
ABSTRACT
Chronic obstructive pulmonary disease (COPD) is the fourth cause of death in the world and it is currently presenting a major global public health challenge, causing premature death from pathophysiological complications and rising economic and social burdens. COPD develops from a combination of factors following exposure to pollutants and cigarette smoke, presenting a combination of both emphysema and chronic obstructive bronchitis, which causes lung airflow limitations that are not fully reversible by bronchodilators. Oxidative stress plays a key role in the maintenance and amplification of inflammation in tissue injury, and also induces DNA damages. Once the DNA molecule is damaged, enzymatic mechanisms act in order to repair the DNA molecule. These mechanisms are specific to repair of oxidative damages, such as nitrogen base modifications, or larger DNA damages, such as double-strand breaks. In addition, there is an enzymatic mechanism for the control of telomere length. All these mechanisms contribute to cell viability and homeostasis. Thus, therapies based on modulation of DNA repair and genomic stability could be effective in improving repair and recovery of lung tissue in patients with COPD.
Introduction
According to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) Citation(1), chronic obstructive pulmonary disease (COPD) is the fourth cause of death in the world and it is currently a major challenge to public health. COPD causes chronic morbidity and mortality, giving rise to premature death from pathophysiological complications, rising economic and social burdens Citation(2). There is expectation that deaths will increase in the next decades due to continued exposure to COPD risk factors and the aging population.
Chronic obstructive pulmonary disease is estimated to affect about 7 million people only in Brazil Citation(3). About 12% of patients are diagnosed and only 18% of those receive some treatment, including palliatives. COPD is the fifth cause of death and hospitalization of about 290,000 patients annually, and generates excessive costs for the health care system Citation(3). It is estimated that 24 million people in the United States were affected by COPD in 2013 Citation(4).
The factors that trigger COPD have individual characteristics, such as epigenetic alterations, polymorphism or mutation in specific genes, genetic susceptibility, and increase of oxidative stress, which contributes to the inflammatory response Citation(5).
When an inflammatory response initiates the development of COPD, there is an increasing generation of free radicals, establishing oxidative stress, which ultimately damages DNA and other molecules in lung cells Citation(6). Indeed, in addition to direct and indirect DNA damages, free radicals could act as second messengers in specific cell signaling pathways, altering gene expression Citation(7).
Despite a number of different DNA damages caused by free radical attacks, genomic integrity is achieved by a set of enzymatic DNA repair mechanisms, which recognize and repair damages in the DNA molecule Citation(8,9). The main DNA repair mechanisms, to fix DNA damages caused by free radicals, are the base excision repair and nucleotide excision repair.
Chronic obstructive pulmonary disease: Pathophysiology
Chronic obstructive pulmonary disease affects 10–14% of world population, and in 2005 three million people who had COPD died. Estimates show that the total number of deaths in the next 10 years will increase above 30% Citation(1,10,11). Until 2030, COPD will be the fourth cause of death worldwide Citation(11). The increase in mortality will be due to the expanding epidemic of smoking, reduced mortality from other common causes of death (e.g. ischemic heart disease and infectious diseases), and aging of the world population Citation(1)
According to GOLD Citation(1), the morbidity and mortality vary across countries. Tobacco smoking is strongly related to developed countries whereas on developing countries the COPD would begin from the air pollution mainly.
Chronic obstructive pulmonary disease is a common, preventable, and treatable disease Citation(1). Although treatment is available, various host factors could influence the pathogenesis, such as immunologic defects; genetic susceptibility; infections and environmental factors, the latter being assigned as a major risk factor; and smoking, due to a number of harmful chemical compounds present in tobacco Citation(12).
Chronic obstructive pulmonary disease is characterized by persistent airflow limitation that is usually progressive and associated with increased chronic inflammation in airways and lungs in response to harmful particulate matter and gases Citation(1). Also, COPD presents inflammation; fibrosis and deposition of mucus in airways, causing chronic bronchitis Citation(10,13); and alveolar destruction in pulmonary parenchyma, resulting in the loss of elastic recoil capacity which characterizes emphysema Citation(10). Destruction of parenchyma is related to loss of lung tissue elasticity, which occurs as a result of the destruction of supporting structures that feed the alveoli, with the small airways tending to collapse during exhalation, thus reducing airflow, increasing air retention in lungs, and causing reduction of pulmonary capacity Citation(14).
Emphysema is an important component of COPD and it is defined as the presence of permanent increase in distal air space to terminal bronchioles followed by destruction of their walls, without clear fibrosis signals Citation(15).
The inflammatory responses by activating the innate and adaptive immune system have been lightened by chronic exposure to the initiating agent of COPD Citation(16). For example, cessation of smoking cigarettes increases the inflammatory process Citation(17).
The innate response will begin with the change in the barrier between the lung tissue and airspace, due to exposure to risk factors such as smoking Citation(14), recruiting neutrophils, macrophages, T-lymphocytes and natural killer cells to alveolous Citation(14) (). The adaptive immune response is started when particles undergo phagocytosis and presented as antigens by dendritic cells to T- and B-lymphocytes Citation(18). Several cytokines and chemokines, such as interleukins (IL-1β, IL, 6 e IL-8), tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), C-reactive protein (CRP), and matrix-metalloproteinases (MMP-6, MMP-9 and MMP-12), are secreted by inflammatory and epithelial cells, thereby perpetuating inflammation Citation(19,20). These inflammatory mediators, besides perpetuating inflammation, give rise to tissue injury, as well as a range of systemic effects Citation(20). The inflammatory response directly causes airway remodeling and narrowing of lungs in COPD patients. Airway space narrowing is caused by peribronchial fibrosis, build-up of scar tissue from damage of airway space, and over-multiplication of epithelial cells, lining the airway space. Also, mucociliary dysfunction by increase in the number of mucosal glands due to inflammation contributes to excess mucus production in airway space, which blocks the air flow Citation(21).
Figure 1. Schematic representation for the development of COPD until the increase of free radicals and their consequences to DNA molecule. COPD, chronic obstructive pulmonary disease; TNF-α, tumor necrosis alpha factor; INF-γ, interferon gamma; CRP, C-reactive protein; MMPs, matrix-metalloproteinases.
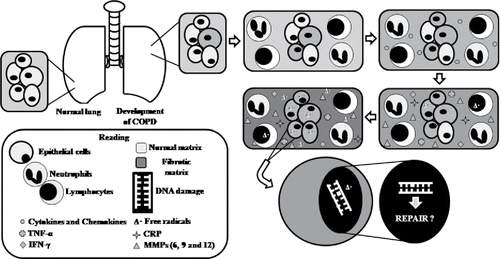
In addition to all these changes, there is imbalance between proteinases and antiproteinases, leading to tissue changes in COPD Citation(22). The increase in the number of neutrophils and macrophages releasing enzymes, such as cathepsin, elastase, proteinase 3, and matrix metalloproteinases 9 and 12, induces proteolysis of extracellular matrix components, resulting in the disruption of alveoli, causing emphysema Citation(23). It was shown that increase in MMP-9 results in increased production of its inhibitor TIMP-1- to repair the damaged tissue, but the increase in inhibitor could contribute to increased deposition of extracellular matrix, and consequently tissue fibrosis Citation(24).
Not only can the imbalance between proteinases and antiproteinases cause tissue destruction, but the oxidative stress could also cause redox imbalances (increase in destructive free radicals levels, as reactive oxygen species—ROS), reducing antioxidant protection pathways, thus perpetuating the inflammatory process Citation(25). Oxidative stress plays a key role in tissue injury and maintenance, and amplification of inflammation. Although the lung epithelium is an important source of antioxidants, individuals with emphysema present imbalance between antioxidant and oxidant agents, with predominance of oxidizing agents, and this imbalance could cause damages to the DNA molecule, in proteins and lipids, as it has been suggested Citation(26). Some clinical studies are listed in correlating COPD with DNA damage or DNA alteration.
Table 1. Summary of clinical studies on the relationship between COPD and DNA damage.
Oxidative stress in inflammation: DNA damage
Oxidative stress acts on a variety of inflammatory mediators, and relationship between oxidative stress and inflammation is in the overproduction of ROS, which represents an important component in the development of several chronic diseases Citation(27). Biomarkers of oxidative stress (e.g., hydrogen peroxide, 8-isoprostane) are increased in exhaled breath condensate, sputum, and in systemic circulation of COPD patients Citation(1).
Inflammation as a natural defense mechanism acts against a variety of harmful factors, as infections, exposure to allergenic, chronic diseases, and smoking. COPD and other chronic diseases are related to increased production of free radicals, which could cause oxidation of proteins, lipids, and nucleic acids, and these are recognized as an inflammatory signal Citation(28,29). These suggest that inflammation worsens clinical aspects of some diseases due to increased oxidative stress, by releasing macrophage-oxidized proteins, such as a peroxiredoxin 2 (PRDX2), which trigger TNF-α production in the extracellular environment, indicating that these mechanisms depend on redox in oxidative cascades and could induce inflammation Citation(28).
Exacerbated acute inflammation also worsens prognosis of patients with COPD, being associated with increased morbidity and mortality Citation(30). The exacerbation is associated with systemic inflammation and physiological changes caused mostly by viral and bacterial infections Citation(31). These exacerbations include breathlessness, cough, increased mucus production, and extreme fatigue, for example, resulting in decline of lung function Citation(32). Also, the infectious conditions cause lung injury and development of sepsis, which manifest as acute respiratory distress syndrome (ARDS), increase in free radicals in lung cells Citation(6,33). COPD exacerbations are considered as aggravations of underlying chronic inflammation in the airways. However, data on inflammatory responses, oxidative stress, and consequently their association with DNA damage in lungs in case of COPD exacerbation are scarce Citation(34). Studies have shown that there is increase in activated neutrophils in airways of COPD patients indicating that they are attracted by cytokines, such as IL-8 and TNF; increased cytokine levels during exacerbations in COPD patients has been reported Citation(35,36), and the frequency of exacerbations has been associated with the severity of COPD Citation(37).
Cell damage caused by oxidative stress alters mitochondrial DNA, protein structure Citation(38), as well cell membranes, such as the plasma lipid peroxidation, in the endoplasmic reticulum and Golgi apparatus Citation(39).
Free radicals are highly reactive atoms or molecules, which contain unpaired electrons in their last electronic layer, and they are produced as byproducts during normal metabolism and exposure to sunlight, ultraviolet light, ionizing radiation and toxic chemicals, such as cigarette smoke Citation(40). Among these, there are the reactive oxygen species (ROS), which are generated in the cells by both oxidizing and reducing agents Citation(41). Hydroxyl radical (OH˙), superoxide (O2˙), hydrogen peroxide (H2O2) ozone (O3), and singlet oxygen (1O2) are examples of ROS Citation(41). Besides ROS, reactive nitrogen species (RNS) are produced in vivo from arginine by nitric oxide synthase (NOS) and play important roles in vascular relaxation, biological defense, neurotransmission modulation, and immune system Citation(41). Nitric oxide (NO) and peroxynitrite (ONOO−) are examples of RNS Citation(41).
Free radicals can act as second messengers in specific cell signaling pathways, in immune function, and signal transductions, thereby affecting cellular homeostasis Citation(42). Some studies have shown that activation of NF-kB (nuclear kappa factor B) and AP-1 (activator protein 1) promotes cell proliferation and survival Citation(43,44). Furthermore, increase in ROS and RNS is related to stabilization of HIF1α (hypoxia-induced factor 1 alpha), which leads to transcription of a number of genes involved in cell metabolism, survival, and death; angiogenesis; invasion; and metastasis Citation(45). Also, ROS regulates serine/threonine AMP-activated protein kinase (AMPK), which contributes to energy metabolism Citation(46).
On the other hand, these free radicals can react with the DNA molecule, mainly causing changes in nitrogenous bases producing oxidized bases or crosslinking between two adjacent bases Citation(47). However, after damage, the DNA molecule can be repaired by enzymatic mechanisms Citation(48), being crucial for maintenance of genome integrity.
In fact, pathogenesis of COPD leads to increased oxidative stress, resulting in damages in lung cells, mucus hypersecretion, inactivation of antiprotease, and exacerbation of inflammation Citation(49). Studies indicate that DNA repair inefficiency is common in COPD patients and correlates with susceptibility to disease development and progression Citation(50), and that in a hypoxic response condition, DNA damage is prominent in lung cells of COPD patients Citation(51). Another study suggests that increased damage and recurrent repair, linked to the condition of hypoxia, could deplete the regenerative capacity of satellite cells in COPD patients, causing muscle atrophy Citation(52). Also, there is an increase in DNA damage under oxygen supplementation conditions, but the response of DNA repair mechanisms is more effective Citation(53). Fetal lung cells exposed to cigarette smoke present breaks in DNA chain, as well as initiation of DNA repair Citation(54). A study carried out with 100 healthy subjects showed that DNA fragmentation is significantly increased in smokers, mainly in women Citation(55). These results were confirmed by Mercken et al. Citation(56), where an immediate increase in DNA damage was observed in patients with COPD who underwent physical exercise tests. Moreover, it has been suggested that oxidative stress may extend beyond the lung to generate systemic manifestations of COPD Citation(57,58) and oxidative stress and DNA damage are increased in peripheral blood cells from COPD patients Citation(59). In addition, there are evidences relating oxidative stress and pro-inflammatory mediators to epigenetic changes, such as nuclear histone acetylation/deacetylation, restricting transcription factors to DNA, and thereby altering the rate of expression of pro-inflammatory genes Citation(60). However, there are no data indicating restriction of transcription factors that alter gene expression related to DNA repair in COPD patients. lists studies on COPD and DNA changes, damage, and repair.
Table 2. Summary of studies on the relationship between COPD, DNA damage and DNA repair mechanisms.
There are different DNA repair pathways, but one of them is mainly related to oxidative damage. The excision repair, also known as “repair in the dark,” was described in 60's by Setlow and Carrier, when they observed DNA repair could carry out by another mechanism, which had not been described yet. This DNA repair occurs when the damaged nitrogenous base is removed and replaced by a normal base in the DNA molecule. Excision repair can be divided into two mechanisms: base excision repair (BER) and nucleotide excision repair (NER) Citation(61).
DNA repair for specific oxidative damages by base excision repair mechanism
Reactive oxygen species (ROS) react with DNA molecule causing simple strand breaks (SSB), especially in sugar bonds Citation(62). 8-oxo-guanine (8-oxoG) and 5-hydroxycytosine, which mispair with adenine and thymine, respectively, are the most commonly oxidized bases Citation(63). As previously described, COPD contributes to increase in free radicals, thus causing oxidative stress, which in turn could damage the DNA molecule and consequently activate base excision repair mechanism Citation(64). Oxidative damage is increased in tumors, thus increasing metabolism, oncogenic signaling, and mitochondrial dysfunction Citation(65). This factor results in 100 times more 8-oxoG in cancer tissues than in normal tissues Citation(65).
Base excision repair acts on single strand breaks, oxidized bases, and little bulky adducts Citation(66), induced by free radicals that produced by endogenous mechanisms and those induced by exogenous agents Citation(64). This mechanism is evolutionarily conserved, with high functional sequence homology from bacteria to mammals presenting two pathways: short-patch BER pathway repairs a single base, and the long BER pathway is able to remove from two–eight bases Citation(67) (). BER is characterized by excision of the damaged base by a series of enzymes called DNA glycosylases Citation(68). 8-oxoguanine DNA glycosylase (OGG1) is a glycosylase, which recognizes oxidized bases, promotes hydrolysis of N-glycosyl bond, and also contains lyase activity Citation(68). Consequently, there is a formation of an abasic site, which is recognized by an apurinic/apyrimidinic endonuclease (APE, in mammalian cells are APE1 and APE2), which in turn produces breaks in the phosphodiester bond 5′ or 3′ in the abasic site Citation(69). After the action of the glycosylase, with the formation of the AP site and the disruption of the DNA strands, the Poly (ADP-ribose) polymerase 1 (PARP1) performs the process known as “PARylation,” in order to modify the surroundings of chromatin proteins, the polarity of the DNA strand, which contributes to the recruitment of other proteins to the BER Citation(69).
Figure 2. Schematic representation for DNA repair by base excision mechanism. (A) Schematic representation for base excision repair mechanism (REB) by short patch (for one damaged base) and long patch (for up to eight damaged bases). (B) Schematic representation for global genome and transcription-coupled nucleotide excision repair mechanism (REN). Endo III (endonuclease III), OGG1 (8-oxoguanine DNA glycosylase) MPG (3-methyladenine-DNA glycosylase), Xth (xyloglucan endo-transglycosylase/hydrolase), XRCC1 (X-ray repair cross-complementing protein 1), PCNA (proliferating cell nuclear antigen), Lig (ligase), XP (xeroderma pigmentosum), ERCC1 (excision repair cross-complementing 1), RPA (replication protein A) and Lig3 (ligase III).
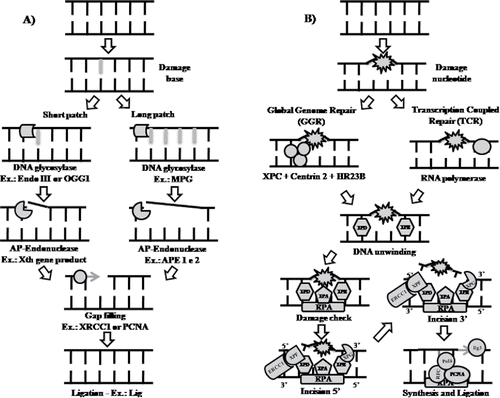
The interaction between Rapamycin and Torin 1 (RAP) protein and PARP1 with X-ray repair cross-complementing (XRCC1) protein stimulates APE1 required for the cleavage of the phosphate backbone, thus generating an AP site, and creating a single-strand break. After cleavage, the polymerase (POL) inserts the new nucleotide Citation(68).
The gap formed is therefore filled with a new nucleotide by action of DNA polymerase β (POL β). Additionally, this enzyme presents phosphodiesterase activity, which hydrolyzes unmodified 5 ends, thereby subsequent connection of the new nucleotide to DNA sequence involved by DNA ligase III that steals the DNA ends Citation(69). BER is not a flexible mechanism for the recognition of DNA damages, since it is not feasible for cells to maintain different enzymes for different types of damages encountered in the DNA Citation(70). The switch of short or long pathway could be linked to the availability of ATP in the vicinity of the AP site Citation(71). In this context, NER is more general and flexible, being able to recognize and repair a large number of damages, not necessarily those related to the DNA structure.
DNA repair for non-specific oxidative damages by base excision repair mechanism
As discussed for BER, COPD contributes to increase in free radicals, which in turn could instigate oxidative stress and consequently damage the DNA molecule; thus nucleotide excision repair mechanisms may be activated and effectively contribute to DNA molecule repair Citation(72). Recognition of DNA damages by NER does not occur by their chemical nature, but by the degree of distortion promoted in the DNA helix. DNA damages, as cyclobutane pyrimidine dimers, 6–4 pyrimidine-pyrimidone chemicals adducts, and certain types of cross-links between the two strands of DNA, caused by substances found in tobacco smoke and food, and intrastrand crosslinks formed by chemotherapeutics like cisplatin are the main cause of DNA deformation Citation(73,74).
Nucleotide excision repair removes 22–30 nucleotides surrounding a helix distorting DNA damages, and depending on the distortion in the double strand, damages are repaired more efficiently or not, but usually occur evenly in the whole genome Citation(75,76). This mechanism could be involved in the repair of some damage types, such as oxidized bases, but its role in the repair of oxidative damages is not completely elucidated Citation(72).
More than 30 different proteins are involved in the NER mechanism, presenting two pathways: global genomic repair, which removes the damages in non-transcribed regions, and transcription coupled repair, which removes damages at transcriped genes Citation(61) (). In the global genomic repair pathway, after the primary recognition of a wide variety of damages in the DNA molecule by the complex XPC/HR23B, factors are recruited, whereas in the transcription coupled repair, the above-mentioned complex is essential for the recognition of the damage Citation(74,77).
However, blocking RNA polymerase II (RNAPII) by damaged DNA, such as those induced by free radicals, is considered as an induction signal for DNA repair mechanisms. After damage recognition, RNAPII enzyme is removed by CSA and CSB proteins (“Cockayne Syndrome” A and B), and DNA repair is started Citation(78). Xeroderma pigmentosum complementation group E (XPE) protein complex, whose subunits are P127/DDB1 and p48/DDB2, recognizes damages not identified by the XPC complex Citation(79). After detection, both NER mechanisms converge to a common pathway for the cleavage of the DNA double strand. RNA polymerase II transcription factor (TFIIH - “Transcription factor II Human”) is recruited to the damage site, the TFIIH may be able to carry out a 5′ to 3′ search for a short distance before anchoring at DNA damage in cooperation with the other protein components of the preincision complex, and provides a catalytic activity to open the DNA Citation(80). TFIIH 9 is composed of subunits of ATP (p62, p52, p44, p34, cdk7, cyclin H and MAT 1). In addition to these subunits, 2 helicases, called XPB and XPD, participate in NER. The role of complex proteins, and the XPD and XPB proteins are present at the opening and unfolding of the DNA double helix at the site of damage, thereby allowing the entry of the NER factors that will succeed Citation(81).
The ATP-dependent helicase 5′ to 3′ (XPD/ERCC2) is required for NER, differently from XPB, whose action is not essential. The role of helicase XPD/ERCC2 is also important to maintain the stability of the complex as a whole, through the interaction of the C-terminal portion with another p44 subunit complex. Some studies suggest that XPD is involved in DNA repair after the recognition of the double helix distortion by the XPC/HR23B complex, checking if there is indeed the presence of damage Citation(76,82).
The complex constituted by Repliacation Protein A (RPA), XPA and XPG proteins conducts the pre-incision, and performs the DNA repair synthesis. The incision in DNA molecule is carried out by a heterodimer constituted by the XPF and Excision Repair Cross-Complementation group 1 (ERCC1) protein, in addition to XPG enzyme. XPF-ERCC1 heterodimer is recruited to the damage site over XPA protein to generate the incision, which occurs in the nucleotide sequence about 15–24 nucleotides at the 5′ side and about 2–8 nucleotides at the 3′ side of the damage Citation(83).
During the 3′ cleavage of the DNA strand by ERCC1-XPF complex, the synthesis of the new DNA fragment is started before a 5′ cut of the DNA strand by the XPG protein Citation(78). The XPF-ERCC1 complex, cited above, is part not only of the NER mechanism, but also of other DNA repair pathways, such as the repair of double breaks of DNA, cross-links, and it is essential for the development in humans as well Citation(76,84).
Nuclear Sirtuin 1 (SIRT1) protein plays a dual role in promoting NER. Firstly, this protein stimulates the recognition of damage by promoting the expression of XPC protein Citation(76) and then, stimulates the excision of damage promoting the NER endonuclease assembly at damage sites by deacetylation of XPA protein Citation(85). DNA synthesis repair is carried out by RPA, RFC, PCNA, and polymerases σ/ϵ, while reclosing of the strand is carried out by the DNA ligase, either LIG3 or LIG1 Citation(61).
Other types of DNA repair in COPD
Some studies suggest that patients with COPD present double-stranded breaks in the DNA molecule Citation(86), and also that tobacco consumption leads to DNA strand double breaks in leukocytes Citation(87). This damage induces phosphorylation of histone γ-H2AX (H2AX), which in turn recruits other DNA repair proteins, such as Breast Cancer-Associated 1 (BRCA1) protein, resulting in the activation of downstream distinctive repair pathways Citation(88).
Starting from the double breaks, DNA will be repaired by homologous recombination (HR) or non-homologous end-joining (NHEJ) repair mechanisms. HR acts preferentially during the S phase, while NHEJ, the main repair mechanism to repair double-strand breaks in higher organisms acts throughout the cell cycle phases Citation(89).
Homologous recombination presents three stages or phases: presynapsis, synapsis, and postsynapsis. Homologous DNA without damages is required to repair the DNA molecule. Citation(90) (). In the presynapsis stage there is a 5′ to 3′ resection process of broken DNA ends by complex to generate to generate a 3′ single ended strand DNA by removing about 100 nucleotides with interaction with the C-teminal protein (CTIP) Citation(91). This MRN heterotrimeric complex is formed by Mre11, NBS 1, and Rad 50 proteins and the MRX heterotrimeric complex is formed by Mre11, Rad 50, and XRS2, which performs the unwinding of the DNA end, while recruiting the BRCA1 protein, whose function is to make an incision and promote activation of CTIP Citation(92).
Figure 3. Schematic representation for DNA repair by recombination mechanism. (A) Schematic representation for homologous recombination. (B) Schematic representation for non-homologous end-joining after DNA double-strand break. DNA PKcs, DNA-dependent protein kinase, catalytic subunit; XRCC4, X-ray repair cross complementing 4; XLF, XRCC4-like factor; ATM, ataxia-telangiectasia mutated; RPA, replication protein A.
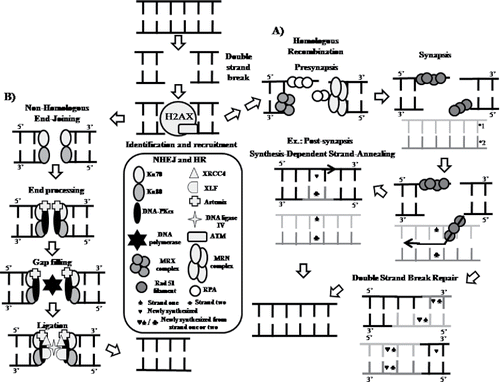
In the synapsis stage, heterotrimeric RPA binds at the single-stranded DNA to prevent internal base pairing Citation(92). Then, RPA and Rad 51 proteins are replaced by recombinase complexing with BRCA2 and paralogs of Rad 51 (RAD51B, C and D, XRCC2 and XRCC3) and the DNA filament will be target of nucleoprotein Rad51 for nucleoprotein Rad51 Citation(93), which identifies homologous sequences in DNA molecule. Thus, Rad51 leaves the 3′ end to reveal a 3′-OH group, permiting that DNA synthesis enzymes access the DNA end Citation(94). From this stage, DNA repair process could take place by the synthesis-dependent strand-annealing (SDSA) or the double-strand break repair (DSBR) pathway.
In the SDSA pathway, after DNA synthesis is performed by polymerase δ, the new DNA fragment is moved and reconnected with the original strand. In the DSBR pathway, two independent strand invasions from both DSB ends are followed by simultaneous DNA synthesis performed by polymerase η Citation(95).
On the other hand, the NHEJ mechanism acts on double-strand breaks reconnecting the two DSB ends. Different from HR, this mechanism can connect any type of DNA end without homology between the sequences. However, NHEJ is more susceptible to errors, once this mechanism can cause small insertions or deletions of nucleotides in the DNA sequence Citation(96).
Double-strand break DNA repair by NHEJ starts with the regulatory subunits binding to DNA, Ku70 (also known as XRCC6), and Ku80 (also known as XRCC5), along with the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) (). DNA-PKcs is a member of the PI3K-related protein kinase (PIKK) family, which also includes ataxia-telangiectasia (ATM), Rad3-related and Mechanistic Target of Rapamycin (mTOR) proteins. Assembly of these proteins form the DNA-PK holoenzyme, and are involved in the recognition of the damage. Pol μ and λ participate in the pathway by binding to Ku proteins by N-terminal domain breast carboxy-terminal cancer (CRCT), providing their gap-filling activity before the end ligation step, which is performed by the XRCC4-ligase IV complex Citation(97). While Pol λ synthesizes new DNA with partially complementary ends, Pol μ synthesizes DNA without the presence of complementary strand Citation(98).
DNA-PK enabled recruit other proteins, such as Artemis (which solves the DNA ends), XRCC4, XLF, DNA ligase IV, and DNA polymerase, which complete the repair Citation(99). Double-strand breaks could trigger permanent cell cycle arrest or apoptosis, leading to important clinical changes, including carcinogenesis.
DNA damages, mainly those caused by oxidative processes, are related to some chronic diseases, such as diabetes mellitus, atherosclerosis, neurodegenerative diseases, and cancer Citation(100). Papi et al. Citation(101) in a case report suggested that COPD, with the exception of chronic bronchitis, increases up to 3 times the risk of patients developing squamous cell carcinoma by the activation of signaling pathways Citation(101). These signaling pathways could be activated by the peripheral distribution of tobacco due to the limitation of the air flow, but other studies reinforce that the activation could also occur by second messengers, as ROS Citation(102). Caramori et al. Citation(103) suggested that inefficiency or imbalance of DNA repair, such as the reduction of expression of one of the Ku86 DNA repair proteins in bronchiolar epithelial cells of patients with COPD, could contribute to increased risk of lung carcinoma Citation(104). Also, a strong correlation between increased oxidative stress and DNA damage and cancer development had been observed, suggesting that molecular studies aiming at new targets for carcinoma prevention are necessary Citation(105). Moreover, molecular targets should take into account not only individuals with COPD, but also at-risk groups, such as smokers, reinforcing that it is necessary to understand the molecular interactions in the development of COPD to achieve molecular targets, which are not present in clinical trials yet Citation(106).
Telomeric and genomic stabilization followed oxidative DNA damage
A number of studies show that DNA damages cause genomic instability, shortening the telomeres, for example. Mammalian telomeres are composed of repeatitions of TTAGGG sequences, as a t-loop, followed by a 3′ end, and they are related to their self-protection and cell longevity Citation(107). During normal cell divisions, enzymes (telomerases) act shortening the telomeres Citation(108). In oxidative stress conditions, such as in those with COPD Citation(109), a drastic telomere shortening has been reported Citation(110). Interestingly, this telomere shortening was related to chronic inflammation in COPD patients Citation(111). Telomere shortening in COPD is related to reduced telomerase activity Citation(111). A study showed that the predisposition to lung emphysema could cause telomere shortening induced by lung injury due to cigarette smoke Citation(112). Another study demonstrated that telomere shortening is associated to increased risk of COPD, as well as to reduction of lung function Citation(113).
In addition to telomerase, telomere integrity relies on shelterin, a DNA-bound protein complex composed by six polypeptides (TRF1, TRF2, RAP1, TIN2, TPP1, and POT1) Citation(114) (). Repeat-binding telomeric factors 1 and 2 (TRF1 and TRF2, respectively) directly bind at the double-stranded telomere repeats, POT1 binds directly at the single-stranded 3′-overhang, and the other three components interconnect these telomere-binding components. TRF1 and TRF2 interact with other proteins Citation(115) in order to protect the telomeres by directly binding to the telomeric duplex Citation(116). The interaction between TRF1 and TRF2, mediated by TRF1-interacting factor-2 (TIN2), contributes to the stabilization of TRF2 on telomere, protecitng DNA ends from degradation Citation(117).
Figure 4. Schematic representation for protein performance in telomeric regulation and genomic stabilization mechanisms. TRF, telomeric repeat-binding factor; TIN2, TRF1-interacting factor-2; POT1, protection of telomeres 1, TPP1, tripeptidyl-peptidase 1; ATM, ataxia telangiectasia mutated; p53, tumor protein 53; MDM2, mouse double minute 2 homolog.
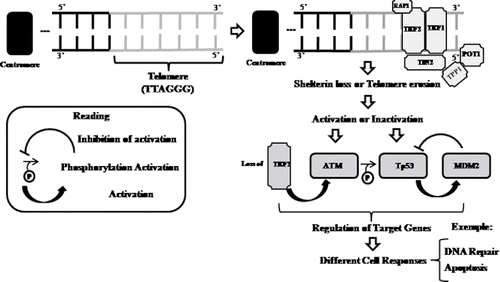
Telomere deprotection, as a consequence of shelterin loss or telomere erosion, induces activation of Ataxia Telangiectasia (ATM) protein and inactivation of Tumor protein p53 (Tp53), which result in chromosome fusions by NHEJ and/or in apoptosis Citation(118). These factors regulate the telomere protection, telomere length, and interact with DNA repair proteins involved in chromosome instability syndromes, which cause premature aging, as well as increased cancer risk Citation(119).
A study suggests that TRF2 levels and telomere protection are subject to modulations by MAP Kinase signaling, such that cell growth could require to be coupled to TRF2-dependent telomere functions in order to ensure proper cell division Citation(120). Other studies have suggested that the loss of TRF2, for example, causes activation of ATM-dependent DNA damage response and DNA repair by NHEJ, resulting in p21-mediated G1/S arrest and downregulation upon p53 activation Citation(119) (). The p53 protein acts as a tumor suppressor, participating in some cellular processes, such as cell cycle regulation, apoptosis, and DNA repair Citation(121). p53 inactivation aborts the TRF2-ATM-p53 feedback regulation Citation(122), inducing cancer-promoting conditions, suach as those in lung cancer. In these conditions, dysfunctional telomeres with supra-physiological amounts of TRF2 are susceptible to chromosomal instability, and senescence checkpoint is impaired Citation(123).
Chronic obstructive pulmonary diseases have a prevalence in the elderly people, and have been related with telomere shortening Citation(124). Additionally, in oxidative stress there is a significant acceleration of telomere shortening Citation(110), and shortness of telomere length was indicated to be associated with a 28-fold increased risk of COPD Citation(113). Although age contributes to the onset of COPD, shortening of telomeres is associated with increased risk of the prevalence of these diseases Citation(94).
There are not studies correlating the development of COPD directly with telomere instability. However, after the initiation of COPD, DNA damage is commonly associated with oxidative stress, and could be used as a diagnostic marker Citation(59). COPD patients have a high incidence of lung cancer as previously mentioned, and DNA modifications by reactive species could be the relationship between these two conditions Citation(59). Also, it is not reported if COPD is induced by total or partial absence of enzymes involved in the repair of DNA molecule. However, due to a series of studies that show mainly oxidative damage to DNA, it is possible to suppose that repair mechanisms have a key role in the correction of damage, thus contributing to a better prognosis in COPD. In fact, a study suggests that deficiency in DNA repair in response to oxidative stress could lead to COPD, lung cancer, or pulmonary fibrosis Citation(5).
Conclusion
In COPD, inflammatory process is intense, increasing the oxidative stress mediated by free radicals and DNA damages. Although there are progresses in treatment for COPD, repair of DNA damages could be important against COPD development, and therapies based on the modulation of DNA integrity and genomic stability could be effective to reduce the mortality and morbidity of patients with COPD.
Declaration of interest
The authors report no conflicts of interest.
Funding
This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Carlos Chagas de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG).
References
- From the Global Strategy for the Diagnosis. Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD). 2016. Available from: http://goldcopd.org/
- Landis SH, Muellerova H, Mannino DM, Menezes AM, Han MK, van der Molen T, et al. Continuing to Confront COPD International Patient Survey: methods, COPD prevalence, and disease burden in 2012–2013. Int J Chron Obstruct Pulmon Dis 2014; 9:597–611.
- Filho GL. Publicação Oficial da Sociedade Brasileira de Pneumologia e Tisiologia. Br J Pneumol 2004; 30:1–55.
- Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013; 187(4):347–365.
- Tzortzaki EG, Dimakou K, Neofytou E, Tsikritsaki K, Samara K, Avgousti M, et al. Oxidative DNA damage and somatic mutations: A link to the molecular pathogenesis of chronic inflammatory airway diseases. Chest 2012; 141(5):1243–1250.
- Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int J Environ Res Public Health 2013; 10(9):3886–3907.
- Newton AC, Bootman MD, Scott JD. Second messengers. Cold Spring Harb Perspect Biol 2016; 8(8):a005926.
- Barzilai A, Yamamoto K. DNA damage responses to oxidative Stress. DNA repair (Amst) 2004; 3(8–9):1109–1115.
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberg T. DNA Repair and Mutagenesis. Washington, DC: ASM Press, 2006.
- Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2007; 176(6):532–555.
- Casey G. COPD: obstructed lung. Nurs N Z 2016; 22(5):20–24.
- Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 2008; 359(22):2355–2365.
- Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 187(3):228–237.
- Lapperre TS, Postma DS, Gosman MME, Snoeck-Stroband JB, Ten Hacken NHT, Hiemstra OS, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax, London 2006; 61(2):115–121.
- Snider GL, Kleinerman J, Thurlbeck WM, Bengali ZH. The definition of emphysema: report of a National Heart, Lung and Blood Institute, Division of Lung Diseases, Workshop. Am Rev Respir Dis 1985; 132(1):182–185.
- R23. Magrone T, Jirillo E. Cigarette smoke-mediated perturbations of the immune response: A new therapeutic approach with natural compounds. Endocr Metab Immune Disord Drug Target 2016; 16:158–167.
- Pan Z, Yu H, Liao JL. Probing cellular and molecular mechanisms of cigarette smoke-induced immune response in the progression of chronic obstructive pulmonary disease using multiscale network modeling. PLoS One 2016; 11:e0163192.
- Upham JW, Xi Y. Dendritic cells in human lung disease: recente advances. Chest 2016; 11(9):e0163192.
- Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153(2):530–534.
- Kalinina EP, Denisenko YK, Vitkina TI, Lobanova EG, Novgorodtseva TP, Antonvuk MV, et al. The mechanisms of the regulation of immune response in patients with comorbidity of chronic obstructive pulmonary disease and asthma. Can Respir J 2016; 2016: 4503267.
- Chung KF. The role of airway smooth muscle in the pathogenesis of airway remodelling in COPD. Proc Am Thorac Soc 2005; 2(4):347–354.
- Martins-Oliveira BT, Almeira-Reis R, Thodoro-Júnior OA, Oliva LV, Neto dos Santos Nunes N, Olivo CR, et al. The plant-derived bauhinia bauhinioides kallikrein proteinase inhibitor (rBbKI) Attenuates Elastase-Induced Emphysema in Mice. Mediators Inflamm 2016; 2016:5346574.
- Macnee W. Pathogenesis of chronic obstructive pulmonar disease. Proc Am Thor Soc 2005; 2(4):258–266.
- Tang LF, Du LZ, Chen ZM, Zou CC. Levels of matrix metalloproteinase-9 and its inhibitor in bronchoalveolar lavage cells of asthmatic children. Fetal Pediatr Pathol 2006; 25(1):1–7.
- Birch-Machin MA, Bowman A. Oxidative stress and aging. Br J Dermatol 2016; 2:26–29.
- Ďuračková Z. Some current insights into oxidative stress. Physiol Res 2010; 59(4):459–469.
- Hussain T, Tan B, Yin Y, Blachier F, Tossou MCB, Rahu N. Oxidative stress and inflammation: what polyphenols can do for us? Oxid Med Cell Longev 2016; 2016:7432797.
- Salzano S, Checconia P, Hanschmannc EM, Lillig CH, Bowler LD, Chan P, et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci USA 2014; 111(33):12157–12162.
- Zhang J, Summah H, Zhu YG, Qu JM. Nicotinic acetylcholine receptor variants associated with susceptibility to chronic obstructive pulmonary disease: A meta-analysis. Respir Res 2011; 12(1):158.
- Wedzicha JA, Seemungal TAR. COPD exacerbations: defining their cause and prevention. Lancet 2007; 370(9589):786–796.
- Seemungal T, Sykes A. Recent advances in exacerbations of COPD. Thorax 2008; 63(10):850–852.
- ICE Clinical Guidance. Management of exacerbations of COPD. Thorax 2004; 59:i131–i156.
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000; 342(18):1334–1349.
- Drost EM, Skwarski KM, Sauleda J, Soler N, Roca J, Agusti A, et al. Oxidative stress and irway inflammationin severe exarcerbations of COPD. Thorax 2005; 60:293–300.
- Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med 1997; 155:449–453.
- Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax 2000; 55:114–120.
- Donaldson GC, Seemungal TA, Patel IS, Lloyd-Owen SJ, Wilkinson TM, Wedzicha JA. Longitudinal changes in the nature, severity and frequency of COPD exacerbations. Eur Respir J 2003; 22:931–936.
- Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res 2013; 21:2003–2014.
- Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 2014; 21:396–413.
- Cerutti PA. Oxidant stress and carcinogenesis. Eur J Clin Invest 1991, 21:1–5.
- Baran CP, Zeigler MM, Tridandapani S, Marsh C. The role of ROS and RNS in regulating life and death of blood monocytes. Curr Pharm Des 2004; 10(8):855–866.
- Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J 1997; 11(2):118–124.
- Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-κB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 2005; 7(3–4):395–403.
- Pantano C, Reynaert NL, van der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappa B signaling pathway. Antioxid Redox Signal 2006; 8(9–10):1791–1806.
- Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol Aspects Med 2003; 24(4–5):149–159.
- Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1α transcription in skeletal muscle cells. Am J Physiol Cell Physiol 2009; 296(1):C116–C123.
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Oxford University Press, USA, 2007.
- Cadet J, Bourdat AG, D'Ham C, Duarte V, Gasparutto D, Romieu A, et al. Oxidative base damage to DNA: specificity of base excision repair enzymes. Mutat Res 2000; 462(2–3):121–128.
- Maluf SW, Margener M, Dalcanale L, Costa CC, Pollo T, Kayser M, et al. DNA damage in peripheral blood of patients with chronic obstructive pulmonary disease (COPD). Genet Toxicol Environ Mutagen 2007; 624:180–184.
- Neofytou E, Tzortzaki EG, Chatziantoniou A, Siafakas NM. DNA damage due to oxidative stress in Chronic Obstructive Pulmonary Disease (COPD). Int J Mol Sci 2012; 13(12):16853–16864.
- Pastukh VM, Zhang L, Ruchko MV, Gorodnya O, Bardwell GC, Tuder RM, et al. Oxidative DNA damage in lung tissue from patients with COPD is clustered in functionally significant sequences. Int J Chron Obstruct Pulmon Dis 2011; 6:209–217.
- Deldicque L, Francaux M. Acute vs. chronic hypoxia: What are the consequences for skeletal muscle mass? Cell Mol Exerc Physiol 2013; 2(1):e5.
- Silva ALG, Karnopp TE, Weber AF, Goulart CL, Scheneiders PB, Cardoso DM, et al. DNA damage and repair capacity in lymphocyte of chronic obstructive pulmonary disease patients during physical exercise with oxygen supplementation. Multidiscip Respir Med 2016; 11:43.
- Kim H, Liu X, Kobayashi T, Conner H, Kohyama T, Wen F, et al. Reversible cigarette smoke extract-induced DNA damage in human lung fibroblasts. Am J Respir Cell Mol Biol 2004; 31:483–490.
- - Betti CT, Davini L, Giannessi N, Loprieno E, Barale R. Microgel electrohoresis assay (comet test) and SCE analysis in human lymphocytes from 100 normal subjets. Mutat Res 1994; 307:323–333.
- Mercken MR, Hageman J, Schols MWJA, Akkermans AM, Bast A. Rehabilitation decreases exercises-induces oxidative stress in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 172:994–1001.
- Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996; 154:1055–1060.
- MacNee W. Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2:50–60.
- Ceylan E, Kocygit A, Gencer M, Aksoy N, Selek S. Increased DNA damage in patients with chronic obstructive pulmonary disease who had once smoked or been exposed to biomass. Respir Med 2006; 100:1270–1276.
- Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol 2003; 36:95–109.
- Costa RMA, Chiganças V, Galhardo RS, Carvalho H, Menck CF. The eukaryotic nucleotide excision repair pathway. Biochimie 2003; 85(11):1083–1099.
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993; 362(6422):709–715.
- van Loon B, Markkanen E, Huübscher U. Oxygen as a friend and enemy: how to combat the mutational potential of 8-oxo-guanine. DNA Repair 2010; 9(6):604–616.
- Mitra S, Hazra TK, Roy R, Ikeda S, Biswas T, Lock J, et al. Complexities of DNA base excision repair in mammalian cells. Mol Cells 1997; 7(3):305–312.
- Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J 1996; 313(Pt 1):17–29.
- - Klungland A, Bjelland S. Oxidative damage to purines in DNA: role of mammalian Ogg1. DNA Repair (Amst) 2007; 6(4):481–488.
- Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci 2009; 66(6):981–993.
- Krokan HE, Bjoras M. Base excision repair. Cold Spring Harb Perspect Biol 2013; 5(4):a012583.
- Satoh MS, Lindahl T. Enzymatic repair of oxidative DNA damage. Cancer Res 1994; 54:1899s–1901s.
- El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucl Acids Res 2003; 31(19):5526–5533.
- Petermann E, Zieler M, Oei SL. ATP-dependent selection between single nucleotide and long patch base excision repair. DNA Repair (Amst) 2003; 2(10):1101–1114.
- Curtis NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Ver Cancer 2012; 12(12):801–817.
- Wood RD. DNA repair in eukaryotes. Annu Ver Biochem 1996; 65:135–167.
- Zhu Q, Wani AA. Nucleotide excision repair: finely tuned molecular orchestra of early pre-incision events. Photochem Photobiol 2016; 93(1):166–177.
- Wood RD. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ Mol Mutagen 2010; 51(6):520–526.
- Scharer OD. Nucleotide excision repair in Eukaryotes. Cold Spring Harb Perspect Biol 2013; 5(10):a012609.
- Krasikova YS, Rechkunova NI, Maltseva EA, Petruseva IO, Silnikov VN, Zatsepin TS, et al. Interaction of nucleotide excision repair factors XPC-HR23B, XPA, and RPA with damaged DNA. Biochemistry (Mosc.) 2008; 73(8):886–896.
- Petruseva IO, Evdikimov NA, Lavrik OI. Molecular mechanism of global genome nucleotide excision repair. Acta Nat 2014; 6(1):23–34.
- Sugawava K, Ng JM, Masutani C, Maekawa T, Uchida A, van der Spek PJ, et al. Two human homologs of Rad23 are functionally interchangeable in complex formation and stimulation of XPC repair activity. Mol Cell Biol 1997; 17(12):6924–6931.
- Sugasawa K, Akagi Ji, Nishi R, Iwai S, Hanaoka F. Two-step recognition of DNA damage for mammalian nucleotide excision repair: directional binding of the XPC complex and DNA strand scanning. Mol Cell 2009; 36(4):642–653.
- Sarasin A, Stary A. New insights for understanding the transcription-coupled repair pathway. DNA Repair (Amst.) 2007; 6(2):265–269.
- Reardon JT, Sancar A. Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev 2003; 17(20):2539–2551.
- Petit C, Sancar A. Nucleotide excision repair: from E. coli to man. Biochimie 1999; 81(1–2):15–25.
- Ahmad A, Enzlin JH, Bhagwat NR, Wijgers N, Raams A, Appledoorn E, et al. Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients. PLoS Genet 2010; 6(3):e1000871.
- Fan W, Luo J. SIRT1 regulates UV-induced DNA repair through deacetylating XPA. Mol Cell 2010; 39(2):247–258.
- Silva ALG, Rosa HT, Karnopp TE, Charlier CF, Ellwanger JH, Moura DJ, et al. Evaluation of DNA damage in COPD patients and its correlation with polymorphisms in repair genes. BMC Med Genet 2013; 14:93.
- Roos WP, Krumm A. The multifaceted influence of histone deacetylases on DNA damage signalling and DNA repair. Nucl Acid Res Adv 2016; 44(21):10017–10030.
- O'Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011; 20(5):606–619.
- Bebenek K, Pedersen LC, Kunkel TA. Structure-function studies of DNA polymerase λ. Biochemistry 2014; 53(17):2781–2792.
- Jeggo PA, Geuting V, Lobrich M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother Oncol: J Eur Soc Ther Radiol Oncol 2011; 101(1):7–12.
- Williams GJ, Williams RS, Williams JS, Moncalian G, Arvai AS, Limbo O, et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat Struct Mol Biol 2011; 18(4):423–431.
- Shibata A, Moiani D, Arvai AS, Perry JJP, Harding SM, Genois MM, et al. DNA double strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 2014; 53(1):7–18.
- Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 2014; 6(9):a016428.
- Dexheimer TS. DNA repair pathways and mechanisms. In: Matthews LA, Cabarcas SM, Hurt EM, editors. DNA Repair of Cancer Stem Cells. Netherlands: Springer, 2013; 19–32.
- Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol 2013; 3:113.
- Mamarche BJ, Orazio NI, Weitzman MD. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett 2010; 584(17):3682–3695.
- Lieber MR, Gu J, Lu H, Shimazaki N, Tsai AG. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in human. Subcell Biochem 2010; 50:279–296.
- Crespan E, Czabany T, Maga G, Hübscher U. Microhomology-mediated DNA strand annealing and elongation by human DNA polymerases λ and β on normal and repetitive DNA sequences. Nucl Acids Res 2012; 40(12):5577–5590.
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39–85.
- Jackson SP, Bartek J. The DNA-damage response in human bi¬ology and disease. Nature 2009; 461:1071–1078.
- Papi A, Casoni G, Caramori G, Guzzinati I, Boschetto P, Ravenna F, et al. COPD increases the risk of squamous histological subtype in smokers who develop non-small cell lung carcinoma. Thorax 2004; 59(8):679–681.
- Iannone MF, Rosales EP, Groppa MD, Benavides MP. Reactive oxygen species formation and cell death in catalase-deficient tobacco leaf disks exposed to cadmium Protoplasma 2010; 245(1–4):15–27.
- Caramori G, Casolari P, Cavallesco GN, Giuffrè S, Adcock I, Papi A. Mechanisms involved in lung câncer development in COPD. Int J Biochem Cell Biol 2011; 43:1030–1044.
- Caramori G, Adcock IM, Casolari P, Ito K, Jazrawi E, Tsaprouni L, et al. Unbalanced oxidant-induced DNA damage and repair in COPD: a link towards lung câncer. Thorax 2011; 66:521–527.
- Caramori G, Papi A. Pathogenic link between chronic obstructive pulmonary disease and squamous cell lung cancer. Expert Rev Respir Med 2007; 1(2):171–175.
- Adcock IM, Caramori G, Barnes PJ. Chronic obstructive pulmonary disease and lung cancer: new molecular insights. Respiration 2011; 81:265–284.
- Donate LE, Blasco MA. Telomeres in cancer and ageing. Philos Trans R Soc Lond B Biol Sci 2011; 366(1561):76–84.
- Shammas MA. Telomeres, lifestyle, cancer, and aging. Curr Opin Clin Nutr Metab Care 2011; 14(1):28–34.
- Amsellen V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, Validire P, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonar disease. Am J Respir Crit Care Med 2011; 184(12):1358–1366.
- Von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci 2002; 27(7):339–344.
- Amsellem V, Gary-Bobo G, Marcos E, Maitre B, Chaar V, Validire P, et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 184(12):1358–1366.
- Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI, et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med 2011; 184(8):904–912.
- Rode L, Bojesen SE, Wischer M, Vestbo J, Nordestgaard BC. Short telomere lenght, lung function and chronic obstructive pulmonar disease in 46.396 individuals. Thorax 2016; 68(5):429–435.
- Giraud-Panis MJ, Pisano S, Benarroch-Popivker D, Pei B, Le Du MH, Gilson E. One identity or more for telomeres? Front Oncol 2013; 3:48.
- Hanaoka S, Nagadoi A, Nishimura Y. Comparison between TRF2 and TRF1 of their telomeric DNA-bound structures and DNA-binding activities. Protein Sci 2005; 14(1):119–130.
- Blanco R, Muñoz P, Flores JM, Klatt P, Blasco MA. Telomerase abrogation dramatically accelerates TRF2-induces epithelial carcinogenesis. Genes Dev 2007; 21(2):206–220.
- O'Connor MS, Safari A, Xin H, Liu D, Songyang Z. A critical role for TPP1 and TIN2 interaction in highorder telomeric complex assembly. Proc Natl Acad Sci USA 2006; 103(32):11874–11879.
- Galati, A, Micheli E, Alicata C, Ingegnere T, Cicconi A, Pusch MC. TRF1 and TRF2 binding to telomeres is modulated by nucleosomal organization. Nucl Acids Res 2015; 43(12):5824–5837.
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19(18):2100–2110.
- Picco V, Coste I, Giraud-Panis MJ, Renno T, Gilson E, Pagès G. ERK1/MAPK pathway-dependent regulation of the telomeric fator TRF2. Oncotarget 2016; 7(29):46615–46627.
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8(4):275–283.
- Horikawa I, Fujita K, Harris CC. p53 governs telomere regulation feedback too, via TRF2. Aging 2011; 3(1):26–32.
- Muñoz P, Blanco R, Blasco MA. Role of the TRF2 telomeric protein in câncer and ageing. Cell Cycle 2006; 5(7):718–721.
- Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest 2009; 135(1):173–180.
- Kim WJ, Lim MN, Hong Y, Silverman EK, Lee JH, Jung BH. Association of Lung function genes with chronic obstructive pulmonary disease. Lung 2014; 192(4):473–480.
- Wu TC, Huang YC, Hsu SY, Wang YC, Yeh SL. Vitamin E and vitamin C supplementation in patients with chronic obstructive pulmonary disease. Int J Vitam Nutr Res 2007; 77(4):272–279.
- Leidinger P, Keller A, Borries A, Huwer H, Rohling M, Huebers J, et al. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung Cancer 2011; 74(4):41–47.
- da Silva ALG, Karnopp TE, Weber AF, Goulart CL, Scheneiders PB, Cardoso DM, et al. DNA damage and repair capacity in lymphocyte of chronic obstructive pulmonary diseases patients during physical exercise with oxygen supplementation. Multidiscip Respir Med 2016; 11:43.
- Avriel A, Rozenberg D, Raviv Y, Heimer D, Bar-Shai A, Gavish R, et al. Prognostic utility of admission cell-free DNA levels in patients with chronic obstructive pulmonary disease exacerbations. Int J Chron Obstruct Pulmon Dis 2016; 11:3153–3161.
- Carpagnano GE, Lacedonia D, Carone M, Soccio P, Cotugo G, Palmiotti GA, et al. Study of mitochondrial DNA alteration in the exhaled breath condensate of patients affected by obstructive lung diseases. J Breath Res 2016; 10(2):026005.
- Xie JG, Xu YJ, Zhang ZX, Ni W, Chen SX. Smoking, the level of DNA adducts and chronic obstructive pulmonary disease. Zhonghua Jie He He Hu Xi Za Zhi 2004; 27(7):469–473.
- ben Anes A, Fetoui H, Bchir S, ben Nasr H, Chahdoura H, Chabchoub E, et al. Increased oxidative stress and altered levels of nitric oxide and peroxynitrite in Tunisian patients with chronic obstructive pulmonary disease: correlation with disease severity and airflow obstruction. Biol Trace Elem Res 2014; 161(1):20–31.
- da Silva AL, da Rosa HT, Karnopp TE, Charlier CF, Ellwanger JH, Moura DJ, et al. Evaluation of DNA damage in COPD patients and its correlation with polymorphisms in repair genes. BMC Med Genet 2013; 14:93.
- Oit-Wiscombe I, Virag L, Soomets U, Altraja A. Increased DNA damage in progression of COPD: a response by poly(ADP-ribose) polymerase-1. PloS One 2013; 8(7):e70333.
- Yang S, Wu H, Zhao J, Wu X, Zhao J, Ning Q, et al. Feasibility of 8-OHdG formation and hOGG1 induction in PBMCs for assessing oxidative DNA damage in the lung of COPD patients. Respirology 2014; 19(8):1183–1190.
- Korytina GF, Akhmadishina LZ, Kochetova OV, Burdiuk luV, Aznabaeva luG, Zagidullin ShZ, et al. Association of genes involved in nicotine and tobacco smoke toxicant metabolism (CHRNA3/5, CYP2A6, and NQO1) and DNA repair (XRCC1, XRCC3, XPC, and XPA) with chronic obstructive pulmonary disease. Mol Biol (Mosk) 2014; 48(6):939–951.