Abstract
Decreased Clara cell secretory protein (CCSP) levels have been found in smokers and chronic obstructive pulmonary disease (COPD) patients, which may be related to the development of COPD. A phosphodiesterase-4 (PDE4) inhibitor, roflumilast, appears to have therapeutic value for COPD. However, its effect on CCSP in cigarette smoke (CS)-exposed lungs has not been investigated. AKR/J mice were treated as follows: air control, CS, roflumilast plus CS, and roflumilast. Mice underwent four weeks of air or CS exposure. Roflumilast was administrated at 5mg/kg via gavage once daily for the duration of the study. CCSP levels in bronchoalveolar lavage (BAL) fluid and ERK1/2 activation in lungs were examined. CS exposure tended to decrease CCSP levels in BAL fluid compared to air controls. Treatment with roflumilast significantly reversed CS-induced downward trend of CCSP in BAL fluid. Roflumilast significantly inhibited CS-induced upward trend of ERK1/2 activation in lungs, and the levels of activated ERK1/2 in lungs negatively correlated with CCSP levels of BAL fluid in CS, and CS plus roflumilast groups. Our results demonstrate that one of the therapeutic mechanisms of roflumilast is to reverse CS-induced downward trend in CCSP levels of BAL fluid, which may be mediated by down-regulating ERK1/2 activity.
INTRODUCTION
Phosphodiesterase type 4 (PDE4) inhibitors may be a treatment option for chronic obstructive pulmonary disease (COPD) due to its efficacy to improve pulmonary function and reduce exacerbations (Citation[1], Citation[2]). Animal studies also demonstrate that PDE4 inhibitors can attenuate lung inflammation and airspace enlargement induced by prolonged cigarette smoke exposure (Citation[3]). However, the mechanism by which PDE4 inhibitors ameliorate these pathologic processes in COPD patients is not clear. Since cigarette smoke (CS) is a main risk factor in the development of COPD, in the present study, a CS-exposed murine model was used to investigate the effects of a PDE4 inhibitor on CS-induced changes in lung epithelial biology, and to explore the underlying potential mechanisms of PDE4 inhibition.
Clara cell secretory protein (CCSP) is mainly secreted by Clara cells in the airway epithelium, and represents one of most abundant secretory proteins within the lung epithelial lining fluid. Increased inflammatory responses have been observed in CCSP-deficient mice after bacterial or viral infection (Citation[4], Citation[5]). Previous studies have also demonstrated that CCSP concentrations in sera and bronchoalveolar lavage (BAL) fluid in healthy smokers were significantly lower than those in healthy nonsmokers (Citation[6], Citation[7]). The expression of CCSP in the airway epithelium in healthy smokers and COPD patients was also significantly decreased as compared with that of non-smokers (Citation[7], Citation[8]). Together, these studies suggest that impaired CCSP production in human smokers may contribute to the disease process.
Prior studies have demonstrated that caffeine and theophylline exert PDE4 inhibitor activity, thus promoting the proliferation of lung pulmonary adenocarcinoma cell line with Clara cell characteristics (Citation[9], Citation[10]). Whether caffeine and theophylline can induce CCSP production was not addressed in this publication. The regulation of PDE4 inhibition on CCSP is still under investigation. A downstream gene of activated ERK1/2, c-Jun, was demonstrated to participate in hypoxia-induced suppression of CCSP (Citation[11]). CS exposure and PDE4 inhibition have been reported to increase and attenuate ERK1/2 activation, respectively (Citation[12], Citation[13], Citation[14]). Thus, it is possible that a specific PDE4 inhibition may increase CCSP through attenuating ERK1/2 activation, but this needs to be determined.
The purpose of the present study is to investigate whether a PDE4 inhibitor, roflumilast, can reverse CS-induced CCSP reduction, and whether ERK1/2 activation and airway epithelial alteration are related to CCSP levels in a murine model.
MATERIAL AND METHODS
Animals
Male AKR/J mice, aged 8 weeks (Jackson Laboratories, Bar Harbor, ME) were used. CS exposure protocol was approved by the Institutional Animal Care and Use Committee at the National Jewish Health.
CS exposure and roflumilast treatment
2R4F research cigarettes (Kentucky Tobacco Research & Development Center, University of Kentucky, Lexington, KY) were used to generate CS. The mice were treated as follows: air, CS alone, roflumilast (a PDE4 inhibitor, Altana Pharma, Konstanz, Germany) plus CS, and roflumilast alone. Animals were exposed to room air or CS for 4 weeks, 2.5 hour each day, 5 days a week. The TE-10c smoking machine (Teague Enterprises, Davis, CA) was maintained to keep the total suspended particulate (TSP) concentration between 85 and 120 mg/m3, carbon monoxide (CO) concentration between 190 and 300 ppm and nicotine concentration at 6 mg/m3. In the roflumilast plus CS, and roflumilast alone groups, roflumilast treatment began one week prior to CS or air exposure. The roflumilast was administered by oral gavage one hour prior to CS exposure once daily. The daily dose of roflumilast was 5 mg/kg of body weight. All mice were sacrificed 24 hours after the last air or CS exposure.
Bronchoalveolar lavage (BAL) and lung tissue processing
The mice were anesthetized and the trachea was exposed in situ. A small incision was made in the trachea, and a plastic catheter attached to a 1ml syringe was inserted. BAL was performed by instillation, and then withdrawal of 1 ml of saline. The BAL fluid was centrifuged and the cell free supernatant was collected for CCSP measurement. One lobe of the right lung was used for the detection of ERK1/2. The left lungs were fixed in 4% paraformaldehyde and embedded in paraffin. Lung sections at 4 μ m thickness were cut for H&E staining, immunohistochemistry and immunofluorescence.
Western blotting
Western blotting was used to assess CCSP levels and ERK1/2 activation. Since mouse CCSP ELISA kit is not available, Western blotting serves as a reliable quantitative method to detect CCSP. BAL fluid (for CCSP, 20 μ l/mouse) or lung homogenate supernatant (for ERK1/2, 100 μ g protein/mouse) was separated in SDS-polyacrylamide gel and transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat milk for 1 hour at room temperature, and then incubated with rabbit anti-CCSP antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or a mixture of mouse anti-phosphorylated ERK1/2 (p-ERK1/2, Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit anti-total ERK1/2 antibodies (t-ERK1/2, Cell Signaling Technology, Danvers, MA) overnight at 4°C. For CCSP detection, the blots were incubated with horseradish peroxidase-conjugated donkey anti-rabbit antibody (Amersham Biosciences, Buckinghamshire, UK) for 1 hour on a rocker at room temperature. Immunoreactive bands were visualized by incubating the membrane with ECL Western blotting detection reagents (Amersham Biosciences, Buckinghamshire, UK) for 3 min and by exposing the membranes to hyperfilms (Amersham Biosciences, Buckinghamshire, UK).
For ERK1/2, the membranes were incubated with a mixture of IRDye 680-conjugated goat anti-rabbit and IRDye 800CW-conjugated goat anti-mouse antibodies (1:10,000, LI-COR Biosciences, Lincoln, Nebraska) for 1 hour on a rocker at room temperature. Immunoreactive bands were visualized by using Odyssey infrared imager (LI-COR Biosciences, Lincoln, Nebraska). Immunoreactive bands were quantified by densitometry analysis using the NIH Scion image 1.63 software. ERK1/2 activation was assessed by the ratio of p-ERK1/2 to t-ERK1/2. The expression of CCSP and ERK1/2 in CS, roflumilast and combinational groups was normalized to that of air control group.
Immunohistochemistry of proliferating cell nuclear antigen (PCNA)
PCNA immunostaining was performed to assess the proliferation state of airway epithelial cells. The lung sections were deparaffined in xylene and rehydrated through graded ethanol series. Antigen retrieval was achieved by heat treatment in 10 mM citrate buffer (pH 6.0). Endogenous peroxidase was blocked with 0.3% H2O2. Lung sections were blocked for 1 hour at room temperature in normal horse serum and incubated with mouse anti-PCNA primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. Slides were washed with Tris-buffered saline (TBS), and then the biotinylated secondary antibody (Vectastain ABC kit, Vector Laboratories, Burlingame, CA) was applied and the sections were incubated at room temperature for 60 minutes. After washes with TBS, slides were incubated with avidin-biotin peroxidase complex (Vectastain ABC kit, Vector Laboratories, Burlingame, CA) at room temperature for 60 minutes. 3-Amino-9-ethylcarbazole (AEC, Sigma, St. Louis, MO) was applied, followed by rinsing the slides using distilled water to stop color development. The NIH Scion image 1.63 software was used to measure the perimeter of the airway epithelial basement membrane. Airway PCNA positive epithelial cells were quantified and presented as the number of positive cells per mm of basement membrane.
Measurement of airway epithelial thickness
The NIH Scion image 1.63 software was used to measure airway epithelial thickness in H&E-stained lung sections. The thickness was calculated by dividing the epithelial area by the perimeter of the corresponding epithelial basement membrane. All airways with maximal diameter/minimum diameter ≤2 (Citation[15]) in lung sections were chosen for the measurement of epithelial thickness.
Immunofluorescent staining of CCSP
Deparraffinization and antigen retrieval were performed as described in PCNA immunohistochemistry. Lung sections were blocked with normal goat serum at room temperature for 1 hour and then were incubated with rabbit anti-CCSP antibody (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. After washes, the lung sections were incubated with FITC-labeled goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) for 1 hour at room temperature, followed by incubation with 4′, 6-diamidino-2-phenylindole (DAPI) to counterstain the cell nuclei. The expression of CCSP in airway epithelial cells was observed under a fluorescent microscope. The percentage of CCSP positive airway epithelial cells in each group was quantified.
Statistical analysis
Normally distributed data are presented as means ± SEM and compared between groups using Student's t-test. Non-normally distributed data are expressed as medians with quartile ranges and comparison between the groups is performed using the Wilcoxon rank-sum test. The Bivariate fit test is used for correlation analysis. p < 0.05 is considered statistically significant.
RESULTS
Roflumilast reverses CS-induced downward trend of CCSP in BAL fluid
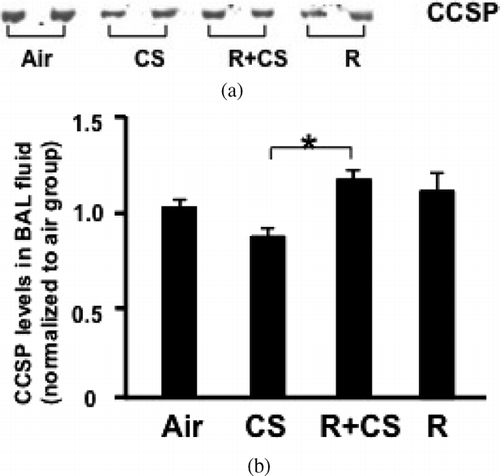
A trend for decreased level of CCSP in BAL fluid was found in CS-exposed mice compared to air control group (p = 0.12). The level of CCSP in BAL fluid of mice treated with roflumilast plus CS was significantly higher compared with CS-exposed mice (p < 0.05). However, the CCSP level was similar between roflumilast alone group and air control group (p > 0.05) (). The lavage total white blood cell counts and differential were similar between the groups ().
Table 1 Bronchoalveolar lavage cell counts
Figure 1 Roflumilast reverses CS-induced downward trend of CCSP in BAL fluid. A: Representative image of CCSP Western blotting. Air: air control; CS: cigarette smoke alone; R+CS: roflumilast plus CS; R: roflumilast alone. B: Densitometry analysis of CCSP. Data are expressed as mean ± SEM normalized to the average CCSP protein level of air control group. n = 12–14 per group. *: p < 0.05 versus CS alone.
Roflumilast suppresses CS-induced upward trend of ERK1/2 activation in lungs
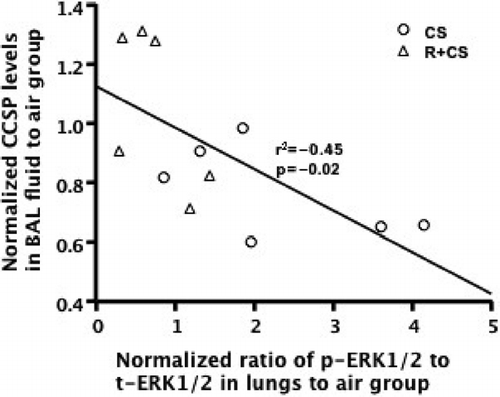
CS exposure tended to increase the ratio of p-ERK1/2 to t-ERK1/2 in lungs compared to air control group (p = 0.06). The phosphorylation of ERK1/2 in lungs was significantly attenuated in roflumilast plus CS group compared to CS alone group (p < 0.05) ( and ). The phosphorylation of ERK1/2 in lungs from the roflumilast alone group was significantly reduced compared to air, CS alone, and roflumilast plus CS-treated mice (p < 0.05). A negative correlation between the phosphorylation of EKR1/2 in lungs and CCSP levels in BAL fluid was observed in CS alone and roflumilast plus CS groups (r2 = −0.45, p = 0.02, ). No correlation (r2 = 0.11, p = 0.11) was found when all groups were analyzed.
Figure 2 Roflumilast suppresses CS-induced upward trend of ERK1/2 activation in lungs. A: Representative image of ERK1/2 Western blotting. Air: air control; CS: cigarette smoke alone; R + CS: roflumilast plus CS; R: roflumilast alone. B: ERK1/2 activation was assessed by the ratio of p-ERK1/2 to t-ERK1/2. Densitometry analysis of ERK1/2 activation is presented in the chart diagram. Data are expressed as median (25%–75% range) normalized to the median ERK1/2 activation of air control group. n = 5–6 per group. *:p < 0.05 versus CS, #: p < 0.05 versus Air, CS, and R + CS.
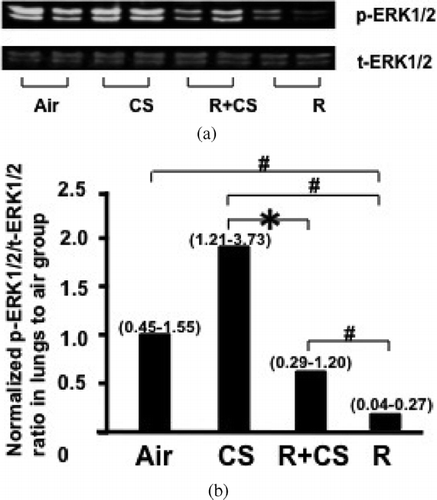
Figure 3 The activation level of ERK1/2 in lung tissues negatively correlated with CCSP levels of BAL fluid in CS alone, and roflumilast plus CS groups. The phosphorylation levels of ERK1/2 in lungs were plotted against CCSP levels in BAL fluid. The activation of ERK1/2 in lungs negatively correlated with CCSP levels of BAL fluid. (r2 = 0.45, p = 0.02).
CS and roflumilast increase PCNA expression in airway epithelium
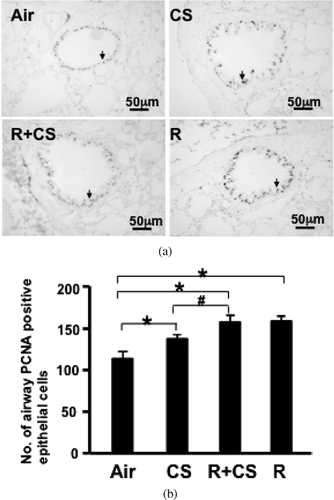
PCNA positive cells were significantly increased in CS alone, roflumilast plus CS, and roflumilast alone group compared to the air control group (p < 0.05). The number of PCNA positive cells was significantly greater in the roflumilast plus CS group than the CS alone group (p < 0.05). No significant difference of PCNA expression in airway epithelium was seen between the roflumilast plus CS group and the roflumilast alone group (p > 0.05) (, ).
Figure 4 CS and roflumilast increase PCNA expression in airway epithelium. PCNA expression in airway epithelial cells was detected by immunohistochemistry in paraffin-embedded lung sections. (A) Representative lung photomicrographs of PCNA immunostaining. Air: air control; CS: cigarette smoke alone; R+CS: roflumilast plus CS; R: roflumilast alone. Arrows indicate positive cells. Internal scale bar = 50 μ m. (B) PCNA expression was assessed as number of positive epithelial cells per mm airway basement membrane. Values are presented as mean± SEM. *: p < 0.05 versus Air. #: p < 0.05 versus CS.
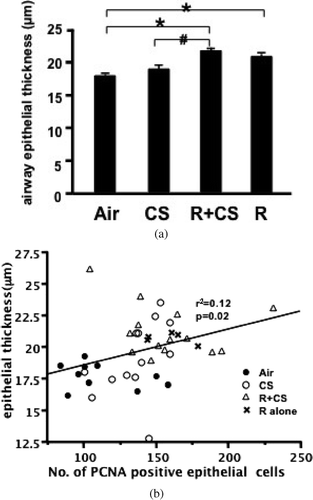
Roflumilast increases thickness of airway epithelium
The airway epithelial thickness was significantly increased in the roflumilast plus CS group and roflumilast alone group compared with the air control group (p < 0.05). The thickness of airway epithelium in mice treated with roflumilast plus CS was significantly greater than that in CS alone-treated mice (p < 0.05). CS exposure did not increase airway epithelial thickness compared with air controls (p > 0.05). No significant difference in thickness of airway epithelium was observed between the roflumilast plus CS and the roflumilast alone group (p > 0.05) (). There was a positive correlation between airway epithelial thickness and airway PCNA positive cell number (r2 = 0.12, p = 0.02) ().
Figure 5 Roflumilast increases thickness of airway epithelium. (A) The airway epithelial thickness was calculated by dividing the epithelial area by the perimeter of corresponding basement membrane. Air: air control; CS: cigarette smoke alone; R+CS: roflumilast plus CS; R: roflumilast alone. Values are presented as mean ± SEM. *: p < 0.05 versus Air. #: p < 0.05 versus CS. (B) Airway epithelial PCNA level was plotted against the epithelial thickness. The airway PCNA expression positively correlated with the epithelial thickness (r2 = 0.12, p = 0.02).
The percentage of CCSP positive airway epithelial cells is not increased in any group
The purpose of immunofluorescent staining of CCSP is to investigate whether increased airway epithelial PCNA levels in roflumilast plus CS, and roflumilast alone groups were accompanied by an up-regulation of airway CCSP positive epithelial cells. Cell quantification results showed that more than 90% of airway epithelial cells stained with CCSP in mouse lungs regardless of treatment. The percentage of CCSP positive airway epithelial cells is 92.87 ± 0.88, 94.64 ± 0.77, 93.57 ± 0.87 and 93.48 ± 0.50 in air, CS, roflumilast plus CS and roflumilast group, respectively. No difference in the percentage of CCSP positive airway epithelial cells was found between groups (p > 0.05).
DISCUSSION
In this study, we have found that roflumilast can reverse CS-induced downward trend of CCSP, which may be mediated by the down-regulation of ERK1/2 activity since there is an inverse correlation between CCSP protein levels in BAL fluid and ERK1/2 activation in lung tissues. To our knowledge, this is the first report evaluating the effect of a PDE4 inhibitor on CCSP.
Our results demonstrated that 1 month of CS exposure tended to decrease CCSP levels in BAL fluid. Roflumilast administration significantly prevented the downward trend of BAL fluid CCSP levels following CS exposure. However, roflumilast alone did not cause an increase in CCSP levels in mouse BAL fluid compared to air control group, suggesting that roflumilast only up-regulated CCSP levels after CS exposure.
To explain why roflumilast increased CCSP levels in BAL fluid only in CS-exposed mice, the EKR1/2 activation in lungs was investigated. We observed a trend of increased phosphorylated ERK1/2 in lungs after CS exposure, which was significantly attenuated by roflumilast. There was an inverse correlation between ERK1/2 activation in lungs and CCSP levels in BAL fluid in all CS-exposed mice, including CS alone and roflumilast plus CS groups. These results suggest that roflumilast may regulate CCSP levels by inhibiting the activation of ERK1/2. c-Jun, a target protein of activated ERK1/2, may be involved in the down-regulation of CCSP expression (Citation[11]). c-Jun is a member of activator protein-1 (AP-1) family. The 5′-flanking region of the CCSP promoter has been reported to contain an AP-1 consensus sequence (Citation[16]).
One published paper found that c-Jun was at least, in part, responsible for the repression of CCSP levels induced by hyperoxia (Citation[11]). This study observed that hyperoxia increased the binding of c-Jun at the AP-1 site of the CCSP promoter by electrophoretic mobility shift assay (EMSA) and Supershift analyses, and demonstrated a dose-dependent decrease in the transcriptional activity of CCSP promoter after c-Jun expression plasmids transfection. Further studies are needed in order to investigate the possible involvement of c-Jun for roflumilast-induced CCSP secretion in airway epithelial cells. In addition, we found that roflumilast alone caused a marked decrease in ERK1/2 activation of lungs compared with any other group. Even though a very low ERK1/2 activation was observed in roflumilast alone group, the level of CCSP in roflumilast alone group wasn't significantly higher than air control group. Moreover, we did not find a correlation between CCSP levels and ERK1/2 activation when all four groups were analyzed.
This suggests that the decrease in ERK1/2 activation caused by roflumilast may initiate a different signal pathway in the presence and absence of CS. Roflumilast is a cAMP-elevating agent, and many studies have reported that biological effects and downstream signals of cAMP could vary depending on extracellular environment, exposure time and cell type (Citation[17], Citation[18], Citation[19]). In our animal model, an interaction between CS and roflumilast may account for the different effect of roflumilast on CCSP in roflumilast plus CS group from roflumilast alone group. Detailed signal pathways initiated by roflumilast with and without CS exposure remain to be investigated.
We also found that CS induced a significant increase in airway epithelial proliferation, as detected by PCNA immunostaining. Terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) staining showed no increase in the number of apoptotic airway epithelial cells in CS-exposed mice compared to air control group (data not shown). These results demonstrate that CS-induced downward trend of CCSP in mouse BAL fluid may not result from a decrease in the number of airway epithelial cells. The therapeutic effects of roflumilast are thought to be mediated via increased levels of intracellular cAMP (Citation[20]). cAMP-mediated signaling has been reported to be involved in retinoic acid, β-carotene, dexamethasone, caffeine or theophylline-induced proliferation in normal or transformed small airway epithelial cells (Citation[9], Citation[10], Citation[21], Citation[22], Citation[23]).
We have also found increased airway epithelial thickness and airway PCNA immunostaining in roflumilast plus CS, and roflumilast alone groups compared with air control group. In addition, airway epithelial thickness and PCNA level were higher in roflumilast plus CS group than those in CS alone group. However, immunofluorescent staining of CCSP showed that there was no significant difference in the percentage of CCSP positive airway epithelial cells between groups. Additionally, there was an increase in airway epithelial PCNA expression in roflumilast alone group compared with air control group, but no corresponding up-regulation in CCSP levels in BAL fluid from roflumilast alone group was observed. Moreover, we did not find any correlation between CCSP and PCNA levels. Therefore, the increase in airway epithelial cell proliferation induced by roflumilast may be not enough to induce a detectable up-regulation in the number of CCSP positive epithelial cells and cannot explain the increase of CCSP induced by roflumilast in CS-exposed mice. An increase in the secretion of CCSP in airway epithelial cells, not the number of airway CCSP positive epithelial cells, may account for the up-regulation of CCSP by roflumilast in smoked mice.
Although CS exposure alone, roflumilast alone, or combination of both were shown to increase airway epithelial cell proliferation in the current study, the role of proliferating epithelial cells remains to be determined. We speculate that the enhanced epithelial cell proliferation indicates physiologic responses to substances (e.g., roflumilast and CS) causing an increase in cAMP levels (Citation[17], Citation[18], Citation[19], Citation[24]), as we did not observe any squamous metaplasia or any premalignant lesions in any lungs of our 4-week mouse model. Nevertheless, the long-term effects of roflumilast on airway epithelial phenotypic changes in CS-exposed lungs should be monitored in future studies.
CCSP is believed to function as a modulator of inflammation in the lungs. Previous studies using CCSP-deficient mice demonstrated a role of CCSP in limiting lung inflammation secondary to viral/bacterial infection, oxidative stress or allergen challenge (Citation[4], Citation[5], Citation[25], Citation[26], Citation[27]). Some studies found that CCSP acted as an inhibitor of phospholipase A2 (PLA2) and phospholipase C (PLC), which provides additional mechanisms of an anti-inflammatory role for CCSP (Citation[28], Citation[29], Citation[30]).
In our 1-month CS-exposure model, lung inflammation was not increased (data not shown), but the decrease in CCSP levels induced by CS may cause severe inflammatory response once animals are subject to viral or bacterial types of infections. This can be supported by one of our previously published papers, which has demonstrated that significant higher neutrophil staining and metalloproteinase-9 mRNA expression in lungs from smoking plus mycoplasma-treated mice compared with mycoplasma alone or CS alone-exposed mice (Citation[31]). Thus, our results suggest that the increase in CCSP concentrations in BAL fluid induced by roflumilast after CS exposure may be one of the mechanisms for its use in COPD patients.
In summary, our findings demonstrate that roflumilast can increase CCSP levels in a CS-exposed murine model. This process is potentially mediated by the inhibition of ERK1/2 activation. Additionally, we found that roflumilast induced an up-regulation in the number of airway PCNA positive epithelial cells regardless of CS exposure, but this did not cause an increase in the percentage of CCSP positive epithelial cells. So the regulation of CCSP secretion in airway epithelial cells, probably via ERK1/2, may be a potential mechanism underlying the therapeutic effect of roflumilast on CCSP levels in CS-exposed setting.
ACKNOWLEDGEMENTS
The authors greatly appreciate the excellent technical assistance of Qun Wu, Taylor Moss, Jyoti Thaikoottathil, Spencer Lafasto, and Nicole Michels, and for manuscript preparation Mandy Comeau.
This study was financially supported by ALTANA Pharma AG (now part of Nycomed Group), Konstanz, Germany. Additionally, R.J. Martin serves on advisory boards for AstraZeneca, Schering, and Teva.
REFERENCES
- Rabe K F, Bateman E D, O'Donnell D, Witte S, Bredenbroker D, Bethke T D. Roflumilast- an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomized controlled trial. Lancet 2005; 366: 563–571
- Grootendorst D C, Gauw S A, Verhoosel R M, Sterk P J, Hospers J J, Bredenbröker D, Bethke T D, Hiemstra P S, Rabe K F. The PDE4 inhibitor roflumilast reduces sputum neutrophil and eosinophil numbers in patients with COPD. Thorax 2007; 62: 1081–1087
- Martorana P A, Beume R, Lucattelli M, Wollin L, Lungarella G. Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. Am J Respir Crit Care Med 2005; 172: 848–853
- Hayashida S, Harrod K S, Whitsett J A. Regulation and function of CCSP during pulmonary Pseudomonas aeruginosa infection in vivo. Am J Physiol Lung Cell Mol Physiol 2000; 279: L452–L459
- Harrod K S, Mounday A D, Stripp B R, Whitsett J A. Clara cell secretory protein decreases lung inflammation after acute virus infection. Am J Physiol 1998; 275: L924–L930
- Shijubo N, Honda Y, Itoh Y, Yamaguchi T, Kuroki Y, Akino T, Kawai T, Abe S. BAL surfactant protein A and Clara cell 10-kDa protein levels in healthy subjects. Lung 1998; 176: 257–265
- Shijubo N, Itoh Y, Yamaguchi T, Shibuya Y, Morita Y, Hirasawa M, Okutani R, Kawai T, Abe S. Serum and BAL Clara cell 10 kDa protein (CC10) levels and CC10-positive bronchiolar cells are decreased in smokers. Eur Respir J 1997; 10: 1108–1114
- Pilette C, Godding V, Kiss R, Delos M, Verbeken E, Decaestecker C, De Paepe K, Vaerman J P, Decramer M, Sibille Y. Reduced epithelial expression of secretory component in small airways correlates with airflow obstruction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 163: 185–194
- Al-Wadei H A, Takahashi T, Schuller H M. Caffeine stimulates the proliferation of human lung adenocarcinoma cells and small airway epithelial cells via activation of PKA, CREB and ERK1/2. Oncol Rep 2006; 15: 431–435
- Hussein A, Al-Wadei N, Takahashi T, Schuller H M. Theophylline stimulates cAMP-mediated signaling associated with growth regulation in human cells from pulmonary adenocarcinoma and small airway epithelia. Int J Oncol 2005; 27: 155–160
- Ramsay P L, Luo Z, Major A, Park M S, Finegold M, Welty S E, Kwak I, Darlington G, Demayo F J. Multiple mechanisms for oxygen-induced regulation of the Clara cell secretory protein gene. FASEB J 2003; 17: 2142–2144
- Luppi F, Aarbiou J, van Wetering S, Rahman I, de Boer W I, Rabe K F, Hiemstra P S. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respir Res 2005; 6: 140
- Zhong C Y, Zhou Y M, Douglas G C, Witschi H, Pinkerton K E. MAPK/AP-1 signal pathway in tobacco smoke-induced cell proliferation and squamous metaplasia in the lungs of rats. Carcinogenesis 2005; 26: 2187–2195
- Mata M, Sarria B, Buenestado A, Cortijo J, Cerda M, Morcillo E J. Phosphodiesterase 4 inhibition decreases MUC5AC expression induced by epidermal growth factor in human airway epithelial cells. Thorax 2005; 60: 144–152
- Chu H W, Rino J G, Wexler R B, Campbell K, Harbeck R J, Martin R J. Mycoplasma pneumoniae infection increases airway collagen deposition in a murine model of allergic airway inflammation. Am J Physiol Lung Cell Mol Physiol 2005; 289: L125–L133
- Sawaya P L, Stripp B R, Whitsett J A, Luse D S. The lung-specific CC10 gene is regulated by transcription factors from the AP-1, octamer, and hepatocyte nuclear factor 3 families. Mol Cell Biol 1993; 13: 3860–3871
- Stevens B. Cross-talk between growth factor and purinergic signalling regulates Schwann cell proliferation. Novartis Found Symp 2006; 276: 162–175
- Hu A, Nino G, Grunstein J S, Fatma S, Grunstein M M. Prolonged heterologous beta2-adrenoceptor desensitization promotes proasthmatic airway smooth muscle function via PKA/ERK1/2-mediated phosphodiesterase-4 induction. Am J Physiol Lung Cell Mol Physiol 2008, Epub
- Al-Wadei H A, Schuller H M. Cyclic adenosine monophosphate-dependent cell type-specific modulation of mitogenic signaling by retinoids in normal and neoplastic lung cells. Cancer Detect Prev 2006; 30: 403–411
- Huang Z, Mancini J A. Phosphodiesterase 4 inhibitors for the treatment of asthma and COPD. Curr Med Chem 2006; 13: 3253–3262
- AI-Wadei H A, Schuller H M. Cyclic adenosine monophosphate-dependent cell type-specific modulation of mitogenic signaling by retinoids in normal and neoplastic lung cells. Cancer Detect Prev 2006; 30: 403–411
- Al-Wadei H A, Takahashi T, Schuller H M. Growth stimulation of human pulmonary adenocarcinoma cells and small airway epithelial cells by beta-carotene via activation of cAMP, PKA, CREB and ERK1/2. Int J Cancer 2006; 118: 1370–1380
- Al-Wadei H A, Takahasi T, Schuller H M. PKA-dependent growth stimulation of cells derived from human pulmonary adenocarcinoma and small airway epithelium by dexamethasone. Eur J Cancer 2005; 41: 2745–2753
- Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds P N. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2007; 37: 748–755
- Mango G W, Johnston C J, Reynolds S D, Finkelstein J N, Plopper C G, Stripp B R. Clara cell secretory protein deficiency increases oxidant stress response in conducting airways. Am J Physiol 1998; 275: L348–L356
- Johnston C J, Mango G W, Finkelstein J N, Stripp B R. Altered pulmonary response to hyperoxia in Clara cell secretory protein deficient mice. Am J Respir Cell Mol Biol 1997; 17: 147–155
- Hung C H, Chen L C, Zhang Z, Chowdhury B, Lee W L, Plunkett B, Chen C H, Myers A C, Huang S K. Regulation of TH2 responses by the pulmonary Clara cell secretory 10-kd protein. J Allergy Clin Immunol 2004; 114: 664–670
- Facchiano A, Cordella-Miele E, Miele L, Mukherjee A B. Inhibition of pancreatic phospholipase A2 activity by uteroglobin and antiflammin peptides: possible mechanism of action. Life Sci 1991; 48: 453–464
- Lesur O, Bernard A, Arsalane K, Lauwerys R, Bégin R, Cantin A, Lane D. Clara cell protein (CC-16) induces a phospholipase A2-mediated inhibition of fibroblast migration in vitro. Am J Respir Crit Care Med 1995; 152: 290–297
- Okutani R, Itoh Y, Yamada T, Yamaguchi T, Singh G, Yagisawa H, Kawai T. Preparation and characterization of human recombinant protein 1/Clara cell M(r) 10,000 protein. Eur J Clin Chem Clin Biochem 1996; 34: 691–696
- Martin R J, Wexler R B, Day B J, Harbeck R J, Pinkerton K E, Chu H W. Interaction between cigarette smoke and mycoplasma infection: a murine model. COPD 2006; 3: 3–8