Figure 1 Survival of patients according to cTnI level.
Study population characteristics are shown in . The mean (± SD) age of the study population was 71.2 ± 10.5 (range, 42–93 years). Most patients (135/182, 74%) were males. Fifty-two percent had ischemic heart disease and 36% had diabetes mellitus. Fifty-two percent were current smokers. The study subjects were followed for a mean ± SD of 50.1 ± 45.6, median 35 months. The 1- and 3-year survival rates were 84%, and 54% %, respectively (). Overall, 66 (36.2%) patients died during the observation period. Differences between survivors and non-survivors during follow-up are listed in . Patients who died were more likely to have IHD. In addition, patients who died were significantly more hypercapnic (mean PaCO2 ± SD in mmHg was 60.3 ± 22.1) than those who survived (53.3 ± 18.6, p = 0.024). cTnI level was significantly higher in non-survivors (mean cTnI ± SD in μ g· L− 1 was 1.35 ± 3.17) compared to survivors (0.53 ± 2.08, p = 0.0033).
Table 2 Comparison of data between survivors and non-survivors
Figure 2 Receiver operator curve for predictive value of cardiac-specific troponin I for mortality following COPD exacerbation.
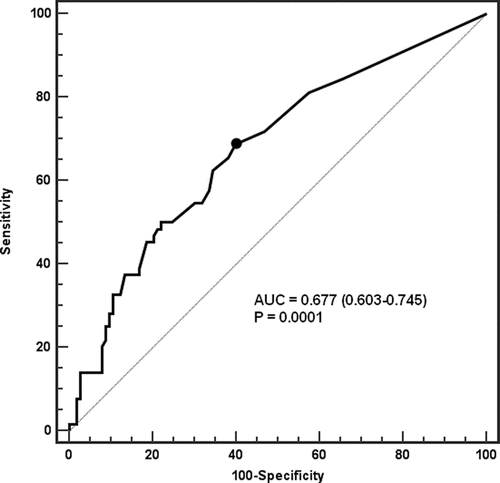
ROC analysis () identified cTnI levels > 0.03μ g· L−1 as the optimal cut-off level to discriminate survivors from deceased subjects. (Area under the ROC = 0.677, standard error = 0.043, 95% confidence interval (CI) = 0.603–0.745, p (area = 0.5) = 0.0001). There were 99 patients with cTnI levels < 0.03 μ g· L− 1 and 83 with cTnI levels > 0.03 μ g· L−1. There was no significant difference in baseline clinical or laboratory parameters between the groups apart from PaCO2 that was significantly higher in the latter group ().
Table 3 Comparison of clinical and physiological parameters between patients with cTnI levels ≤ 0.03 μ g· L−1 or > 0.03 μ g· L−1
Figure 3 Survival of patients stratified according to cTnI levels above and below 0.03 μg·L−1.
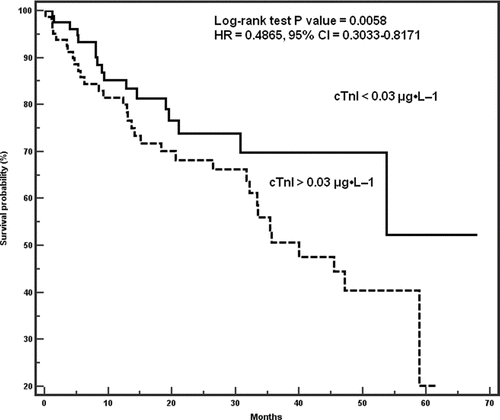
On the other hand, the survival of patients with cTnI levels < 0.03 μ g· L− 1 and those with cTnI levels > 0.03 μ g· L−1 differed significantly. A Kaplan–Meier plot of survival probability by cTnI levels < 0.03 μ g· L−1 and > 0.03 μ g· L−1 is shown in . The probability of survival at 1, 3 and 5 years in patients with cTnI Levels < 0.03 μ g· L−1 was 91%, 78% and 70%, while the corresponding probabilities of survival in patients with cTnI levels > 0.03 μ g· L−1 were 75%, 70% and 58%, respectively (log-rank test p-value = 0.0058, HR = 0.4865, 95% CI = 0.3033–0.8171).
A Cox proportional hazards model utilizing all potential predictors of survival was performed (). Age, IHD and total cTnI (HR = 1.0653, p = 0.0006) levels were found to be significant predictors of mortality, while PaCO2 on admission was marginally predictive for mortality.
Table 4 Risk factors and associated unadjusted hazard ratios for mortality probability using the Cox proportional-hazards model
The multiple Cox proportional-hazards model () revealed that only IHD (HR = 2.3350, 95% CI = 1.3796–3.9520, p = 0.0017) and total cTnI Levels (HR = 1.31541, 95% CI = 1.0753–2.2512, p = 0.2513) were independent strong predictors of mortality.
Table 5 Risk factors and associated adjusted hazard ratios for mortality probability using multivariate analysis
CONCLUSION
The main finding of the current study is that elevated cTnI measured during hospitalization for AECOPD is a strong independent prognostic factor for long-term mortality. Even after adjusting for multiple well-accepted determinants of mortality, cTnI level remained a strong predictor of mortality during up to 6 years of follow-up.
Cardiac troponins are the most sensitive and specific biochemical markers of myocardial damage, and elevated levels of troponin are poor prognostic markers in patients admitted with acute coronary syndrome (Citation[9], Citation[10]). Other medical conditions, not necessarily associated with ischemic cardiac injury such as sepsis (Citation[15]), pulmonary embolism (Citation[16]), viral myocarditis (Citation[17]) and renal failure (Citation[22]) may be associated with increased troponin levels also. Possible mechanisms include a mismatch between myocardial oxygen demand and supply in the absence of flow-limiting epicardial stenosis, imbalance of the autonomic nervous system with resulting excess sympathetic activity (Citation[11]) and an increased catecholamine effect on the myocardial cells, myocardial cell injury by traumatic or inflammatory processes, and volume and pressure overload resulting in an excessive increase in wall tension with secondary myofibrillary damage (Citation[12]).
The literature on troponin in AECOPD is spare and only three studies have addressed this issue to date. Harvey and Hancox (Citation[18]) first noted raised serum troponin levels in 58/238 patients with AECOPD and concluded that it reflected the severity of the exacerbation. Raised troponin was detected mostly in older patients and in subjects who were more hypoxemic, hypercapnic and acidotic. The authors noted that patients with raised troponin had longer admissions but they did not seek a correlation between troponin levels and either short- or long-term survival.
Baillard et al. (Citation[19]) found that elevated cTnI is a strong and independent predictor of in-hospital death in 71 patients admitted for AECOPD. No data was available regarding the long-term follow-up. Recently, Brekke et al (Citation[20]) retrospectively identified 396 patients with AECOPD in whom cardiac troponin T levels during hospitalization were available. The authors found that elevated Troponin T levels were significantly associated with all-cause post-discharge mortality. It is noteworthy that the authors used a cut-off level for cardiac troponin T of 0.04 μ g· L− 1 that is identical to the local laboratory's cut-off level to identify myocardial infarction.
Our observation that mildly elevated cTnI levels have negative prognostic implications in patients following AECOPD is novel and extends current knowledge is several aspects. Firstly, contrary to all three previous reports that used the traditional laboratory cut-off level used to diagnose obvious myocardial injury, by using an ultra-sensitive detection assay, we proved that even minor cTnI elevation (> 0.03 μ g· L−1) identified patients with AECOPD at high risk for death later on. A potential source of concern would be that the assay used in the current report (AxSYM troponin-I ADV - Abbott Diagnostics, Redwood City CA, USA) has a high coefficient of variation (CV) at that level according to the manufacturer's manual.
However, a recent report (Citation[23]) has shown that when tested in a large reference population, the CV of the assay at the range of 0.03 μ g· L−1 is significantly smaller providing high analytical precision. Secondly, the observation time in the current study was longer (median 2.9 years compared to 1.9 years, Brekkel (Citation[20])). Hence, we had the opportunity to draw unequivocal conclusions regarding the long-term significance of elevated troponin in patients with AECOPD. Thirdly, by using multivariate analysis, we demonstrated that, after adjusting for well-known risk factors for mortality, elevated cTnI is strongly and independently associated with long-term mortality. cTnI level was second only to IHD as the most powerful prognostic factor for mortality (stronger than age, diabetes, FEV1, and arterial blood gas values obtained on admission).
There are several alternative explanations for cTnI elevation, which should be considered in the study patient population. There is increasing awareness that IHD and cardiac co-morbidity are major contributors to mortality in patients with COPD (Citation[8], Citation[9], Citation[10]). The majority of patients with COPD are current or former smokers and are at increased risk of developing atherosclerosis and IHD. Similar to the proposed mechanism of troponin release in pulmonary embolism (Citation[16]), during AECOPD, hypoxia and elevation of pulmonary vascular resistance might cause right ventricular distension, increased burden on the myocardium, non-ischemic myocardial injury and release of cardiac-specific biomarkers (Citation[20]).
COPD exacerbation by itself does not produce sufficient strain on the heart to induce myocardial necrosis. Alternately, increased inflammatory response during AECOPD may increase the ongoing inflammatory process associated with atherosclerosis and result in escalation of atherosclerosis and coronary artery luminal narrowing, thereby increasing the likelihood of atheroma plaque rupture and subsequent ischemia and myocardial events, the frequent cause of death in this patient population.
Another possible mechanism for increased cTnI levels during AECOPD is myocardial injury induced by elevated circulating levels of cytokines, such as tumor necrosis factor (TNF) and interleukin-1β, as previously reported for example in sepsis (Citation[15]). These cytokines may increase the permeability of the myocyte membrane with the leakage of free cTnI from the cytoplasm to the serum while the myocyte-contraction complex remains intact (Citation[15]). In accordance with Harvey et al. (Citation[18]), we did not detect any significant difference in baseline severity of COPD between groups of patients with elevated versus normal cTnI levels.
As previously mentioned, for the purpose of long-term survival analysis, we used a cut-off level for cTnI that is markedly lower than the level used to diagnose myocardial injury in subjects with acute coronary syndrome. Accordingly, we might have identified a group of patients with COPD who are not sicker than other patients as judged by common clinical and laboratory parameters, but the magnitude of the response of their myocardium during AECOPD (sub-clinical myocardial injury manifested as mild elevation of cTnI) provide data regarding their risk for death later on.
Our study has several limitations. The survival analysis in this retrospective study was performed using all-cause mortality as the outcome. The correct coding of cause of death in out-patients in general and in COPD particularly is problematic because death certificates do not necessarily reflect the exact cause of death. Nevertheless, all-cause mortality is considered to be an acceptable end-point in most long-term COPD survival studies.
Due to the nature of this retrospective non-randomized study, one obvious limitation is the selection of patients who underwent cTnI examination. Of 1135 COPD patients in the initial cohort, cTnI was analyzed in 192, 10 of whom were patients with chronic renal failure who were excluded. The policy in our hospital is to perform cTnI testing in patients admitted to the emergency room for suspected acute coronary syndromes, myocarditis or pulmonary embolism. We may assume that the study population included mainly COPD patients with active cardiac conditions.
The baseline clinical and laboratory characteristics and hospital outcome were similar between patients in whom cTnI levels were obtained and those not, reducing to a minimum the risk of selection bias. In any event, the results of this study have clinical significance even if elevated cTnL levels predict death from unmasked coronary artery disease. Prospective studies delving into the cardiac status of patients with elevated cTnL during hospitalization for AECOPD are expected to highlight this topic.
In conclusion, assessment of cardiac-specific troponin I levels in patients with AECOPD provides an important prognostic factor by identifying patients with mildly elevated cTnI who are at increased risk for death independent of other well-known prognostic factors. This unique opportunity to identify such patients has important clinical implications as it may create a new risk-stratification scheme for patients following AECOPD that might set a base for further clinical interventions aimed at reducing death.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- Halpern M T, Stanford R H, Borker R. The burden of COPD in the U.S.A.: results from the Confronting COPD survey. Respir Med 2003; 97: S81–S89
- Chapman K R, Mannino D M, Soriano J B, Vermeire P A, Buist A S, Thun M J, Connell C, Jemal A, Lee T A, Miravitlles M, Aldington S, Beasley R. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J 2006; 27: 188–207
- Almagro P, Calbo E, Ochoa de Echaguen A, Barreiro B, Quintana S, Heredia J L, Garau J. Mortality after hospitalization for COPD. Chest 2002; 121: 1441–1448
- Miravitlles M, Guerrero T, Mayordomo C, Sánchez-Agudo L, Nicolau F, Segú J L. Factors associated with increased risk of exacerbation and hospital admission in a cohort of ambulatory COPD patients: a multiple logistic regression analysis. The EOLO Study Group. Respiration 2000; 67: 495–501
- Groenewegen K H, Schols A M, Wouters E F. Mortality and mortality-related factors after hospitalization for acute exacerbation of COPD. Chest 2003; 124: 459–467
- Hansell A, Walk J A, Soriano J B. What do chronic obstructive pulmonary disease patients die from? A multiple cause coding analysis. Eur Respir J 2003; 22: 809–814
- Sin D D, Man S F. Impact of cancers and cardiovascular diseases in chronic obstructive pulmonary disease. Curr Opin Pulm Med 2008; 14: 115–121
- Sin D D, Anthonisen N R, Soriano J B, Agusti A G. Mortality in COPD: Role of comorbidities. Eur Respir J 2006; 28: 1245–1257
- Tsai S H, Chu S J, Hsu C W, Cheng S M, Yang S P. Use and interpretation of cardiac troponins in the ED. Am J Emerg Med 2008; 26: 331–341
- Antman E M, Tanasijevic M J, Thompson B, Schactman M, McCabe C H, Cannon C P, Fischer G A, Fung A Y, Thompson C, Wybenga D, Braunwald E. Cardiac-specific troponin I levels to predict the risk of mortality in patients with acute coronary syndromes. N Engl J Med 1996; 335: 1342–1349
- Alcalai R, Planer D, Culhaoglu A, Osman A, Pollak A, Lotan C. Acute coronary syndrome vs nonspecific troponin elevation: clinical predictors and survival analysis. Arch Intern Med 2007; 167: 276–281
- Jeremias A, Gibson C M. Narrative review: alternative causes for elevated cardiac troponin levels when acute coronary syndromes are excluded. Ann Intern Med 2005; 142: 786–791
- Roongsritong C, Warraich I, Bradley C. Common causes of troponin elevations in the absence of acute myocardial infarction: incidence and clinical significance. Chest 2004; 125: 1877–1884
- Mahajan N, Mehta Y, Rose M, Shani J, Lichstein E. Elevated troponin level is not synonymous with myocardial infarction. Int J Cardiol 2006; 111: 442–449
- Fernandes C J, Jr, Akamine N, Knobel E. Cardiac troponin: a new serum marker of myocardial injury in sepsis. Intensive Care Med 1999; 25: 1165–1168
- Giannitsis E, Muller-Bardorff M, Kurowski V, Weidtmann B, Wiegand U, Kampmann M, Katus H A. Independent prognostic value of cardiac troponin T in patients with confirmed pulmonary embolism. Circulation 2000; 102: 211–217
- Smith S C, Ladenson J H, Mason J W, Jaffe A S. Elevations of cardiac troponin I associated with myocarditis: experimental and clinical correlates. Circulation 1997; 95: 163–168
- Harvey M G, Hancox R J. Elevation of cardiac troponins in exacerbation of chronic obstructive pulmonary disease. Emerg Med Australas 2004; 16: 212–215
- Baillard C, Boussarsar M, Fosse J P, Girou E, Le Toumelin P, Cracco C, Jaber S, Cohen Y, Brochard L. Cardiac troponin I in patients with severe exacerbation of chronic obstructive pulmonary disease. Intensive Care Med 2003; 29: 584–589
- Brekke P H, Omland T, Holmedal S H, Smith P, Søyseth V. Troponin T elevation and long-term mortality after chronic obstructive pulmonary disease exacerbation. Eur Respir J 2008; 31: 563–570
- Rabe K F, Hurd S, Anzueto A, Barnes P J, Buist S A, Calverley P, Fukuchi Y, Jenkins C, Rodriguez-Roisin R, van Weel C, Zielinski J, Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176: 532–555
- Frankel W L, Herold D A, Ziegler T W, Fitzgerald R L. Cardiac troponin T is elevated in asymptomatic patients with chronic renal failure. Am J Clin Pathol 1996; 106: 118–123
- Tate J R, Ferguson W, Bais R, Kostner K, Marwick T, Carter A. The determination of the 99th centile level for troponin assays in an Australian reference population. Ann Clin Biochem 2008; 45: 275–288