Abstract
Emphysema is characterized by the destruction of alveolar parenchymal tissue and the concordant loss of lung epithelial cells, endothelial cells, and interstitial mesenchymal cells. Key features in the pathobiology of emphysema include inflammation, alveolar epithelial cell injury/apoptosis, and excessive activation of extracellular matrix (ECM) proteases. Mesenchymal cells are versatile connective tissue cells that are critical effectors of wound-repair. The excessive loss of connective tissue and the destruction of alveolar septae in emphysema suggest that the mesenchymal cell reparative response to epithelial injury is impaired. Yet, the mechanisms regulating mesenchymal cell (dys)function in emphysema remain poorly understood. We propose that mesenchymal cell fate, modulated by transforming growth factor beta-1 (TGF-β 1) and the balance of ECM proteases and antiproteases, is a critical determinant of the emphysema phenotype. We examine emphysema in the context of wound-repair responses, with a focus on the regulation of mesenchymal cell fate and phenotype. We discuss the emerging evidence supporting that genetic factors, inflammation and environmental factors, including cigarette smoke itself, collectively impair mesenchymal cell survival and function, thus contributing to the pathogenesis of emphysema.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) refers to a heterogeneous group of clinical syndromes characterized physiologically by chronic airflow obstruction that is not completely reversible. The two main clinical phenotypes of COPD represent varying degrees of airway abnormality and emphysema (Citation[1]), although overlapping features are seen in individual patients. Chronic bronchitis, defined clinically by the presence of productive cough for 3 months in 2 consecutive years, has been considered a common clinical phenotype. Emphysema, defined pathologically by airspace enlargement and destruction of alveolar septae in the absence of significant fibrosis, accounts for about 20–25% of COPD cases and is estimated to affect 4–5 million patients in the United States (Citation[2], Citation[3], Citation[4], Citation[5]).
COPD is currently a leading cause of death worldwide and in the United States (Citation[6]). Of the 6 leading causes of death in the United States, only COPD has been associated with increasing mortality since 1970 (Citation[7]). While reports of prevalence vary based on country and methodology, studies have estimated that 9–10% of the worldwide population over 40 years of age has COPD and that COPD affects approximately 14% of the adult population in the United States (Citation[3], Citation[8]). The principal risk factor for COPD is exposure to tobacco smoke, which accounts for as much as 90% of cases (Citation[9]). Environmental exposures such as pollution and biomass fuels are an increasingly appreciated cause of COPD worldwide (Citation[10]).
There is remarkable heterogeneity in an individual's susceptibility to COPD. It is widely reported that only 15–20% of cigarette smokers develop COPD, but more recent literature estimates that up to 50% of long-term smokers actually have COPD (Citation[9], Citation[11]). The mechanisms underlying the variable susceptibility to tobacco smoke-induced COPD are unclear.
In this review, we will discuss emerging concepts of the pathobiology of the emphysema phenotype of COPD. Emphysema is unique in that it primarily involves the terminal alveolar units with relative sparing of the airways. It is characterized by the degradation of elastic fibers and loss of parenchymal “tethering,” which lead to increased lung compliance, hyperinflation, air trapping and airflow obstruction. These pathophysiologic changes result in dyspnea, the cardinal clinical manifestation of emphysema (Citation[12], Citation[13]).
Importantly, greater degrees of emphysema may be associated with impaired prognosis in COPD (Citation[14]). Currently, management of emphysema relies on risk factor modification, prevention and treatment of acute exacerbations, and symptom relief with anti-inflammatory approaches, bronchodilators, and oxygen supplementation (Citation[13]). In a subpopulation of patients with upper-lobe predominant emphysema, lung volume reduction surgery may improve pulmonary mechanics, quality of life, and survival (Citation[15]). Novel therapeutic strategies for emphysema require improved understanding of the fundamental pathophysiologic mechanisms involved. Recent reviews have provided in-depth discussion of the roles of chronic inflammation, protease activation, oxidative stress and alveolar epithelial cell apoptosis in the pathogenesis of emphysema (Citation[4], Citation[5], Citation[10], Citation[16]).
It has been proposed that emphysema results from failed lung maintenance and repair following lung injury (Citation[4], Citation[16], Citation[17]). The goal of this paper is to expand on this concept of failed lung repair and regeneration with a focus on mesenchymal cells, including fibroblasts and myofibroblasts, key effectors of wound-repair responsible for extracellular matrix (ECM) synthesis and remodeling (Citation[18], Citation[19]). We propose that mesenchymal cell fate, modulated by transforming growth factor-β 1 (TGF-β 1) and ECM proteases/antiproteases, may be a key determinant of the emphysema phenotype.
EMPHYSEMA PATHOGENESIS: A PARADIGM OF INEFFECTIVE WOUND REPAIR
The histopathology of emphysema is characterized by chronic inflammation and the destruction of lung alveolar-capillary units. This is typically associated with the loss of alveolar epithelial and endothelial cells in concordance with the loss of reparative interstitial mesenchymal cells. The loss of all three of these cell types suggests that the mechanisms underlying parenchymal cell injury may also lead to mesenchymal cell dysfunction and/or death, thereby imparing the reparative response and promoting emphysema.
The prevailing hypothesis of emphysema pathogenesis suggests that it begins with exposure to toxic substances in tobacco smoke (or other toxic inhalational substances) which induce a chronic inflammatory response (Citation[10], Citation[12]). Neutrophils, macrophages and lymphocytes are recruited to the alveolar environment where they release elastases, cytokines and oxidants which may then perpetuate the cycle of epithelial injury and inflammation. Unless neutralized by antiproteases, elastases promote proteolysis of the ECM directly and through activation of collagenases. Elastin degradation products further amplify the inflammatory response (Citation[20], Citation[21]).
Interestingly, alveolar epithelial cell injury and apoptosis are observed in both emphysema and pulmonary fibrosis; yet, in emphysema there is an apparent loss of interstitial mesenchymal cells and matrix components while in fibrosis there is accumulation of activated myofibroblasts and ECM (Citation[5], Citation[22]). Thus, while pulmonary fibrosis may represent an exuberant mesenchymal cell response to alveolar epithelial injury and apoptosis, emphysema may be conceptualized as a deficiency in mesenchymal cell responses following epithelial cell injury. Despite the recognized role of mesenchymal cells in the reparative responses to tissue injury, little is known about the role of these cells in the pathobiology of emphysema.
Myofibroblasts in lung development and repair
There is better understanding of the role of interstitial mesenchymal cells in normal lung development. During fetal development, epithelial-mesenchymal communication is critical for branching morphogenesis and alveolar septal formation (Citation[23], Citation[24]). The completion of alveolarization requires lung fibroblast apoptosis (Citation[25]). Mesenchymal cell responses following epithelial injury recapitulate, in many ways, their roles in lung development (Citation[26]). General paradigms of wound-repair propose that recruited fibroblasts, derived from several different potential origins, infiltrate the wound microenvironment following injury (Citation[27], Citation[28]). Soluble factors and the extracellular matrix (ECM) induce differentiation into “activated” myofibroblasts which synthesize, secrete, and remodel new ECM.
The activated myofibroblast also serves as a source of cytokines, growth factors, and reactive oxygen species that function as autocrine and paracrine signaling mediators while the newly synthesized matrix serves as a functional scaffold for reepithelialization (Citation[18], Citation[29], Citation[30], Citation[31]). Establishing normal tissue architecture, however, requires the precise spatial and temporal regulation of myofibroblast function and appropriate myofibroblast apoptosis, which may herald the termination of the wound-repair response (Citation[26], Citation[32], Citation[33]). On the one hand, prolonged or excessive myofibroblast activation is thought to contribute to pathologic scar formation, or fibrosis. On the other hand, a lack (or loss) of appropriate myofibroblast activity might result in ineffective or insufficient wound repair (Citation[28], Citation[34]). Thus, mesenchymal cell fate represents a critical determinant of physiologic and pathologic repair.
The role of alveolar interstitial mesenchymal cells in homeostasis and disease
In the adult lung, mesenchymal cells are present within the subepithelial and subendothelial matrix of the airways and alveoli. The subepithelial/subendothelial matrix and its cellular components become progressively attenuated at the level of the alveolus, and this space between the alveolar epithelium and the capillary endothelium is commonly referred to as the ‘interstitium’ of the lung. There is significant diversity and heterogeneity of mesenchymal cells that normally reside in the lung interstitium; these mesenchymal cells include fibroblasts, myofibroblast-like contractile cells, smooth muscle cells, pericytes, and some fibroblastic cells that appear less differentiated (Citation[35], Citation[36]). While the relationships between these cell types are not well understood, mesenchymal cells are likely to play major roles in maintaining homeostasis and in repair responses of the adult lung. Homeostatic roles are supported by the finding that mesenchymal cells form direct contacts with alveolar epithelial cells and capillary endothelial cells within the alveolar wall of the normal adult lung (Citation[37], Citation[38], Citation[39]). Loss of homeostatic relationships between these cell types during reparative responses to injury may give rise to specific disease phenotypes.
Sirriani and colleagues reported that the normal fibroblast-mediated linkage between type 2 pneumocytes and the capillary endothelium through basal lamina apertures was lost in emphysema (Citation[37]). In fibrotic lung diseases, there is an expansion of the interstitium that may involve the activation and persistence of specific mesenchymal cell types such as myofibroblasts (Citation[37], Citation[40]). Our current understanding of the phenotype and function of interstitial mesenchymal cells, and how these phenotypes and functions are affected by cross-talk with other cell types, in different lung remodeling diseases is limited and requires further study.
Transforming growth factor-β 1 in lung injury and repair
The TGF-β gene family encodes for a number of signaling proteins that regulate developmental lung morphogenesis, adult tissue homeostasis and reparative responses to injury. TGF-β 1 is a 25 kDa dimeric polypeptide that regulates mesenchymal cell phenotype and fate (Citation[26], Citation[41]). Secreted as part of a latent complex by a number of cells, including myofibroblasts themselves, TGF-β 1 may be activated through the proteolytic activity of a number of mediators including plasmin, matrix metalloproteinases (MMPs), and thrombospondin (Citation[42]). Recent studies have shown that integrin binding facilitates proteolytic activation of TGF-β 1 and that integrins can activate TGF-β 1 in a protease-independent manner (Citation[42]). Activated TGF-β 1 binds to the TGF-β receptor complex, a heterotetramer composed of two type II receptors and two type I receptors. The type II receptor transphosphorylates the type-1 receptor, inducing phosphorylation of receptor-associated SMADs (R-SMADs; SMAD2 and SMAD3). The R-SMADs dissociate from the receptor complex, partner with the common co-SMAD (SMAD4), and translocate to the nucleus where the SMAD-complex regulates gene transcription. Transcriptional regulation is cell-type and context-specific, and is modified by other transcription factors, co-activators, and co-repressors which cumulatively determine the outcome of TGF-β 1 signaling (Citation[43]). Additional complexity is derived from studies demonstrating that TGF-β 1 also signals via non-SMAD pathways (Citation[44]).
TGF-β 1 typically functions as a tumor-suppressor through growth inhibition and induction of apoptosis in epithelial cells (Citation[45]). Additionally, TGF-β 1 has immunomodulatory actions (Citation[46]). In mesenchymal cells, however, TGF-β 1 is a potent “activator” of myofibroblast differentiation, ECM synthesis, migration, oxidant production, and survival (Citation[18]). In contrast to its typical pro-apoptotic and tumor-supressive actions on epithelial cells, TGF-β 1 can also promote epithelial-mesenchymal transition, a process which may facilitate cancer metastasis and contribute to pulmonary fibrosis (Citation[47]).
A POTENTIAL ROLE FOR TGF-B1 IN THE PATHOGENESIS OF EMPHYSEMA
Evidence from patients with emphysema and from animal models suggests that disruption of TGF-β 1 signaling, at the level of its synthesis/activation, receptor ligation or post-receptor signal transduction, may be critical to the pathobiology of emphysema. TGF-β 1 polymorphisms have been associated with COPD in a number of studies, although the specific effects of these polymorphisms on cellular functions have not been fully elucidated (Citation[48], Citation[49], Citation[50], Citation[51], Citation[52]). One study examined the leucine-proline polymorphism in codon 10 of the TGF-β gene and found that the leucine allele is more common in COPD patients (Citation[50]). Moreover, the proline allele, which results in increased levels of TGF-β 1 protein and mRNA, was shown to be associated with increased resistance to cigarette smoke-induced COPD (Citation[50], Citation[53]). Thus, a “high-producing” TGF-β 1 phenotype appears to confer protection against the development of COPD.
Animal models also support a role for TGF-β 1 signaling in resistance to emphysema. Morris and colleagues showed that mice lacking the β 6 subunit of integrin α vβ 6 develop age-related emphysema due to an inability to activate latent TGF-β 1 and, consequently, increased expression of MMP-12, a matrix metalloproteinase that is a critical mediator of cigarette smoke-induced emphysema in mice (Citation[54], Citation[55]). Consistent with a role for TGF-β 1 signaling in the prevention of emphysema, two studies have found that mice deficient in SMAD3 develop emphysema (Citation[56], Citation[57]). Additionally, mice deficient in latent TGF-β -binding protein 4, which is involved in latent TGF-β 1 activation, have impaired nuclear localization of SMADs and defective alveolarization (Citation[58]). It is currently unknown if emphysema in these studies results from defective alveolarization, impaired post-natal maintenance of alveolar septal structure, or a combination of these mechanisms. Finally, recent evidence supports the presence of impaired TGF-β 1 signaling in fibroblasts from humans with COPD (Citation[17]). Collectively, these studies support a critical role for TGF-β 1 signaling in the maintenance of lung architecture and the prevention of emphysema.
ECM PROTEOLYSIS IN EMPHYSEMA AND MESENCHYMAL CELL PHENOTYPES
TGF-β 1 and the ECM are both critical to the maintenance of mesenchymal cell phenotypes. Recent studies demonstrate that plasmin-mediated proteolysis of ECM fibronectin induces fibroblast apoptosis, while TGF-β 1 upregulation of the antiprotease plasminogen activator-inhibitor 1 (PAI-1) inhibits fibronectin proteolysis and prevents apoptosis (Citation[59]). Protease-antiprotease balance is important in the regulation of wound-repair, and excess antiprotease activity has been implicated in the pathogenesis of pulmonary fibrosis (Citation[60]). The protease/antiprotease hypothesis posits that emphysema results from protease (particularly elastase) activity in excess of anti-protease (α 1 antitrypsin) activity. Indeed, animal models show that the intratracheal administration of elastase induces emphysema and that elastase is increased following cigarette smoke exposure in humans and animals (Citation[61]).
Cigarette smoke induces airway and alveolar inflammation. Recruited neutrophils and macrophages are the primary sources of elastase, which proteolytically cleaves elastin and other ECM components (Citation[10]). The consequences of unopposed ECM proteolysis, however, extend beyond the loss of connective tissue. Cleaved elastin-fragments act as biologically active “matrikines” to further promote inflammation (Citation[20], Citation[21]). Thus, a vicious cycle of inflammation and ECM proteolysis contributes to the pathobiology of emphysema.
Mesenchymal cells are responsible for the maintenance and repair of ECM components, including elastin (Citation[18]). Fibroblasts repair elastase-digested matrices through de novo elastin synthesis and by repair of degraded elastin fibers (Citation[62]), suggesting that they may reduce inflammation by limiting matrikine generation. TGF-β 1 stimulates elastin production by myofibroblasts through transcriptional mechanisms (Citation[63], Citation[64]). In inflammatory states, however, neutrophil elastase inhibits elastin synthesis by suppressing expression of the elastin precursor, tropoelastin (Citation[65]). Recent studies have elucidated the mechanism, demonstrating that elastin proteolysis liberates EGF and EGF-like peptides (Citation[66]), which induce expression of the TGF-β co-repressor, TGIF, thereby reducing mesenchymal cell responsiveness to TGF-β 1 signals (Citation[67], Citation[68]).
A large body of evidence supports the role of MMPs in the pathobiology of emphysema (recently reviewed in ref (Citation[10])). Of the MMPs, there is compelling support for the pathogenic roles of elastolytic MMPs-9 and 12 (Citation[54], Citation[55], Citation[57], Citation[69], Citation[70], Citation[71], Citation[72], Citation[73]). In humans, polymorphisms in MMP-9 are associated with the development of cigarette-smoke induced emphysema (Citation[71]). Additionally, alveolar macrophages from patients with emphysema generate greater amounts of MMP-9 with higher levels of activity than those of normal subjects (Citation[72]). Consistently, MMP-9 levels are increased in the BAL of smoke-exposed mice (Citation[73]). A recent study has shown that transgenic overexpression of MMP-9 induces late onset emphysema (Citation[70]).
Mice deficient in the β 6 subunit of the integrin α vβ 6 are unable to activate TGF-β 1 from its latent form and develop spontaneous emphysema associated with increased macrophage expression of MMP-12. However, when these mice transgenically overexpress active TGF-β 1 (obviating the need for activation by α vβ 6), MMP-12 expression is normal and emphysema fails to develop (Citation[54]). MMP-12 deficient mice, consistently, are protected from cigarette smoke-induced emphysema (Citation[54], Citation[55]). SMAD-3 knockout mice, which develop emphysema, have increased levels of MMP-9 and MMP-12 (Citation[57]). In guinea pigs subjected to cigarette smoke inhalation for 6 months, a pharmacologic inhibitor of MMP-9 and MMP-12 attenuates the pulmonary physiologic effects of cigarette smoke while reducing emphysema and BAL evidence of elastolysis (Citation[69]). This is consistent with another study demonstrating that a broad-spectrum MMP inhibitor attenuates tobacco smoke induced inflammation and emphysema in guinea pigs (Citation[74]). In humans with COPD, several studies have shown increased expression of MMP-12 (Citation[75], Citation[76]). Some studies, however, have failed to identify increased MMP-12 in humans with emphysema, suggesting additional mechanisms independent of MMP-12 (Citation[77]).
Myofibroblasts secrete MMP inhibitors (TIMPs), which would be expected to limit the proteolytic actions of MMPs and alter the protease-antiprotease balance. However, few studies have reported on the specific regulation of MMPs and TIMPs by mesenchymal cells in the context of emphysema. Elastase promotes cleavage/inactivation of TIMPs while inducing fibroblast production of MMP-2 (Citation[78], Citation[79]). Moreover, pro-MMP-1, MMP-2 and MT1-MMP are increased in fibroblasts exposed to cigarette smoke extract (CSE) and high concentrations of CSE are sufficient to activate MMP-1 (Citation[80], Citation[81]). Collectively, these studies suggest that CSE and elastase induce ECM proteolysis, in part, by regulating mesenchymal cell function.
A direct effect of elastase on mesenchymal cell survival has not been demonstrated; however, recent studies suggest that elastases may regulate fibroblast survival. While elastase does not directly function as a collagenase, it induces collagen degradation and augments collagen gel contraction when added to fibroblasts in culture (Citation[79], Citation[82]). Moreover, elastase-mediated collagen degradation is enhanced in the presence of inflammatory cytokines (Citation[79], Citation[83], Citation[84]). Inflammatory cytokines induce fibroblast synthesis of MMPs which are activated by elastase, thereby promoting collagen degradation (Citation[79]). An additive or synergistic role for inflammation in fibroblast-mediated collagen degradation is further supported by a recent study that showed conditioned media from stimulated CD4+ T-lymphocytes induces collagen degradation by fibroblasts (Citation[85]). As both ECM proteolysis and collagen gel contraction have been shown to induce fibroblast apoptosis, these studies support a mechanism whereby elastases may promote fibroblast apoptosis (Citation[59], Citation[86], Citation[87]).
While the deleterious effects of elastase in emphysema pathogenesis extend beyond elastin proteolysis, the “protective” actions of antiproteases extend beyond inhibition of proteolysis. α 1-AT has been shown to stimulate fibroblast proliferation and procollagen production (Citation[88]). Additionally, serine protease inhibitors block cigarette smoke-induced fibroblast secretion of inflammatory cell chemoattractants (Citation[89]). Finally, although the role of α 1-AT on mesenchymal cell survival/apoptosis has not been investigated, α 1-AT protects endothelial cells from apoptosis by inhibition of caspase-3 (Citation[90], Citation[91]). These findings suggest that anti-proteases (α 1-AT, serpins and TIMPs) promote apoptosis-resistant cellular phenotypes and support the role of an alveolar microenvironment that promotes emphysema not only through ECM proteolysis, but also through inhibition of mesenchymal cell repair functions.
CIGARETTE SMOKE INHIBITS FIBROBLAST FUNCTION
Cigarette smoke is the leading cause of emphysema and is known to induce inflammation and alveolar epithelial cell injury/apoptosis (Citation[10]). Additionally, a number of studies have shown that cigarette smoke adversely affects the ability of mesenchymal cells to respond appropriately to epithelial injury. In a series of studies by Rennard and colleagues, volatile compounds in cigarette smoke extract (CSE) were found to inhibit fibroblast proliferation and fibronectin synthesis (Citation[92], Citation[93]). Functionally, CSE impairs the ability of fibroblasts to contract 3-D collagen gels (Citation[93], Citation[94]). The anti-oxidant, glutathione, restores contractile capabilities to the CSE-exposed fibroblasts, suggesting a role for oxidative stress as a mechanism of CSE actions on fibroblast function (Citation[95]). Moreover, cigarette smoke stimulates collagen degradation by fibroblasts and inhibits fibroblast-mediated collagen cross-linking. In human fetal lung fibroblasts, CSE induces expression and activation of MMP-1, MMP-2, and MT1-MMP (Citation[80], Citation[81]). Finally, cigarette smoke condensate impairs the transcription, stability, and promoter activity of lysyl oxidase, a collagen cross-linking enzyme (Citation[96]).
Epithelial cell death has been implicated in the pathogenesis of COPD. Although the precise mechaninsms of cell death have not been clearly defined, a number of studies have shown activation of apoptosis pathways (Citation[97]). More recently, CSE has been found to activate autophagy, a pro-survival mechanism that can ultimately lead to apoptosis, in epithelial cells (Citation[98], Citation[99]). In some cases, however, components in cigarette smoke have been shown to inhibit cellular apoptosis. In one example, acrolein was found to inhibit neutrophil apoptosis through inhibition of caspase-3, providing another potential mechanism for an ongoing inflammatory response in COPD. Moreover, nicotine and NNK (a carcinogen contained within tobacco) were shown to rapidly activate pro-survival signaling in human bronchial epithelial cells, a mechanism which would favor carcinogenesis (Citation[100]).
Few studies have examined the effects of cigarette smoke on mesenchymal cell survival. Ishii et al. found that low concentrations of CSE induce fibroblast apoptosis while higher concentrations of CSE induce necrosis (Citation[101]). CSE-stimulated fibroblast apoptosis is attributable, at least in part, to oxidative stress as overexpression of glutathione-S-transferase protected fibroblasts from apoptosis (Citation[101]). Similarly, the glutathione precursor, N-acetyl cysteine, attenuates CSE-induced fibroblast apoptosis while glutathione depletion promotes fibroblast apoptosis (Citation[102]).
Interestingly, Baglole and colleagues used four different commercially available normal adult lung fibroblast cell lines to demonstrate significant heterogeneity in apoptotic susceptibility to CSE (Citation[103]). This marked variation, with fibroblast viability ranging from 33% to 90%, highlights the potential role for genetic and epigenetic contributions to the apoptosis-susceptibility of fibroblasts. Collectively, these studies support the concept that cigarette smoke activates inflammatory pathways, promotes epithelial cell death and directly impairs the mesenchymal cell reparative response through inhibition of fibroblast activity and induction of apoptosis.
FIBROBLAST PHENOTYPES IN HUMANS WITH EMPHYSEMA
Several studies comparing normal mesenchymal cells with those isolated from the lungs of humans with emphysema support the hypothesis that an impaired mesenchymal cell reparative capacity contributes to the pathobiology of emphysema. Although there are differences between studies, several investigators report that emphysema fibroblasts have reduced proliferative capacity compared with normal lung fibroblasts (Citation[104], Citation[105], Citation[106]). Moreover, the reduction in fibroblast proliferation due to CSE is more pronounced in emphysema fibroblasts than in normal lung fibroblasts (Citation[105]). Emphysema fibroblasts have decreased proliferation on exposure to the pro-inflammatory cytokine, interleukin-1β (IL-1β) (Citation[106]). Similarly, Plantier and colleagues found that emphysema fibroblasts have decreased basal- and IL-1β -mediated production of hepatocyte growth factor and decreased IL-1β -stimulated production of keratinocyte growth factor (KGF) compared with normal lung fibroblasts (Citation[107], Citation[108]).
As KGF attenuates elastase-induced emphysema in mice, these findings are consistent with impaired reparative capacity of emphysema fibroblasts (Citation[107]). In further support of this concept, a recent study by Rennard and colleagues found that fibroblasts from COPD patients have decreased contractility and migration compared to normal fibroblasts (Citation[17]). In this study, the COPD fibroblasts demonstrated increased generation of PGE2, which has been shown to suppress myofibroblast differentiation, proliferation, and collagen expression. Additionally, these COPD fibroblasts have increased expression of the PGE2 receptors, EP2 and EP4 and decreased responsiveness to exogenous TGF-β 1 (Citation[17], Citation[109], Citation[110]). Importantly, this study also showed that impaired fibroblast contraction and migration correlate with declines in pulmonary function (Citation[17]).
Decreased TGF-β 1 responsiveness in emphysema fibroblasts was also observed in another study in which fibroblasts from patients with severe emphysema showed a decline in TGF-β 1-induced decorin synthesis (Citation[111]). Together, these studies demonstrate that mesenchymal cells from emphysema patients manifest decreased proliferative capacity, decreased ability to elaborate epithelial cell-protective growth factors, and decreased ECM production in response to TGF-β 1. These phenotypic alterations favor epithelial dysrepair and loss of connective tissue, key features of emphysema.
Age-associated alterations in the phenotype of epithelial cells, endothelial cells and fibroblasts may also contribute to the emphysema phenotype. A higher percentage of alveolar epithelial and endothelial cells in patients with emphysema express p16INK4a, a marker of cellular senescence, compared to asymptomatic smokers and nonsmokers (Citation[112]). This may be attributable, at least in part, to environmental stressors linked to emphysema as exemplified by the observation that cigarette smoke extract can directly induce cellular senescence (Citation[113]). Genetic and epigenetic factors are also likely to play important roles in lung senescence phenotypes. Genetic deletion of the Klotho gene, which functions to limit oxidative stress and cellular senescence, leads to emphysema in addition to a number of other age-related manifestations in mice (Citation[114]).
Mice deficient in senescence marker protein-30 (SMP30 knockout mice) demonstrate increased susceptibility to emphysema in association with increased oxidative stress and apoptosis of lung cells in response to cigarette smoke exposure (Citation[115]). While these studies support the importance of host stress-repair responses to combat environmental toxins such as cigarette smoke, mechanisms for the deficiency of such responses in sporadic emphysema remain unclear. Additionally, the varying responses to cellular stress and senescent phenotypes of different cell types (e,g., mesenchymal vs. epithelial/endothelial cells) in the lung are not well understood. Future studies in these areas will provide novel insights into the divergent tissue remodeling responses (e.g., fibrosis vs. emphysema) that can result from exposure even to same environmental insult.
CONCLUSION
Emphysema, a sub-type of COPD characterized by loss of alveolar parenchyma, results from epithelial cell death in combination with failed reparative responses by mesenchymal cells. Specifically, the failure of mesenchymal cell accumulation, activation, contraction, synthetic function, and survival have all been shown. Evidence suggests that the same factors that drive inflammation and epithelial cell death, namely ECM proteases and cigarette-smoke, directly contribute to these dysregulated mesenchymal cell phenotypes ().
Figure 1 Loss of cellular homeostasis in emphysema pathogenesis. Exposure to inhaled toxins (such as cigarette smoke) leads to epithelial cell death, inflammation, and extracellular matrix proteolysis. In susceptible individuals, mesenchymal cell survival and reparative functions are impaired by direct effects of inhaled toxic substances, inflammatory mediators, and by the loss of the peri- and extracellular matrix. The result is the loss of structural cells of the alveolar wall and the associated matrix components.
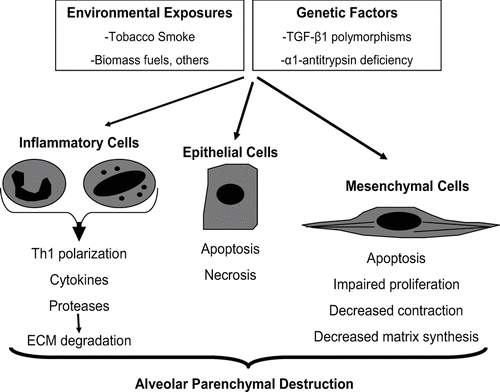
The loss of interstitial mesenchymal cells in emphysema stands in contrast with the accumulation of mesenchymal cells associated with bronchocentric fibrotic processes such as asthma, chronic bronchitis, and obliterative bronchiolitis (Citation[116], Citation[117], Citation[118]). Further studies are required to characterize the phenotype of mesenchymal cells within the airway vs. alveolar compartments and to determine if these differences may account for varying clinical phenotypes in COPD. Emphysema is a complex disease, and the substantial heterogeneity in disease susceptibility and severity support a role for genetic and/or epigenetic regulation of cellular responses. Future studies of mesenchymal cell fates and phenotypes in emphysema will shed light on the role of these cells in maintaining cellular homeostasis of the alveolar wall.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Grant support: This work was supported by the National Institutes of Health grants, K08 HL081059 (J.C.H), R01 HL67967 (V.J.T.), and the American Lung Association Dalsemer Award (J.C.H.). This article is not subject to United States' copyright law.
Related Research Data
REFERENCES
- Friedlander A L, Lynch D, Dyar L A, Bowler R P. Phenotypes of chronic obstructive pulmonary disease. COPD 2007; 4: 355–384
- Celli B R, Roger S. Mitchell Lecture. Chronic obstructive pulmonary disease phenotypes and their clinical relevance. Proc Am Thorac Soc 2006; 3: 461–465
- Halbert R J, Natoli J L, Gano A, Badamgarav E, Buist A S, Mannino D M. Global burden of COPD systematic review and meta-analysis. Eur Respir J 2006; 28: 523–532
- Tuder R M, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc 2006; 3: 503–510
- Taraseviciene-Stewart L, Voelkel N F. Molecular pathogenesis of emphysema. J Clin Invest 2008; 118: 394–402
- Mannino D M, Buist A S. Global burden of COPD: Risk factors, prevalence, and future trends. Lancet 2007; 370: 765–773
- Jemal A, Ward E, Hao Y, Thun M. Trends in the leading causes of death in the United States, 1970–2002. JAMA 2005; 294: 1255–1259
- Pauwels R A, Buist A S, Calverley P M, Jenkins C R, Hurd S S. Global Strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Nhlbi/Who Global Initiative for Chronic Obstructive Lung Disease (Gold) Workshop Summary. Am J Respir Crit Care Med 2001; 163: 1256–1276
- Chapman K R, Mannino D M, Soriano J B, Vermeire P A, Buist A S, Thun M J, Connell C, Jemal A, Lee T A, Miravitlles M, et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J 2006; 27: 188–207
- Yoshida T, Tuder R M. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 2007; 87: 1047–1082
- Lundback B, Lindberg A, Lindstrom M, Ronmark E, Jonsson A C, Jonsson E, Larsson L G, Andersson S, Sandstrom T, Larsson K. Not 15 but 50% of smokers develop COPD?—Report from the Obstructive Lung Disease in Northern Sweden Studies. Respir Med 2003; 97: 115–122
- Hogg J C. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet 2004; 364: 709–721
- Sutherland E R, Cherniack R M. Management of chronic obstructive pulmonary disease. N Engl J Med 2004; 350: 2689–2697
- Martinez F J, Foster G, Curtis J L, Criner G, Weinmann G, Fishman A, DeCamp M M, Benditt J, Sciurba F, Make B, et al. Predictors of mortality in patients with emphysema and severe airflow obstruction. Am J Respir Crit Care Med 2006; 173: 1326–1334
- Fishman A, Martinez F, Naunheim K, Piantadosi S, Wise R, Ries A, Weinmann G, Wood D E. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348: 2059–2073
- Churg A, Cosio M, Wright J L. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol 2008; 294: L612–631
- Togo S, Holz O, Liu X, Sugiura H, Kamio K, Wang X, Kawasaki S, Ahn Y, Fredriksson K, Skold C M, et al. Lung fibroblast repair functions in patients with chronic obstructive pulmonary disease are altered by multiple mechanisms. Am J Respir Crit Care Med 2008; 178: 248–260
- Hinz B, Phan S H, Thannickal V J, Galli A, Bochaton-Piallat M L, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol 2007; 170: 1807–1816
- Singer A J, Clark R A. Cutaneous wound healing. N Engl J Med 1999; 341: 738–746
- Houghton A M, Quintero P A, Perkins D L, Kobayashi D K, Kelley D G, Marconcini L A, Mecham R P, Senior R M, Shapiro S D. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest 2006; 116: 753–759
- Adair-Kirk T L, Senior R M. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol 2008; 40: 1101–1110
- Thannickal V J, Horowitz J C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc 2006; 3: 350–356
- Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, Hedstrand H, Pekna M, Hellstrom M, Gebre-Medhin S, Schalling M, et al. Pdgf-a signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 1996; 85: 863–873
- Hogan B L, Yingling J M. Epithelial/mesenchymal interactions and branching morphogenesis of the lung. Curr Opin Genet Dev 1998; 8: 481–486
- Bruce M C, Honaker C E, Cross R J. Lung fibroblasts undergo apoptosis following alveolarization. Am J Respir Cell Mol Biol 1999; 20: 228–236
- Horowitz J C, Thannickal V J. Epithelial-mesenchymal interactions in pulmonary fibrosis. Semin Respir Crit Care Med 2006; 27: 600–612
- Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen 2005; 13: 7–12
- Tomasek J J, Gabbiani G, Hinz B, Chaponnier C, Brown R A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002; 3: 349–363
- Thannickal V J, Aldweib K D, Rajan T, Fanburg B L. Upregulated expression of fibroblast growth factor (fgf) receptors by transforming growth factor-beta-1 (Tgf-Beta1) mediates enhanced mitogenic responses to fgfs in cultured human lung fibroblasts. Biochem Biophys Res Commun 1998; 251: 437–441
- Thannickal V J, Fanburg B L. Activation of an H2O2-generating Nadh oxidase in human lung fibroblasts by transforming growth factor beta-1. J Biol Chem 1995; 270: 30334–30338
- Thannickal V J, Lee D Y, White E S, Cui Z, Larios J M, Chacon R, Horowitz J C, Day R M, Thomas P E. Myofibroblast differentiation by transforming growth factor-beta-1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 2003; 278: 12384–12389
- Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995; 146: 56–66
- Horowitz J C, Thannickal V J. Idiopathic pulmonary fibrosis : new concepts in pathogenesis and implications for drug therapy. Treat Respir Med 2006; 5: 325–342
- Gauldie J, Kolb M, Ask K, Martin G, Bonniaud P, Warburton D. Smad3 signaling involved in pulmonary fibrosis and emphysema. Proc Am Thorac Soc 2006; 3: 696–702
- Bradley K H, Kawanami O, Ferrans V J, Crystal R G. The fibroblast of human lung alveolar structures: A differentiated cell with a major role in lung structure and function. Methods Cell Biol 1980; 21A: 37–64
- Crapo J D, Barry B E, Gehr P, Bachofen M, Weibel E R. Cell number and cell characteristics of the normal human lung. Am Rev Respir Dis 1982; 126: 332–337
- Sirianni F E, Milaninezhad A, Chu F S, Walker D C. Alteration of fibroblast architecture and loss of basal lamina apertures in human emphysematous lung. Am J Respir Crit Care Med 2006; 173: 632–638
- Evans M J, Van Winkle L S, Fanucchi M V, Plopper C G. The attenuated fibroblast sheath of the respiratory tract epithelial-mesenchymal trophic unit. Am J Respir Cell Mol Biol 1999; 21: 655–657
- Sirianni F E, Chu F S, Walker D C. Human alveolar wall fibroblasts directly link epithelial type 2 cells to capillary endothelium. Am J Respir Crit Care Med 2003; 168: 1532–1537
- Kuhn C, 3rd, Boldt J, King T E, Jr., Crouch E, Vartio T, McDonald J A. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis 1989; 140: 1693–1703
- Shi Y, Massague J. Mechanisms of Tgf-beta signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700
- Wipff P J, Hinz B. Integrins and the activation of latent transforming growth factor Beta-1—an intimate relationship. Eur J Cell Biol 2008; 87(8–9)601–615
- Massague J, Wotton D. Transcriptional control by the Tgf-Beta/Smad signaling system. Embo J 2000; 19: 1745–1754
- Derynck R, Zhang Y E. Smad-dependent and Smad-independent pathways in Tgf-beta family signalling. Nature 2003; 425: 577–584
- Blobe G C, Schiemann W P, Lodish H F. Role of transforming growth factor beta in human disease. N Engl J Med 2000; 342: 1350–1358
- Letterio J J, Roberts A B. Regulation of immune responses by Tgf-Beta. Annu Rev Immunol 1998; 16: 137–161
- Willis B C, Borok Z. Tgf-beta-induced Emt: Mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 2007; 293: L525–534
- Celedon J C, Lange C, Raby B A, Litonjua A A, Palmer L J, DeMeo D L, Reilly J J, Kwiatkowski D J, Chapman H A, Laird N, et al. The transforming growth factor-beta1 (Tgfb1) gene is associated with chronic obstructive pulmonary disease (COPD). Hum Mol Genet 2004; 13: 1649–1656
- Hersh C P, Demeo D L, Lazarus R, Celedon J C, Raby B A, Benditt J O, Criner G, Make B, Martinez F J, Scanlon P D, et al. Genetic association analysis of functional impairment in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006; 173: 977–984
- Wu L, Chau J, Young R P, Pokorny V, Mills G D, Hopkins R, McLean L, Black P N. Transforming growth factor-beta-1 genotype and susceptibility to chronic obstructive pulmonary disease. Thorax 2004; 59: 126–129
- Ogawa E, Ruan J, Connett J E, Anthonisen N R, Pare P D, Sandford A J. Transforming growth factor-beta-1 polymorphisms, airway responsiveness and lung function decline in smokers. Respir Med 2007; 101: 938–943
- DeMeo D L, Hersh C P, Hoffman E A, Litonjua A A, Lazarus R, Sparrow D, Benditt J O, Criner G, Make B, Martinez F J, et al. Genetic determinants of emphysema distribution in the national emphysema treatment trial. Am J Respir Crit Care Med 2007; 176: 42–48
- Suthanthiran M, Li B, Song J O, Ding R, Sharma V K, Schwartz J E, August P. Transforming growth factor-beta 1 Hyperexpression in African-American hypertensives: a novel mediator of hypertension and/or target organ damage. Proc Natl Acad Sci U S A 2000; 97: 3479–3484
- Morris D G, Huang X, Kaminski N, Wang Y, Shapiro S D, Dolganov G, Glick A, Sheppard D. Loss of integrin alpha(V)beta6-mediated Tgf-beta activation causes Mmp12-dependent emphysema. Nature 2003; 422: 169–173
- Hautamaki R D, Kobayashi D K, Senior R M, Shapiro S D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997; 277: 2002–2004
- Chen H, Sun J, Buckley S, Chen C, Warburton D, Wang X F, Shi W. Abnormal mouse lung alveolarization caused by Smad3 deficiency is a developmental antecedent of centrilobular emphysema. Am J Physiol Lung Cell Mol Physiol 2005; 288: L683–691
- Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts P J, Roberts A B, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to Tgf-Beta-mediated pulmonary fibrosis. J Immunol 2004; 173: 2099–2108
- Sterner-Kock A, Thorey I S, Koli K, Wempe F, Otte J, Bangsow T, Kuhlmeier K, Kirchner T, Jin S, Keski-Oja J, et al. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (Ltbp-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev 2002; 16: 2264–2273
- Horowitz J C, Rogers D S, Simon R H, Sisson T H, Thannickal V J. Plasminogen activation induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am J Respir Cell Mol Biol 2008; 38: 78–87
- Chapman H A. Disorders of lung matrix remodeling. J Clin Invest 2004; 113: 148–157
- Janoff A. Elastases and emphysema. Current assessment of the protease-antiprotease hypothesis. Am Rev Respir Dis 1985; 132: 417–433
- Stone P J, Morris S M, Thomas K M, Schuhwerk K, Mitchelson A. Repair of elastase-digested elastic fibers in acellular matrices by replating with neonatal rat-lung lipid interstitial fibroblasts or other elastogenic cell types. Am J Respir Cell Mol Biol 1997; 17: 289–301
- Kuang P P, Zhang X H, Rich C B, Foster J A, Subramanian M, Goldstein R H. Activation of elastin transcription by transforming growth factor-beta in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2007; 292: L944–952
- McGowan S E, McNamer R. Transforming growth factor-beta increases elastin production by neonatal rat lung fibroblasts. Am J Respir Cell Mol Biol 1990; 3: 369–376
- Foster J A, Rich C B, Miller M F. Pulmonary fibroblasts: an in vitro model of emphysema. regulation of elastin gene expression. J Biol Chem 1990; 265: 15544–15549
- DiCamillo S J, Carreras I, Panchenko M V, Stone P J, Nugent M A, Foster J A, Panchenko M P. Elastase-released epidermal growth factor recruits epidermal growth factor receptor and extracellular signal-regulated kinases to down-regulate tropoelastin Mrna in lung fibroblasts. J Biol Chem 2002; 277: 18938–18946
- DiCamillo S J, Yang S, Panchenko M V, Toselli P A, Naggar E F, Rich C B, Stone P J, Nugent M A, Panchenko M P. Neutrophil elastase-initiated EGFR/MEK/ERK signaling counteracts stabilizing effect of autocrine Tgf-Beta on Tropoelastin Mrna in Lung Fibroblasts. Am J Physiol Lung Cell Mol Physiol 2006; 291: L232–243
- Yang S, Nugent M A, Panchenko M P. Egf antagonizes Tgf-{Beta} induced tropoelastin expression in lung fibroblasts via stabilization of Smad corepressor Tgif. Am J Physiol Lung Cell Mol Physiol Jul, 2008; 295(1)L143–L151
- Churg A, Wang R, Wang X, Onnervik P O, Thim K, Wright J L. Effect of an Mmp-9/Mmp-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 2007; 62: 706–713
- Foronjy R, Nkyimbeng T, Wallace A, Thankachen J, Okada Y, Lemaitre V, D'Armiento J. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Physiol Lung Cell Mol Physiol 2008; 294: L1149–1157
- Minematsu N, Nakamura H, Tateno H, Nakajima T, Yamaguchi K. Genetic polymorphism in matrix metalloproteinase-9 and pulmonary emphysema. Biochem Biophys Res Commun 2001; 289: 116–119
- Russell R E, Culpitt S V, DeMatos C, Donnelly L, Smith M, Wiggins J, Barnes P J. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2002; 26: 602–609
- Seagrave J, Barr E B, March T H, Nikula K J. Effects of cigarette smoke exposure and cessation on inflammatory cells and matrix metalloproteinase activity in mice. Exp Lung Res 2004; 30: 1–15
- Selman M, Cisneros-Lira J, Gaxiola M, Ramirez R, Kudlacz E M, Mitchell P G, Pardo A. Matrix metalloproteinases inhibition attenuates tobacco smoke-induced emphysema in guinea pigs. Chest 2003; 123: 1633–1641
- Demedts I K, Morel-Montero A, Lebecque S, Pacheco Y, Cataldo D, Joos G F, Pauwels R A, Brusselle G G. Elevated Mmp-12 protein levels in induced sputum from patients with COPD. Thorax 2006; 61: 196–201
- Molet S, Belleguic C, Lena H, Germain N, Bertrand C P, Shapiro S D, Planquois J M, Delaval P, Lagente V. Increase in macrophage elastase (Mmp-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res 2005; 54: 31–36
- Imai K, Dalal S S, Chen E S, Downey R, Schulman L L, Ginsburg M, D'Armiento J. Human collagenase (Matrix Metalloproteinase-1) expression in the lungs of patients with emphysema. Am J Respir Crit Care Med 2001; 163: 786–791
- Fredriksson K, Liu X D, Lundahl J, Klominek J, Rennard S I, Skold C M. Red blood cells increase secretion of matrix metalloproteinases from human lung fibroblasts in vitro. Am J Physiol Lung Cell Mol Physiol 2006; 290: L326–333
- Zhu Y K, Liu X D, Skold C M, Umino T, Wang H J, Spurzem J R, Kohyama T, Ertl R F, Rennard S I. Synergistic neutrophil elastase-cytokine interaction degrades collagen in three-dimensional culture. Am J Physiol Lung Cell Mol Physiol 2001; 281: L868–878
- Ning W, Dong Y, Sun J, Li C, Matthay M A, Feghali-Bostwick C A, Choi A M. Cigarette smoke stimulates matrix metalloproteinase-2 activity Via Egr-1 in human lung fibroblasts. Am J Respir Cell Mol Biol 2007; 36: 480–490
- Kim H, Liu X, Kohyama T, Kobayashi T, Conner H, Abe S, Fang Q, Wen F Q, Rennard S I. Cigarette smoke stimulates Mmp-1 production by human lung fibroblasts through the Erk1/2 pathway. COPD 2004; 1: 13–23
- Skold C M, Liu X, Umino T, Zhu Y, Ohkuni Y, Romberger D J, Spurzem J R, Heires A J, Rennard S I. Human neutrophil elastase augments fibroblast-mediated contraction of released collagen gels. Am J Respir Crit Care Med 1999; 159: 1138–1146
- Zhu Y, Skold C M, Liu X, Wang H, Kohyama T, Wen F Q, Ertl R F, Rennard S I. Fibroblasts and monocyte macrophages contract and degrade three-dimensional collagen gels in extended co-culture. Respir Res 2001; 2: 295–299
- Kohyama T, Liu X, Wen F Q, Zhu Y K, Wang H, Kim H J, Takizawa H, Cieslinski L B, Barnette M S, Rennard S I. Pde4 inhibitors attenuate fibroblast chemotaxis and contraction of native collagen gels. Am J Respir Cell Mol Biol 2002; 26: 694–701
- Mikko M, Fredriksson K, Wahlstrom J, Eriksson P, Grunewald J, Skold C M. Human T cells stimulate fibroblast-mediated degradation of extracellular matrix in vitro. Clin Exp Immunol 2008; 151: 317–325
- Horowitz J C, Rogers D S, Sharma V, Vittal R, White E S, Cui Z, Thannickal V J. Combinatorial Activation of Fak and Akt by Transforming Growth Factor-Beta1 Confers an Anoikis-Resistant Phenotype to Myofibroblasts. Cell Signal 2007; 19: 761–771
- Zhu Y K, Umino T, Liu X D, Wang H J, Romberger D J, Spurzem J R, Rennard S I. Contraction of fibroblast-containing collagen gels: initial collagen concentration regulates the degree of contraction and cell survival. In Vitro Cell Dev Biol Anim 2001; 37: 10–16
- Dabbagh K, Laurent G J, Shock A, Leoni P, Papakrivopoulou J, Chambers R C. Alpha-1-antitrypsin stimulates fibroblast proliferation and procollagen production and activates classical map kinase signalling pathways. J Cell Physiol 2001; 186: 73–81
- Numanami H, Koyama S, Nelson D K, Hoyt J C, Freels J L, Habib M P, Amano J, Haniuda M, Sato E, Robbins R A. Serine protease inhibitors modulate smoke-induced chemokine release from human lung fibroblasts. Am J Respir Cell Mol Biol 2003; 29: 613–619
- Petrache I, Fijalkowska I, Medler T R, Skirball J, Cruz P, Zhen L, Petrache H I, Flotte T R, Tuder R M. Alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol 2006; 169: 1155–1166
- Petrache I, Fijalkowska I, Zhen L, Medler T R, Brown E, Cruz P, Choe K H, Taraseviciene-Stewart L, Scerbavicius R, Shapiro L, et al. A novel antiapoptotic role for Alpha-1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med 2006; 173: 1222–1228
- Nakamura Y, Romberger D J, Tate L, Ertl R F, Kawamoto M, Adachi Y, Mio T, Sisson J H, Spurzem J R, Rennard S I. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. Am J Respir Crit Care Med 1995; 151: 1497–1503
- Wang H, Liu X, Umino T, Kohyama T, Zhu Y K, Wen F Q, Spurzem J R, Romberger D J, Kim H J, Rennard S I. Effect of cigarette smoke on fibroblast-mediated gel contraction is dependent on cell density. Am J Physiol Lung Cell Mol Physiol 2003; 284: L205–213
- Carnevali S, Nakamura Y, Mio T, Liu X, Takigawa K, Romberger D J, Spurzem J R, Rennard S I. Cigarette smoke extract inhibits fibroblast-mediated collagen gel contraction. Am J Physiol 1998; 274: L591–598
- Kim H J, Liu X, Wang H, Kohyama T, Kobayashi T, Wen F Q, Romberger D J, Abe S, MacNee W, Rahman I, et al. Glutathione prevents inhibition of fibroblast-mediated collagen gel contraction by cigarette smoke. Am J Physiol Lung Cell Mol Physiol 2002; 283: L409–417
- Gao S, Chen K, Zhao Y, Rich C B, Chen L, Li S J, Toselli P, Stone P, Li W. Transcriptional and posttranscriptional inhibition of lysyl oxidase expression by cigarette smoke condensate in cultured rat fetal lung fibroblasts. Toxicol Sci 2005; 87: 197–203
- Demedts I K, Demoor T, Bracke K R, Joos G F, Brusselle G G. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 2006; 7: 53
- Kim H P, Wang X, Chen Z H, Lee S J, Huang M H, Wang Y, Ryter S W, Choi A M. Autophagic proteins regulate cigarette smoke-induced apoptosis: protective role of heme Oxygenase-1. Autophagy 2008; 4: 887–895
- Ryter S W, Chen Z H, Kim H P, Choi A M. Autophagy in chronic obstructive pulmonary disease: homeostatic or pathogenic mechanism?. Autophagy 2009; 5(2)235–237
- West K A, Brognard J, Clark A S, Linnoila I R, Yang X, Swain S M, Harris C, Belinsky S, Dennis P A. Rapid Akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest 2003; 111: 81–90
- Ishii T, Matsuse T, Igarashi H, Masuda M, Teramoto S, Ouchi Y. Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am J Physiol Lung Cell Mol Physiol 2001; 280: L1189–1195
- Carnevali S, Petruzzelli S, Longoni B, Vanacore R, Barale R, Cipollini M, Scatena F, Paggiaro P, Celi A, Giuntini C. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2003; 284: L955–963
- Baglole C J, Bushinsky S M, Garcia T M, Kode A, Rahman I, Sime P J, Phipps R P. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: implications for emphysema. Am J Physiol Lung Cell Mol Physiol 2006; 291: L19–29
- Holz O, Zuhlke I, Jaksztat E, Muller K C, Welker L, Nakashima M, Diemel K D, Branscheid D, Magnussen H, Jorres R A. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J 2004; 24: 575–579
- Nobukuni S, Watanabe K, Inoue J, Wen F Q, Tamaru N, Yoshida M. Cigarette smoke inhibits the growth of lung fibroblasts from patients with pulmonary emphysema. Respirology 2002; 7: 217–223
- Noordhoek J A, Postma D S, Chong L L, Vos J T, Kauffman H F, Timens W, van Straaten J F. Different proliferative capacity of lung fibroblasts obtained from control subjects and patients with emphysema. Exp Lung Res 2003; 29: 291–302
- Plantier L, Marchand-Adam S, Antico V G, Boyer L, De Coster C, Marchal J, Bachoual R, Mailleux A, Boczkowski J, Crestani B. Keratinocyte growth factor protects against elastase-induced pulmonary emphysema in mice. Am J Physiol Lung Cell Mol Physiol 2007; 293: L1230–1239
- Plantier L, Marchand-Adam S, Marchal-Somme J, Leseche G, Fournier M, Dehoux M, Aubier M, Crestani B. Defect of hepatocyte growth factor production by fibroblasts in human pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol 2005; 288: L641–647
- Huang S, Wettlaufer S H, Hogaboam C, Aronoff D M, Peters-Golden M. Prostaglandin E(2) Inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E Prostanoid 2 receptor and camp signaling. Am J Physiol Lung Cell Mol Physiol 2007; 292: L405–413
- Kolodsick J E, Peters-Golden M, Larios J, Toews G B, Thannickal V J, Moore B B. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid Receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol 2003; 29: 537–544
- Noordhoek J A, Postma D S, Chong L L, Menkema L, Kauffman H F, Timens W, van Straaten J F, van der Geld Y M. Different modulation of decorin production by lung fibroblasts from patients with mild and severe emphysema. COPD 2005; 2: 17–25
- Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med 2006; 174: 886–893
- Nyunoya T, Monick M M, Klingelhutz A, Yarovinsky T O, Cagley J R, Hunninghake G W. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol 2006; 35: 681–688
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390: 45–51
- Sato T, Seyama K, Sato Y, Mori H, Souma S, Akiyoshi T, Kodama Y, Mori T, Goto S, Takahashi K, et al. Senescence marker Protein-30 protects mice lungs from oxidative stress, aging, and smoking. Am J Respir Crit Care Med 2006; 174: 530–537
- Araya J, Cambier S, Markovics J A, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus V C, Sheppard D, et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest 2007; 117: 3551–3562
- Wang C H, Huang C D, Lin H C, Lee K Y, Lin S M, Liu C Y, Huang K H, Ko Y S, Chung K F, Kuo H P. Increased circulating fibrocytes in asthma with chronic airflow obstruction. Am J Respir Crit Care Med 2008; 178: 583–591
- Ramirez A M, Shen Z, Ritzenthaler J D, Roman J. Myofibroblast transdifferentiation in obliterative bronchiolitis: tgf-beta signaling through smad3-dependent and -independent pathways. Am J Transplant 2006; 6: 2080–2088