
ABSTRACT
The purpose of this study was to determine whether expression of connective tissue growth factor (CTGF) protein in chronic obstructive pulmonary disease (COPD) is consistent in humans and animal models of COPD and to investigate the role of this protein in lung epithelial cells. CTGF in lung epithelial cells of ex-smokers with COPD was compared with ex-smokers without COPD by immunofluorescence. A total of twenty C57Bl/6 mice and sixteen non-human primates (NHPs) were exposed to cigarette smoke (CS) for 4 weeks. Ten mice of these CS-exposed mice and eight of the CS-exposed NHPs were infected with H3N2 influenza A virus (IAV), while the remaining ten mice and eight NHPs were mock-infected with vehicle as control. Both mRNA and protein expression of CTGF in lung epithelial cells of mice and NHPs were determined. The effects of CTGF overexpression on cell proliferation, p16 protein, and senescence-associated β-galactosidase (SA-β-gal) activity were examined in cultured human bronchial epithelial cells (HBECs). In humans, CTGF expression increased with increasing COPD severity. We found that protein expression of CTGF was upregulated in lung epithelial cells in both mice and NHPs exposed to CS and infected with IAV compared to those exposed to CS only. When overexpressed in HBECs, CTGF accelerated cellular senescence accompanied by p16 accumulation. Both CTGF and p16 protein expression in lung epithelia are positively associated with the severity of COPD in ex-smokers. These findings show that CTGF is consistently expressed in epithelial cells of COPD lungs. By accelerating lung epithelial senescence, CTGF may block regeneration relative to epithelial cell loss and lead to emphysema.
Introduction
Cigarette smoking is a major risk factor for chronic obstructive pulmonary disease (COPD), a disease characterized by an irreversible airflow obstruction Citation(1). Ning et al. Citation(2) conducted a comprehensive analysis of gene expression in the lungs (i.e., both airway and alveolar epithelial cells) of healthy smokers versus smokers with moderate COPD and identified multiple differentially expressed candidate genes. One of those candidate genes, connective tissue growth factor (CTGF), was confirmed to be increased at both the transcript and protein expression levels in both airway and alveolar epithelial cells of COPD subjects, but not in healthy smokers. CTGF has multiple cell biological functions, including cell proliferation, cell adhesion, and wound repair Citation(3). However, little is known about the triggers that increase CTGF in lung epithelial cells and its biological relevance.
Smoking makes susceptibility to common viral infections (e.g., Influenza A Virus—IAV). Both the prevalence of and mortality from IAV infection are significantly increased among smokers Citation(4–7). In a mouse model, two hits consisting of short-term exposure to cigarette smoke (CS) and a single IAV infection cause emphysema, a pathological phenotype of COPD, within 1 month Citation(8,9). However, effects of the two hits of CS exposure and IAV infection on CTGF expression in the lung have not been reported.
Cynomolgus macaques (Macaca fascicularis) are a species of Old World monkeys that have been widely used as a non-human primates (NHP) model for biomedical research Citation(10). NHPs closely mimic the physiological and biological changes in response to human pathogens, likely due to the high degree of genetic homology to humans Citation(11). We have previously shown that exposure of NHPs to CS for 12 weeks causes extensive chronic bronchitis, but not emphysematous changes Citation(12). In the present study, we wanted to determine the effects of the two-hit challenge model (CS + IAV) on the development of emphysema in NHPs and examine CTGF expression in lung epithelial cells. We further tested expression of CTGF in mice exposed to a similar two-hit challenge model.
Markers of aging, another major risk factor for COPD, are strongly associated with accumulation of senescent cells that are metabolically active but permanently unable to divide Citation(13,14). Senescence of alveolar epithelial cells is observed in smokers with COPD as compared with smokers without COPD Citation(15), suggesting a potential role of cellular senescence in the pathogenesis of COPD. Cellular senescence can occur by telomere shortening through cell division (referred to as replicative senescence) Citation(16) or by various noxious stimuli (referred to as stress-induced premature senescence) Citation(17–19). The two canonical senescence-inducing pathways emanate from either the p53 protein or the p16-retinoblastoma (Rb) pathway Citation(20,21). These two canonical pathways can be activated in response to CS exposure Citation(19), thereby inducing senescence through the suppressing activity of cyclin-dependent kinases (CDKs), such as CDK4 and CDK6 Citation(20,22). As long-term cigarette smoking causes alveolar cell apoptosis and impaired tissue repair, compensatory proliferation of alveolar type 2 cells would be needed to close the wound gap Citation(15). However, because cellular senescence limits the proliferative capacity of alveolar cells, the dying cells cannot be replaced, which ultimately may lead to the development of emphysema Citation(23,24).
The present study evaluated expression of CTGF in the lungs of NHPs and mice following combined CS exposure and IAV infection to determine a potential role of CTGF as a biomarker for COPD. We also tested the biological effects of CTGF in lung epithelial cells. We propose that CTGF overexpression may be a key signaling protein that contributes to pulmonary epithelial cell senescence.
Materials and methods
Human lung tissue samples
The use of human subject samples was approved by the New Mexico VA Healthcare System institutional review board (#11–056). Formalin-fixed lung slide sections obtained from ex-smokers without COPD (n = 6) or with moderate (n = 3), or severe/very severe COPD (n = 8) were provided by the Lung Tissue Research Consortium.
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee and were performed at Lovelace Respiratory Research Institute (LRRI), a facility approved by the Association for the Assessment and Accreditation for Laboratory Animal Care International. Sixteen female cynomolgus macaques (2–5 year-old, their body weights ranging from 2.2 to 3.8 kg) from a colony maintained at LRRI were utilized for this study. C57Bl/6 mice, 8–10 weeks of age, were purchased from the Jackson Laboratory.
Cigarette smoke exposure and instillation of influenza virus (H3N2) and bronchial brushing for NHPs
NHPs were exposed to smoke aerosol generated from type 3R4F research cigarettes (Kentucky Tobacco Research and Development Center) at concentrations of 100 mg/m3 total particulate matter (TPM) for the first week and 200 mg/m3 TPM for subsequent 3 weeks. All sixteen NHPs were exposed for 6 hours/day, 5 days/week in Hazelton 1000 whole body exposure chambers for 4 weeks. In order to obtain bronchial brushing samples, animals received ketamine (10 mg/kg, IM) followed by general anesthesia with inhaled isoflurane to permit passage of the BF-XP40 (2.8 mm) bronchoscope. A 2 mm × 6 mm sterile cytology brush (Olympus model BC-203D-2006) was introduced through the bronchoscope to obtain the bronchial brushings from four sites in airway generations 3–5. After 4 weeks of CS exposure, eight NHPs received a single instillation of 106 plaque forming units (pfu) of IAV, strain HKX31 (H3N2), or vehicle (phosphate-buffered saline—PBS) into the right diaphragmatic/caudal lobe. Bronchial brushing samples were collected at 2 weeks after IAV or mock infection. All NHPs were euthanized 2 weeks after IAV or mock infection.
Cigarette smoke exposure and instillation of influenza virus (H3N2) for mice
A total of thirty mice were exposed to CS (n = 20) or filtered air (FA) (n = 10) for 6 hours/day, 5 days/week in Hazelton 1000 whole body exposure chambers at CS concentrations of 100 mg/m3 TPM for the first week followed by 250 mg/m3 for subsequent 3 weeks as previously described Citation(25). After 4 weeks of CS exposure, mice were randomized to receive a nasal instillation of 5 × 103 pfu of IAV (H3N2) or vehicle (PBS) as mock infection (n = 10 per group).
Lung histology
The lungs of animals were fixed under a constant pressure (25 cm H2O) in neutral buffered formalin, embedded in paraffin, and sectioned at 5-μm thickness as we previously described Citation(26).
RNA isolation and quantitative RT-PCR
RNA was isolated from bronchial brushing samples using Trizol RNA extraction buffer (Molecular Research Center, INC, Cincinnati, OH) as previously described Citation(27). Quantitative RT-RCR analysis for CTGF and CDKN1B mRNA was performed using TaqMan One-Step Reverse transcription polymerase chain reaction (RT-PCR) Master Mix Reagents (Applied Biosystems, Carlsbad, CA) as previously described Citation(28). The following sets of probes CTGF (Cat#:Hs01026927_g1) and CDKN1B (Hs01597588_ml) were purchased from Applied Biosystems (Foster City, CA). RT-PCR reactions were performed using real-time ABI PRISM 7900HT PCR system.
Immunofluorescent staining and image analysis
Lung tissue sections from humans and NHPs were deparaffinized, hydrated, and washed in 0.05% Brij-35/PBS (pH 7.4). The CTGF antigens were retrieved using citrate buffer (pH 6.0) and probed by overnight incubation with anti-CTGF antibody (Santa Cruz Technologies, CA) or anti-p16 antibody. The immunolabeled cells were detected using secondary antibodies conjugated either to DyLightTM-549 or DyLightTM-649 (Jackson Immunoresearch, West Grove, PA), and sections were mounted with 4 ,6-diamidino-2-phenylindole (DAPI) containing Fluoromount-G (Southern Biotech, Birmingham, AL) for nuclear staining. Micrographs were captured using a Zeiss LSM 510 Meta confocal microscope (Carl Zeiss MicroImaging, Inc, Thornwood, NY) mounted on an Axiovert 100 scope (Carl Zeiss Microimaging Inc, Thornwood, NY) and analyzed using NIH ImageJ (http://imagej.nih.gov/ij/) software.
CTGF overexpression in cell lines
UNCN3T cells (a Bmi-1/hTERT HBEC cell line, a gift from Dr. Scott Randell) were originally generated by Fulcher et al. Citation(29) and maintained as previously described. UNCN3T cells were transduced with a lentiviral vector (cat#EX-A0312-LV152, pReceiver, GeneCopoeia, Rockville, MD) to overexpress CTGF protein as previously described Citation(30). The transduced cells were selected with 5 μg/ml hygromycin for 14 days, and surviving cells were collected. Experiments to monitor cell growth were performed in twelve-well Costar tissue culture plates at a starting cell density of 15 × 103/cm2. The cell counts were performed at 3 and 6 days by an electric particle counter (Beckman Coulter, Indianapolis, IN).
Immunoblot analysis
CTGF overexpressing or the control cells were cultured in p100 plates (100 mm) at a starting cell density of 15 × 103/cm2 and harvested after 3 days. Cell lysates were prepared in Radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors (Boehringer Mannheim, Ridgefield, CT) and analyzed by immunoblotting as previously described Citation(30). Protein levels were evaluated using anti-CTGF, anti-p53 antibody (Santa Cruz, CA), or anti-p16 and anti-p21 antibodies (Abcam, Cambridge, MA), and equal loading of protein samples from each group was evaluated using anti-β actin antibody (Sigma-Aldrich) after using the Restore WB stripping buffer (Thermo Fisher Scientific, Barrington, IL).
Senescence-associated β-galactosidase (SA β-gal) activity
SA β-Gal staining was performed according to a previously described method Citation(28). Briefly, after washing with PBS, cell samples in 6-well cell culture plates were fixed in PBS containing 2% formaldehyde, 0.2% glutaraldehyde for 15 minutes at room temperature. Fixed cells were washed with PBS and incubated with staining solution mix (40 mM citric acid/sodium phosphate (pH 6.0), 150 mM NaCl, 2 mM MgCl2, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 1 mg/ml 5-bromo-4-chloro-3-indolyl-βD-galactopyranoside) for overnight at 37°C. SA β-Gal activity is presented as the percentage of SA β-Gal-positive cells per number of total cells in randomly selected six fields per well, at a magnification of 20×. Quantification was from three independent experiments.
Analysis of conditioned medium
Culture medium from CTGF-overexpressing and control cells was collected at 48 hours of culture and kept in −80°C until use. For immunoblot analysis, 2 ml of collected medium was evaporated using a SpeedVac Concentrator (Savant, Farmingdale, NY), resuspended in 80 μl of RIPA lysis buffer (Sigma-Aldrich, St. Louis, MO) and analyzed by immunoblotting for CTGF as previously described Citation(18).
Cell proliferation assay
To determine the effect of secreted CTGF, UNCN3T cells were plated at a starting density of 15 × 103/cm2 and maintained either in medium collected from CTGF-overexpressing or from empty vector–infected controls. Medium was changed at 3 days, and cell proliferation was determined at 3 and 6 days by the 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay as previously described Citation(18).
Statistical analysis
We used Student unpaired t tests for the comparison of two groups (e.g., CTGF-overexpressing cells and control cells). For multiple comparisons, we used a one-way analysis of variance with Bonferroni correction with post hoc comparisons of specific pairwise differences. Data were expressed as mean ± SEM, and p < 0.05 was considered statistically significant.
Results
CTGF expression is increased in lung epithelial cells of ex-smokers with increasing COPD severity
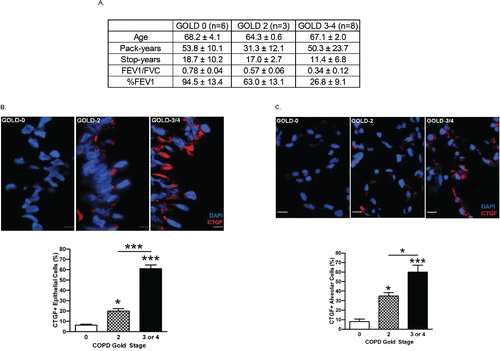
While CTGF has been reported as one of the potential biomarkers for COPD among smokers Citation(2), whether smoking cessation affects expression of this protein in COPD patients was not investigated Citation(2,31). To avoid confounding effects from recent CS exposure, we selected study subjects representing the different stages of COPD severity and who had stopped smoking for >5 years (). Lung tissues from ex-smokers with COPD [The Global Initiative for Chronic Obstructive Lung Disease (GOLD) stage 2 (n = 3) and stage 3 or 4 (n = 8)] were analyzed and compared with ex-smokers without COPD (n = 6). The IF staining data reveal that CTGF expression in both airway () and alveolar () epithelial cells of ex-smokers was increased with increasing severity of COPD. These data suggest that CTGF expression in lung epithelial cells is positively associated with the severity of airway obstruction among ex-smokers and may be a biomarker for COPD.
Figure 1. CTGF expression is increased in lung epithelial cells of ex-smokers with increasing COPD severity. (A) The demographic and clinical data for ex-smokers based on the clinical stage of COPD (GOLD 0: n = 6, GOLD 2: n = 3, GOLD 3–4: n = 8) analyzed in this study (**p < 0.01). GOLD: The Global Initiative for Chronic Obstructive Lung Disease; FEV1: forced expiratory volume in 1 second; FVC: forced vital capacity % FEV1: % predicted value of FEV1. (B) Analysis of CTGF expression in airway epithelium of ex-smokers with COPD. Representative micrographs showing CTGF-immunopositive cells (red) in airway epithelial cells from ex-smokers with COPD at GOLD stage 0, 2 and 3–4. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μm). Quantitative analysis of CTGF-positive airway cells in the three clinical groups are also shown (*p < 0.05; ***p < 0.001). (C) Analysis of CTGF expression in alveolar cells of ex-smokers with COPD. Representative micrographs showing CTGF-immunopositive cells (red) in alveolar cells from ex-smokers with COPD as mentioned above. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μm). Quantitative analysis of CTGF-positive airway cells in the three clinical groups are also shown (*p < 0.05; ***p < 0.001).
Influenza virus infection induces CTGF expression in lung epithelial cells of non-human primates exposed to cigarette smoke
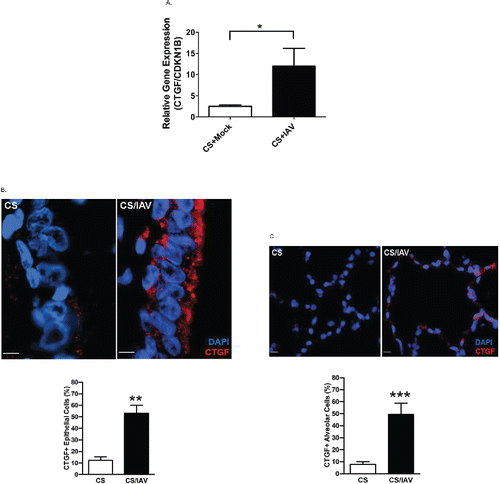
Smoking habits increase the risk for IAV infection and contribute to the higher mortality than that of non-smokers Citation(4–7). Exposure of NHPs to CS alone causes extensive bronchitis throughout the respiratory tract Citation(12) but does not cause emphysema. Because viral infection after 4 weeks of CS causes emphysema in mice Citation(8,9), we investigated whether the same approach causes emphysema in a more relevant NHP. Therefore, we investigated lung tissues from NHPs exposed to a two-hit (CS + IAV) model. A total of 16 NHPs were exposed to CS for 4 weeks, and 8 NHPs each were then either infected with IAV or vehicle. Two weeks post infection, animals were euthanized, and tissues were harvested for analysis. We did not observe a significant enlargement of alveolar diameter in the two-hit-exposed NHPs compared with NHPs exposed to CS only (data not shown). However, qRT-PCR analysis from bronchial brushing samples showed that CTGF mRNA levels were increased in the two-hit exposed NHPs compared with those of CS-exposed NHPs (). In addition, increased CTGF protein levels were detected by IF in airway () and alveolar epithelia () from NHPs exposed to CS and IAV compared with NHPs exposed to CS only. These data suggest that the changes in lung epithelial cells of NHPs exposed to the two-hit (CS and IAV infection) resemble some features that are observed in humans with COPD.
Figure 2. Influenza virus infection induces CTGF expression in lung epithelial cells of non-human primates exposed to cigarette smoke. (A) CTGF mRNA levels in epithelial cells obtained by bronchial brushings of IAV- or mock-infected NHPs following 4 weeks of CS exposure were analyzed by quantitative RT-PCR. Data are shown as mean ± SEM (n = 3 per group; *p < 0.05). (B) Analysis of CTGF expression in airway epithelium of NHPs exposed to CS and IAV infection. Representative micrographs showing CTGF-immunopositive cells (red) in airway tissues from NHPs exposed to CS and IAV infection compared with CS and mock infection. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μM). Lower panel shows quantitative analysis of CTGF-positive cells in the two groups of NHPs (**p < 0.01). (C) Analysis of CTGF expression in alveolar cells of NHPs exposed to CS and IAV infection. Representative micrographs showing CTGF-immunopositive cells (red) in alveolar cells from NHPs exposed to CS + IAV or CS + mock infection. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μM). Lower panel shows quantitative analysis of CTGF-positive cells in the two groups of NHPs (***p < 0.001).
Influenza virus infection induces CTGF expression in lung epithelial cells of mice exposed to cigarette smoke
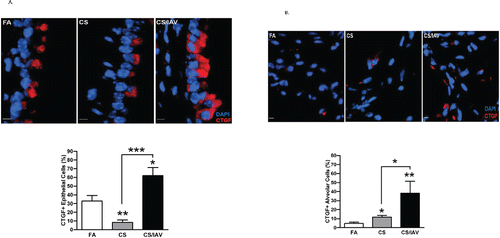
The two-hit (CS + IAV) enhances emphysematous changes in a mouse model Citation(8,9). To validate that the viral infection following the CS exposure augments CTGF expression, 10 mice were exposed to FA, 20 mice to CS for 4 weeks, 10 of the 20 mice were infected with IAV, and the other 10 were mock-infected. CTGF expression was significantly augmented in lung epithelial cells of mice exposed to CS and infected with IAV compared with CS + mock-infected mice ( and ), again resembling the findings in humans and NHPs. Interestingly, compared with filtered air (FA)-exposed mice, CS + mock-infected mice exhibited a significantly reduced CTGF expression in airway epithelial cells () but significantly increased expression in alveolar epithelial cells ().
Figure 3. Influenza virus infection induces CTGF expression in lung epithelial cells of mice exposed to cigarette smoke. (A) Analysis of CTGF expression in airway epithelium of mice exposed to CS and IAV infection. Representative micrographs showing CTGF-immunopositive cells (red) in airway tissues from mice exposed to CS and IAV infection compared with CS and mock infection. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μM). Lower panel shows quantitative analysis of CTGF-positive cells in the two groups of mice (***p < 0.001). (B) Analysis of CTGF expression in alveolar cells of mice exposed to CS and IAV infection. Representative micrographs showing CTGF-immunopositive cells (red) in alveolar cells from mice exposed to CS + IAV or CS + mock infection. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μM). Left panel shows quantitative analysis of CTGF-positive cells in the two groups of mice (*p < 0.05).
CTGF overexpression induces cellular senescence in human airway epithelial cells
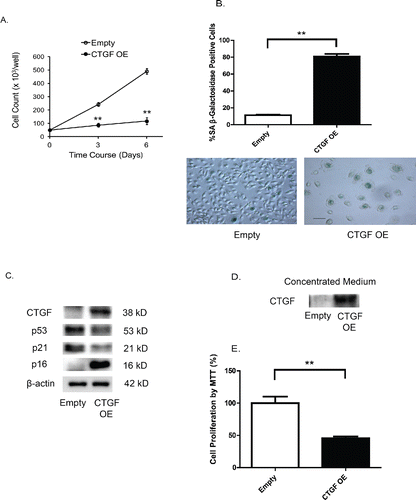
To examine the cellular role of CTGF upregulation, we established a stable cell line expressing high levels of CTGF by transducing HBECs with a lentiviral CTGF expression vector. CTGF overexpression markedly reduced cell growth (), and this arrest in growth was associated with the cells, showing an enlarged morphology and an increase in (SA)-β-gal activity, a marker of senescence (). Furthermore, CTGF-overexpressing cells also displayed highly increased levels of p16, another marker of senescence. While these two markers of cellular senescence were increased, expression of both p53 and p21 was downregulated (). Since CTGF is a protein that is immediately secreted by cells, we next determined whether the medium obtained from CTGF-overexpressing cells reduces cell growth of HBECs. We confirmed CTGF accumulation () in the medium from CTGF-overexpressing cells by immunoblotting, and that cells exposed to this conditioned medium have reduced cell growth (). These findings suggest that secreted CTGF is the main driving factor for cell senescence.
Figure 4. Transgenic overexpression of CTGF induces cellular senescence in human airway epithelial cells. (A) Effect of CTGF overexpression on cell viability. Primary HBECs were transduced with a lentiviral vector (pReceiver) encoding either CTGF cDNA or an empty vector, and transduced cells were selected using hygromycin (5 μg/ml). The viable cell counts monitored at 0, 3 and 6 days post-transduction showed a significant (**p < 0.01) attenuation of cell growth in cells with CTGF overexpression (CTGF-OE). (B) CTGF overexpression induces cellular senescence as measured by SA-β-galactosidase activity. The percentage of SA β-gal positive cells/total cell number was measured for the transduced cells at 6 days. Data are expressed as mean ± SEM for three independent experiments (**p < 0.01). Representative photomicrographs of cells transduced with either empty or CTGF-OE vector and stained for β-gal activity (blue) are shown (scale bar, 10 μm). (C) CTGF-OE induces p16 protein levels. HBECs treated as in A were lysed for immunoblot analysis of p53, p21, and p16 proteins after 3 days of culture. Immunoblotting data are representative of three experiments. (D) HBECs were treated as in (A). Complete medium obtained from the transduced cells were concentrated using a SpeedVac. Immunoblot analysis of CTGF was performed. (E) HBECs were cultured for 3 days in conditioned medium obtained from CTGF-overexpressing cells or control cells. Cell proliferation was determined by MTT assay. Data are expressed as the mean ± SEM for two independent experiments with triplicate samples (**p < 0.01).
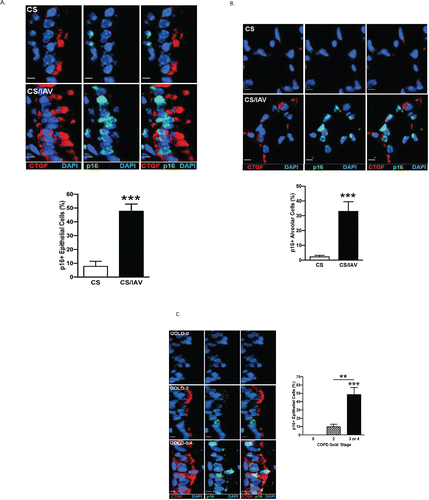
CTGF expression is positively associated with p16 accumulation in lung epithelial cells in vivo
To determine the levels of p16 protein expression in CTGF-positive cells in vivo, we co-immunostained lung tissues of mice exposed to CS + IAV for CTGF and p16 and found that nuclear p16-postivity was accompanied with CTGF expression in both airway and alveolar cells ( and ). Similarly, p16 positivity was co-localized with CTGF expression in lung tissues of ex-smokers with COPD, and the number of p16-positive cells increased with the severity of disease ( and ), suggesting that induced CTGF expression in vivo is also associated with p16 accumulation and may drive cellular senescence of lung epithelial cells.
Figure 5. CTGF expression is positively associated with p16 accumulation in lung epithelial cells in vivo. (A) Analysis of CTGF and p16 co-expression in airway epithelium of mice exposed to CS and IAV infection. Representative micrographs showing CTGF- (red) and p16 (green)-positive cells in airway tissues from mice exposed to CS and IAV infection compared with CS and mock infection. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μM). Lower panel shows quantitative analysis of p16-positive cells (***p < 0.001). (B) Analysis of CTGF and p16 co-expression in alveolar cells of mice exposed to CS and IAV infection. Representative micrographs showing CTGF- (red) and p16 (green)-positive cells in alveolar tissues from mice exposed to CS and IAV infection compared with CS and mock infection. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μM). Lower panel shows quantitative analysis of p16-positive cells (***p < 0.001). (C) Analysis of CTGF and p16 co-expression in airway epithelium of ex-smokers with COPD. Representative micrographs showing CTGF- (red) and p16 (green)-positive cells in airway epithelium of ex-smokers with COPD at GOLD stage 0, 2 and 3 or 4. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μm). Right panel shows quantitative analysis of p16-positive cells from the three clinical groups (**p < 0.01; ***p < 0.001). (D) Analysis of CTGF and p16 co-expression in alveolar cells of ex-smokers with COPD. Representative micrographs showing CTGF- (red) and p16 (green)-positive cells in alveolar cells of ex-smokers with COPD at GOLD stage 0, 2 and 3 or 4. Nuclei were counterstained with DAPI (blue) (scale bar, 10 μm). Right panel shows quantitative analysis of p16-positive cells from the three clinical groups (**p < 0.01; ***p < 0.001).
Discussion
The present study demonstrates using human, NHP, and mouse models of COPD that CTGF is expressed in COPD lungs. We show that expression of CTGF in lung epithelial cells positively correlates with the prevalence of epithelial senescence and is associated with the severity of airway obstruction among patients with smoking-induced COPD. In addition, we provide evidence that CTGF is involved in inducing cellular senescence. We propose that CTGF may play a role in the development of emphysema by blocking the renewal potential of lung epithelial cells.
Aging systemically causes progressive decline of vital organs, including the lung, accompanied by accumulation of senescent cells and depletion of stem cells Citation(32). Senescence of lung epithelial cells is strongly associated with COPD Citation(14,15). There are several known modulators of the aging process including oxidative stress and telomere shortening. For example, genetic deletion of Nrf2, a master regulator for antioxidant enzymes Citation(33), or short length of telomere Citation(34) augments the susceptibility to smoking-induced emphysema in vivo, suggesting a potential role of cellular senescence in the development of COPD. However, the causative role of cellular senescence in COPD still remains to be elucidated.
In this study, we found that protein levels of CTGF are increased in lung epithelial cells of ex-smokers with increasing severity of COPD. In a previous study, a comprehensive analysis of gene expression revealed multiple genes, including CTGF, that are differentially expressed in the lung between smokers without COPD and smokers with moderate COPD Citation(2). Ning et al. Citation(2) also confirmed that both gene and protein expression of CTGF were elevated in lung epithelial cells of smokers with moderate to severe COPD compared with those of smokers without COPD. However, whether clinical COPD severity and smoking status affect CTGF expression has not been addressed. While our results support the potential utility of CTGF as a biomarker of COPD severity regardless of smoking status, additional studies using larger cohorts are required to define CTGF as a bona fide biomarker.
Several studies showed that oxidative stress (e.g., hydrogen peroxide exposure or mechanical stress) is sufficient to increase CTGF both in vitro and in vivo Citation(35,36). In the present study, we identified that a combined two-hit (CS + IAV) significantly increased CTGF at the protein level in lung epithelial cells of mice and NHPs. These results suggest that viral infections in cigarette smoking patients may limit epithelial cell replication through senescent mechanisms.
Possibly due to differences in pulmonary anatomy between mice and humans, smoking-induced airway and lymphoid pathologies are dissimilar between humans and mice, with mice showing little to no pathology Citation(37,38). To overcome these limitations of the mouse COPD model, we previously developed a smoking-induced COPD model in NHPs Citation(12). Exposure to CS for up to 12 weeks did not show significant emphysematous change but caused airway inflammation associated with extensive mucous cell hyperplasia and metaplasia, bronchial lymphoid aggregates, and alveolar septal cell apoptosis Citation(12). How CTGF overexpression contributes to these early lung pathologies and the subsequent development of emphysematous changes is unclear. Evidence for CTGF being involved in the development of emphysema comes from transgenic mouse that express CTGF in in alveolar type 2 epithelial cells driven by surfactant protein C promoter. These mice exhibited spontaneous enlargement of alveolar diameter associated with increased lung inflammation and decreased vascular development during development Citation(39).
To elucidate the biological relevance of CTGF overexpression, we utilized an in vitro model of lentiviral vector-mediated CTGF overexpression. We found that ectopically expressed CTGF consistently induces cellular senescence accompanied by increased p16 expression. These findings are consistent with those of a previous study, showing that CTGF overexpression induced cellular senescence in cultured immortalized foreskin fibroblasts by activating both p21 and p16 pathways Citation(40). However, we did not observe p21 induction in senescent HBECs overexpressing CTGF. The discrepancy in p21 expression could stem from the cell type-dependent differences (fibroblasts versus epithelial cells) or differences in the stage of senescence (e.g., pre-senescent versus senescent phase). We believe that based on the reduced cell growth, typical morphological changes and increased senescence-associated β-galactosidase activity and overexpression of p16, CTGF overexpression induced cellular senescence. In addition, cells showed decreased expression of p53, a cell death-inducing protein. Therefore, we do not believe that CTGF transfection caused toxicity and cell death.
It is known that either CS or IAV infection causes both oxidative stress and DNA damage Citation(41–43), which may contribute to the development of cellular senescence in lung epithelial cells. In addition, both oxidative stress and DNA damage are associated with increased expression of CTGF Citation(36,44). The pathway by which CTGF induces p16 expression is not known, but we speculate that CTGF either by affecting cell cycle regulatory proteins or together with CS and IAV-induced oxidative stress and DNA damage may facilitate activation of cell cycle arrest through the p16 pathway rather than the p53 pathway. More detailed investigations to determine the mechanism of inducing p16 expression and cellular senescence are beyond the scope of the present study.
Collectively, these findings suggest a potentially critical role for CTGF in the development of COPD by causing accelerated senescence of epithelial cells. While we failed to observe any significant emphysematous changes in NHPs exposed to CS and IAV, the finding that CTGF was highly increased in our two-hit model supports the idea that the short (4 weeks) CS exposure combined with IAV infection may have set the stage for the development of emphysema. We believe that longer exposure to CS and/or viral infection with a higher titer may result in discernible emphysematous changes in the NHP model. Based on our findings that CTGF overexpression causes senescence in epithelial cells, we speculate that lack of compensatory proliferation of alveolar type 2 epithelial cells will ultimately contribute to alveolar wall destruction and emphysema following CS-induced alveolar septal cell apoptosis. The mouse model of two-hit exposure clearly supports our hypothesis that there were significant emphysematous changes in mice following CS exposure and viral infection that resulted in increased CTGF and p16 expression in lung epithelial cells. When analyzed for the senescence marker, p16 levels, only fraction of the CTGF-positive cells showed p16-positivity. This could be due to CTGF being an upstream effector that activates p16 and induces cellular senescence as we observed in our in vitro studies with CTGF-overexpressing cells. It is possible that expression of p16 may occur over a series of time course, and more time points need to be taken to observe increased number of cells expressing both CTGF and p16. It is also possible that in vivo CTGF may require another factor to induce p16 expression. Nonetheless, the in vitro studies suggest that the two-hit CS/IAV-induced CTGF may contribute to the development of cell senescence in vivo.
Although airway and alveolar epithelial cells were carefully differentiated by experienced pulmonary pathologist based on the morphology and adjunct structures, such as interstitium, double staining with cell-specific markers can further strengthen these findings in future studies.
CS exposure activates diverse age-associated molecular pathways in circulating lymphocytes and induces cellular senescence of lung fibroblasts, and pulmonary epithelial and endothelial cells in smokers with compared with smokers without COPD Citation(13,14,45,46). Cellular senescence also alters the secretory phenotype (e.g., growth factors and inflammatory mediators) that contributes to chronic inflammation in COPD Citation(46,47). A previous in vitro study demonstrated that CTGF is upregulated in cultured senescent fibroblasts, suggesting that CTGF is also a member of the senescence-associated secretory phenotype in dermal fibroblasts Citation(48). By contrast, we also found that CTGF is one of the driving factors for cell senescence in cultured human lung epithelial cells. These results suggest that CTGF can be either a biomarker or an inducer for cell senescence.
In conclusion, we observed increased CTGF levels in lung epithelial cells in humans diagnosed with COPD 5 years after smoking cessation and in both NHPs and mice exposed to CS and infected with IAV. Expression of this protein in HBECs caused senescence, providing a potential mechanism by which CTGF upregulation leads to CS-induced lung pathology. We propose that CTGF is not only a severity-dependent biomarker but also a potential therapeutic target for COPD.
Declaration of interest
All authors state that there are no conflicts of interest.
Funding
This manuscript was supported, in part, by the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development by a Merit Review award CX001048 and CTSC003-2 (to TN). This work was also supported by HL068111 and ES015482 (to YT), by LRRI Institutional Funds (to YT and HSC); National Institute of Health (AI117560) and American Lung Association Biomedical Research Grant (RG306208) (to HSC); Flight Attendant Medical Research Institute (to RM).
References
- Han MK. Update in chronic obstructive pulmonary disease in 2010. Am J Respir Crit Care Med 2011; 183(10):1311–1315.
- Ning W, Li CJ, Kaminski N, Feghali-Bostwick CA, Alber SM, Di YP, et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A 2004; 101(41):14895–14900.
- Leask A, Parapuram SK, Shi-Wen X, Abraham DJ. Connective tissue growth factor (CTGF, CCN2) gene regulation: a potent clinical bio-marker of fibroproliferative disease? J Cell Commun Signal 2009; 3(2):89–94.
- Kark JD, Lebiush M, Rannon L. Cigarette smoking as a risk factor for epidemic a(h1n1) influenza in young men. N Engl J Med 1982; 307(17):1042–1046.
- Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med 2004; 164(20):2206–2216.
- Rogot E, Murray JL. Smoking and causes of death among U.S. veterans: 16 years of observation. Public Health Rep 1980; 95(3):213–222.
- Wong CM, Yang L, Chan KP, Chan WM, Song L, Lai HK, et al. Cigarette smoking as a risk factor for influenza-associated mortality: evidence from an elderly cohort. Influenza Other Respir Viruses 2012.
- Kang MJ, Lee CG, Lee JY, Dela Cruz CS, Chen ZJ, Enelow R, et al. Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J Clin Invest 2008; 118(8):2771–2784.
- Mebratu YA, Smith KR, Agga GE, Tesfaigzi Y. Inflammation and emphysema in cigarette smoke-exposed mice when instilled with poly (I:C) or infected with influenza A or respiratory syncytial viruses. Respir Res 2016; 17(1):75.
- Carlsson HE, Schapiro SJ, Farah I, Hau J. Use of primates in research: a global overview. Am J Primatol 2004; 63(4):225–237.
- Messaoudi I, Estep R, Robinson B, Wong SW. Nonhuman primate models of human immunology. Antioxid Redox Signal 2011; 14(2):261–273.
- Polverino F, Doyle-Eisele M, McDonald J, Wilder JA, Royer C, Laucho-Contreras M, et al. A novel nonhuman primate model of cigarette smoke-induced airway disease. Am J Pathol 2015; 185(3):741–755.
- Holz O, Zuhlke I, Jaksztat E, Muller KC, Welker L, Nakashima M, et al. Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. Eur Respir J 2004; 24(4):575–579.
- Aoshiba K, Zhou F, Tsuji T, Nagai A. DNAdamage asamolecular link in the pathogensis ofCOPDin smokers. Eur Respir J 2012; 39(6):1368–1376.
- Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in pulmonary 510 emphysema patients. Am J Respir Crit Care Med 2006; 174(8):886–893.
- Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol 2001; 13(6):748–753.
- Chen JH, Stoeber K, Kingsbury S, Ozanne SE, Williams GH, Hales CN. Loss of proliferative capacity and induction of senescence in oxidatively stressed human fibroblasts. J Biol Chem 2004; 279(47):49439–49446.
- Klimova TA, Bell EL, Shroff EH, Weinberg FD, Snyder CM, Dimri GP, et al. Hyperoxia-induced premature senescence requires p53 and pRb, but not mitochondrial matrix ROS. Faseb J 2009; 23(3):783–794.
- Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, Hunninghake GW. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol 2006; 35(6):681–688.
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120(4):513–522.
- Satyanarayana A, Rudolph KL. p16 and ARF: activation of teenage proteins in old age. J Clin Invest 2004; 114(9):1237–1240.
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell 2002; 2(2):103–112.
- Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 2005; 11(5):491–498.
- Aoshiba K, Nagai A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2009; 6(7):596–601.
- Awji EG, Seagrave JC, Tesfaigzi Y. Correlation of cigarette smoke-induced pulmonary inflammation and emphysema in C3H and C57Bl/6 mice. Toxicol Sci 2015; 147(1):75–83.
- Nyunoya T, March TH, Tesfaigzi Y, Seagrave J. Antioxidant diet protects against emphysema, but increases mortality in cigarette smoke-exposed mice. COPD 2011; 8(5):362–368.
- Schwalm K, Stevens JF, Jiang Z, Schuyler MR, Schrader R, Randell SH, et al. Expression of the proapoptotic protein Bax is reduced in bronchial mucous cells of asthmatic subjects. Am J Physiol Lung Cell Mol Physiol 2008; 294(6):L1102–L1109.
- Jang JH, Bruse S, Huneidi S, Schrader RM, Monick MM, Lin Y, et al. Acrolein-exposed normal human lung fibroblasts: cellular senescence, enhanced telomere erosion, and degradation of Werner's syndrome protein. Environ Health Perspect 2014; 122(9):955.
- Fulcher ML, Gabriel SE, Olsen JC, Tatreau JR, Gentzsch M, Livanos E, et al. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am J Physiol Lung Cell Mol Physiol 2009; 296(1):L82–L91.
- Jang JH, Bruse S, Liu Y, Duffy V, Zhang C, Oyamada N, et al. Aldehyde dehydrogenase 3A1 protects airway epithelial cells from cigarette smoke-induced DNA damage and cytotoxicity. Free Radic Biol Med 2014; 68:80–86.
- Rangasamy T, Misra V, Zhen L, Tankersley CG, Tuder RM, Biswal S. Cigarette smoke-induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol Lung Cell Mol Physiol 2009; 296(6):L888–L900.
- Mercado N, Ito K, Barnes PJ. Accelerated ageing of the lung in COPD: new concepts. Thorax 2015; 70(5):482–489.
- Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 2004; 114(9):1248–1259.
- Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI, et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med 2011; 184(8):904–912.
- Matsuda S, Gomi F, Katayama T, Koyama Y, Tohyama M, Tano Y. Induction of connective tissue growth factor in retinal pigment epithelium cells by oxidative stress. Jpn J Ophthalmol 2006; 50(3):229–234.
- Branchetti E, Poggio P, Sainger R, Shang E, Grau JB, Jackson BM, et al. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc Res 2013; 100(2):316–324.
- Cosio MG, Guerassimov A. Chronic obstructive pulmonary disease. Inflammation of small airways and lung parenchyma. Am J Respir Crit Care Med 1999; 160(5 Pt 2):S21–S25.
- Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol 2008; 294(4):L612–L631.
- Chen S, Rong M, Platteau A, Hehre D, Smith H, Ruiz P, et al. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: implication in the pathogenesis of severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 2011; 300(3):L330–L340.
- Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 2012; 11(12):2272–2284.
- Nyunoya T, Mebratu Y, Contreras A, Delgado M, Chand HS, Tesfaigzi Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am J Respir Cell Mol Biol 2014; 50(3):471–482.
- Schwarz KB. Oxidative stress during viral infection: a review. Free Radic Biol Med 1996; 21(5):641–649.
- Vijaya Lakshmi AN, Ramana MV, Vijayashree B, Ahuja YR, Sharma G. Detection of influenza virus induced DNA damage by comet assay. Mutat Res 1999; 442(1):53–58.
- Kushwaha S, Vikram A, Jena GB. Protective effects of enalapril in streptozotocin-induced diabetic rat: studies of DNA damage, apoptosis and expression of CCN2 in the heart, kidney and liver. J Appl Toxicol 2012; 32(9):662–672.
- Rutten EP, Gopal P, Wouters EF, Franssen FM, Hageman GJ, Vanfleteren LE, et al. Various mechanistic pathways representing the aging process are altered in COPD. Chest 2016; 149(1):53–61.
- Amsellem V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, Validire P, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 184(12):1358–1366.
- Adnot S, Amsellem V, Boyer L, Marcos E, Saker M, Houssaini A, et al. Telomere dysfunction and cell senescence in chronic lung diseases: therapeutic potential. Pharmacol Ther 2015; 153:125–134.
- Kim KH, Park GT, Lim YB, Rue SW, Jung JC, Sonn JK, et al. Expression of connective tissue growth factor, a biomarker in senescence of human diploid fibroblasts, is up-regulated by a transforming growth factor-beta-mediated signaling pathway. Biochem Biophys Res Commun 2004; 318(4):819–825.