
ABSTRACT
IL-6 and IL-33 are involved in the inflammatory process in obstructive lung diseases. In contrast to IL-6, few data on the expression of IL-33 in different biological samples from asthma and COPD patients are available. The aim was to evaluate the expressions of IL-33 and IL-6 in bronchial mucosa and to compare these expressions with the concentrations of both cytokines in various respiratory samples from patients with mild-to-moderate asthma and COPD. Serum, induced sputum and exhaled breath condensate IL-6 and IL-33 levels, as well as their expression in bronchial mucosa were evaluated in 22 asthma and 33 COPD patients. There were significant differences between bronchial mucosa IL-6, but not IL-33 expression in asthma and COPD. Serum and IS IL-6 concentrations were higher in COPD than in asthma (3.4 vs. 2.02 pg/mL, p = 0.002 and 16.5 vs. 12.7 pg/mL, p = 0.007, respectively); IL-33 levels reached similar values in asthma and COPD in all investigated samples. In both diseases, the lowest levels of IL-6 and IL-33 were found in EBC. EBC levels of both cytokines did not correlate with their expression in other materials. The IL-33 and IL-6 are detectable in serum, IS and EBC not only in asthma but also in COPD patients. In the COPD group, serum and IS IL-6 concentrations were statistically higher than in the asthma group. The tissue expression of IL-33 and IL-33 concentrations in the investigated biological samples were on a comparable level in both diseases. Our findings may suggest that IL-33 activation is a common pathway in asthma and COPD.
| Abbreviations | ||
| ACT | = | Asthma Control Test |
| ATS | = | American Thoracic Society |
| BMI | = | Body mass index |
| CAT | = | COPD Assessment Test |
| COPD | = | Chronic obstructive pulmonary disease |
| EBC | = | Exhaled breath condensate |
| ELISA | = | Enzyme-linked immunosorbent assay |
| ERS | = | European Respiratory Society |
| FEV1 | = | Forced expiratory volume at first second |
| FVC | = | Forced vital capacity |
| GOLD | = | Global Initiative for Chronic Obstructive Lung Disease |
| IL | = | Interleukin |
| ILC2 | = | Innate lymphoid cells type 2 |
| IS | = | Induced sputum |
| RV | = | Residual volume |
| TLC | = | Total lung capacity |
| TSLP | = | Thymic stromal lymphopoietin |
Introduction
Although the nature of the airway inflammation in asthma and chronic obstructive pulmonary disease (COPD) differs considerably (Citation1,Citation2), some common features of inflammatory process can be seen in both diseases. This was demonstrated in several studies which showed similarities between the mechanisms of airway inflammation, at least in mild-to-moderate stages of asthma and COPD (Citation3–5).
Determination of the inflammatory phenotype of both diseases can be useful not only in terms of more precise differentiation between both entities but also in the prediction of response to specific treatment (Citation6). According to a new interesting concept on therapeutic strategies in obstructive airway diseases, the future management of asthma and COPD will be based on clinical, functional, biological and imaging features of the disease and focus on ‘treatable traits’ reflecting the needs of the individual patient rather than numerical indices such as forced expiratory volume in one second (FEV1) (Citation4). In the above context, studies evaluating different inflammatory biomarkers in asthma and COPD may have potential practical applications.
In recent years, a group of epithelium-derived cytokines that include thymic stromal lymphopoietin (TSLP), interleukin (IL)-33 and IL-25, has attracted great attention because of its function in immune responses and inflammatory airway diseases. IL-33 is produced by a variety of cells and tissues including human and mouse lung tissue, lung stromal cells, airway epithelial and muscle cells, as well as alveolar macrophages (Citation7). IL-33 together with TSLP and IL-25 provide a signal for innate lymphoid cells type 2 (ILC2), which in turn initiate and amplify allergic inflammation by orchestrating type 2 immune response (Citation8). Due to their significant contribution in Th2 response these biomarkers are intensively studied and they are viewed as potential novel therapeutic targets (Citation9–11). Elevated IL-33 expression was demonstrated in epithelial and smooth muscle cells from asthma patients (Citation12,Citation13). The role of IL-33 in patients with COPD is unclear. Some data suggest that IL-33 might promote chronic airway inflammation contributing to COPD, by inducing and enhancing the expression of IL-6 and IL-8 in human bronchial epithelial cells and peripheral blood mononuclear cells (Citation14).
In contrast to IL-33, IL-6 is one of the best characterized proinflammatory cytokine. Although serum and induced sputum concentrations of IL-6 have been shown to be elevated in both asthma (Citation15,Citation16) and COPD (Citation3,Citation17), IL-6 is rather viewed as typical for COPD. Rincon and Irvin suggested that the presence of IL-6 in the airways of asthma and COPD patients may not only be associated with ongoing inflammation, but may also result from the “activated” state of pulmonary epithelial cells (Citation18).
The nature and severity of airway inflammation can be evaluated using various biological materials. The most reliable data come from studies that used bronchial biopsy samples. However, as obtaining in vivo tissue samples from the lower airways requires relatively invasive procedures, numerous studies investigated the feasibility to assess airway inflammation based on indirect airway samples. These mainly include induced sputum (IS) and exhaled breath condensate (EBC). It is believed that EBC reflects the molecular environment of the lower respiratory tract, whereas IS consists of cells and secretions derived mainly from the large airways (Citation19). To our knowledge, the available data on airway expression of IL-33 in obstructive lung diseases are mainly based on cell cultures and animal models (Citation20,Citation21). The relationships between the tissue expression of IL-33 and its levels in other respiratory specimens have not been adequately evaluated.
Therefore, we undertook a study aimed at evaluating the expression of IL-33 in bronchial mucosa and to compare this expression with IL-33 concentrations and mRNA expressions in various respiratory samples from patients with mild-to-moderate asthma and COPD. In the same samples, tissue expression, mRNA and protein concentration of IL-6 were measured to compare patterns of expression of this well-studied proinflammatory cytokine with the patterns of IL-33 expression.
Materials and methods
This was a prospective, cross-sectional study, which included 22 patients with mild-to-moderate asthma and 33 patients with mild-to-moderate COPD (GOLD stage 1–2). The patients were recruited from an outpatient department of the University Hospital in Warsaw.
The study project was approved by the institutional review board and registered at ClinicalTrial.gov (NCT02069054). All participants signed an informed consent.
Definitions and exclusion/inclusion criteria
The diagnosis of asthma and its severity were established in accordance with the Global Initiative for Asthma (Citation22), the diagnosis of COPD was made in accordance with the Global Initiative for Chronic Obstructive Lung Disease (Citation23).
The following criteria had to be met for inclusion to the asthma study group: (1) signs and symptoms consistent with asthma, (2) spirometric features of airway obstruction with positive bronchial reversibility test, (3) a positive result of methacholine challenge test (PC20< 8 mg/mL). Inclusion criteria for the COPD group were as follows: (1) signs and symptoms consistent with COPD with onset after the age of 40 years, (2) a positive smoking history (>10 pack-years), (3) persistent airway obstruction (FEV1/FVC <5th percentile) in post-bronchodilator spirometry. A common inclusion criterion in asthma and COPD groups was stable disease defined as no exacerbations requiring treatment escalation for at least 6 weeks before the study enrolment.
The number of exacerbations in the past year was recorded and disease control was assessed by Asthma Control Test (ACT) in asthmatics and by the COPD Assessment Test (CAT) in COPD patients. Bronchial obstruction reversibility and airway hyper responsiveness were tested according to the respective European Respiratory Society (ERS), and American Thoracic Society (ATS) recommendations, respectively (Citation24,Citation25). Reversible airflow limitation was defined as post-bronchodilator FEV1/FVC >5th percentile of predicted value, while persistent (‘fixed’) airway obstruction as post-bronchodilator FEV1/FVC <5th percentile, regardless of positive (i.e. FEV1 and/or FVC increase of both >200 mL and >12% of predicted) or negative bronchial reversibility test. The severity of airflow limitation was evaluated in accordance with ATS/ERS recommendations (Citation26). Patients with cigarette smoke exposure less than 10 pack-years were considered as non-smokers, patients who had smoked in the past but denied smoking in one year before the study onset were defined as ex-smokers.
Exclusion criteria for all subjects were inhaled and/or oral steroid use during 6 weeks before the study enrolment, respiratory tract infection and/or asthma/COPD exacerbation within 6 weeks prior to the onset of the study.
Samples collection and processing
Serum samples were obtained by centrifugation of 10 mL of venous blood collected from the peripheral vein. The serum was separated by centrifuging at 1,000 × g for 15 minutes and stored at –70°C.
Sputum induction was preceded by inhalation of 400 μg of salbutamol. Then, inhalations of sterile hypertonic saline (NaCl) were applied at increasing concentrations (3, 4 and 5% solutions) via an ultrasonic nebulizer (ULTRA-NEB™2000, DeVilbiss, Somerset, PA, USA) in accordance with ERS recommendations (Citation27,Citation28). The sputum samples were processed with 0.1% solution of dithiothreitol (DTT) as described previously (Citation3).
Exhaled breath condensate (EBC) was collected with the use of the TURBO-DECCS 09 system (Medivac, Parma, Italy) in concordance with ATS/ERS recommendations (Citation29). The obtained samples were portioned in 250 µL aliquots and immediately stored at -70°C for subsequent analysis.
Flexible bronchoscopy was performed as described earlier (Citation30, Citation31). Two to four bronchial forceps biopsies were taken from the segmental and sub-segmental bronchi of the right lower lobe. Freshly obtained biopsy specimens from bronchial mucosa were fixed in 10% formalin, routinely processed and embedded in paraffin wax.
None of the above samples were repeatedly thawed/frozen for analysis.
Cytokine concentration measurements
IL-6 and IL-33 concentrations in serum, EBC and IS supernatant were measured using standard quantitative sandwich enzyme-linked immunosorbent assay (ELISA) technique according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, USA). The IL-6 assay range and sensitivity were 3.1–300 pg/mL and 0.7 pg/mL, respectively, and for the IL-33 assay, the respective values were 6.3–400 pg/mL and 1.65 pg/mL. The mean minimal detectable concentrations in the used tests were 0.7 pg/mL and 0.519 pg/mL for for IL-6 and IL-33, respectively. Possible effects of 0.1% solution of DTT on the cytokine concentrations in induced sputum were tested and excluded in a preliminary study (Citation32).
Cytokine expression and cellular composition of IS
The differential cell count in IS was determined in May–Grünwald–Giemsa-stained smears based on the morphology of 300 influx cells from various fields. Samples with ≥3% sputum eosinophils were classified as eosinophilic (Citation33,Citation34).
Gene expression analysis was performed using real-time PCR method. Total RNA was isolated from induced sputum cells, using NucleoSpin RNA isolation kit (Macherey–Nagel GmbH & Co, Düren, Germany) and converted to cDNA using a GoTaq® qPCR Master Mix (Promega Corporation, Madison, WI, USA). Target mRNA was quantified by real-time PCR assay using specific fluorogenic probes and primer sets and the PCR Master Mix system (Applied Biosystems, Foster City, CA, USA). All probes were designed to span introns and did not react with genomic DNA. Samples were assayed with the 7500 Real-Time PCR System and analysed using 7500 System Software (Applied Biosystems, Foster City, CA, USA). For tested samples Ct (threshold cycle) values were normalized to 18sRNA mRNA and expressed as fold change relative to the mean Ct value for the non-COPD non-asthma control samples using the ∆∆Ct method.
Cells and cytokine expression in bronchial biopsy samples
Immunohistochemical staining with monoclonal antibodies for: CD4 clone 4B12 (M731001-2, dilution 1:20, Dako Denmark A/S, Glostrub, Denmark), CD8 clone C8/144B (M7103, dilution 1:20, Dako Denmark A/S, Glostrub, Denmark), and polyclonal antibodies for: IL6 (ab6672, dilution 1:500, Abcam, Cambridge, United Kingdom) and IL33 (ab118503, dilution:5 µg/mL, Abcam, Cambridge, United Kingdom) was applied to 5 µm thick paraffin-embedded sections according to the manufacturer's instructions. The EnVision Detection System (Dako Denmark A/S, Glostrub, Denmark) was used for detection. Human tonsil biopsies were used as positive staining controls for CD4, CD8, IL6 and IL33. Negative (isotype) controls were performed using ready to use FLEX Negative Control Mouse (cocktail of mouse IgG1, IgG2a, IgG2b, IgG3 and IgM; code nr IR750; Dako Denmark A/S, Glostrub, Denmark). All cases were photographed at x200 magnification and analysed with the acquisition software of the CellSens package (Olympus, Tokyo, Japan). To quantify the number of inflammatory cells in bronchial mucosa, all slides were assessed to select the section with the most prominent inflammatory infiltrates and this section was used to evaluate the number of eosinophils, CD4+ and CD8+ cells in an area of 0.948 mm2. The threshold defining eosinophilic inflammation in the bronchial biopsy specimens was ≥2 eosinophils/mm2 (Citation35).
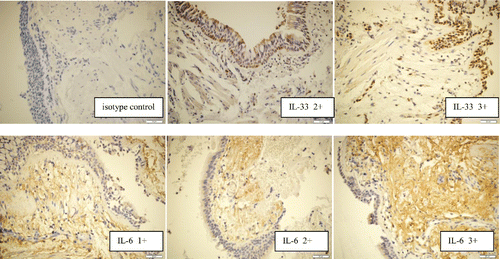
IL-6 and IL-33 protein expressions in the bronchial biopsy specimens were evaluated with a semi-quantitative method based on the intensity of immunostainings (Citation35) on extracellular space and extracellular matrix for IL-6, cell nucleus for IL-33 ().
Figure 1. IL-6 and IL-33 protein expression in bronchial mucosa samples, examples of rating based on the intensity of immunostaining (control – no staining; 1+, weak; 2+, intermediate; and 3+, strong).
Statistical analysis
Estimation of the sample size was based on data from an earlier study by Grubek-Jaworska et al (Citation32). We assumed that the difference between sputum IL-6 level in asthma and COPD which should be detected would be at least 2-fold lower than that found by Grubek-Jaworska et al. To detect the difference with a power of 80% and a significance level of 5%, the sample size was estimated as 47 patients (19 asthmatics and 28 COPD patients).
Data are expressed as medians and interquartile ranges (IQRs, 25th to 75th percentiles) or numbers and percentages. Statistical analysis was performed using Statistica 10.0 (StatSoft Inc., Tulsa, OK, USA) and MedCalc Statistical Software version 13.2.2 (MedCalc Software bvba, Ostend, Belgium). Quantitive data distribution was assessed by using the Shapiro–Wilk test. The differences between continuous variables in two or more groups were tested using the non-parametric Mann–Whitney U-test or Kruskal–Wallis test, respectively. Categorical variables were compared using chi-squared test or Fisher's exact test. The strength and direction of the relationship between two variables was measured with Spearman‘s rank correlation coefficient. Statistical significance was accepted at p-value less than 0.05.
Results
Patient characteristics
Comparative characteristics of patients with asthma and COPD are presented in . Mild persistent asthma was diagnosed in 16 (73%) asthmatics and moderate asthma in the remaining 6 (27%) patients. Mild and moderate airflow limitation (consistent with GOLD stage 1 and 2) was diagnosed in 9 (27%) and 24 (73%) COPD patients, respectively. In the asthma group, 19 patients were treated with a short acting beta-agonist (SABA) and 3 patients with long acting beta2-agonist (formoterol) on demand. In the COPD group, 6 patients regularly received a long acting muscarinic antagonist (LAMA) in monotherapy, 6 – long acting beta2-agonist (LABA) only and 10 – SABA as needed, 8 patients were treated with a combination of LABA and LAMA, and finally 3 patients received LABA and short acting muscarinic antagonists.
Table 1. Clinical characteristics of patients with asthma and COPD.
IL-33 and IL-6 concentration in various samples
In both study groups the highest concentrations of IL-6 and IL-33 were detected in IS (). The median sputum level of IL-6 was 6-fold higher than that found in serum, and 10-fold higher than that in EBC (p < 0.001 for both comparisons). In all specimens, IL-33 concentration and mRNA expression were significantly lower than IL-6 protein level and mRNA expression ().
Table 2. Cytokine expression and cells count in different biological samples from patients with asthma and COPD.
Figure 2. Concentrations of IL-6 and IL-33 in serum, induced sputum (IS), and exhaled breath condensate (EBC) from patients with asthma and chronic obstructive pulmonary disease (COPD).
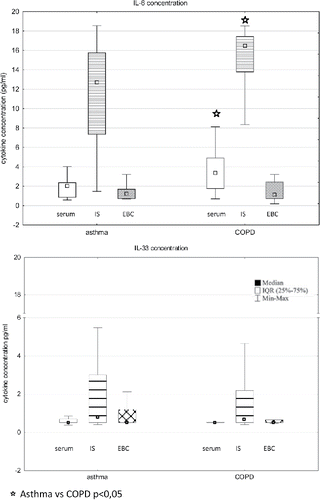
COPD patients showed significantly higher serum and induced sputum IL-6 concentration than asthmatics. The serum, IS and EBC IL-33 concentration was on comparable level in both study groups. A significant correlation between IS IL-6 and IL-33 concentration was found in asthmatics (r = 0.49, p < 0.05). In the COPD group, EBC IL-6 level correlated inversely with EBC IL-33 (r = −0.48, p < 0.05). There were no other significant correlations between IL-6 and IL-33 levels in both studied groups ().
Table 3. Spearman correlation coefficients for relationships between the levels/expression of IL-6 and IL-33 in the investigated materials from patients with asthma.
Table 4. Spearman correlation coefficients for relationships between the levels/expression of IL-6 and IL-33 in the investigated materials from patients with chronic obstructive pulmonary disease (COPD).
Tissue IL-33 and IL-6 protein expression
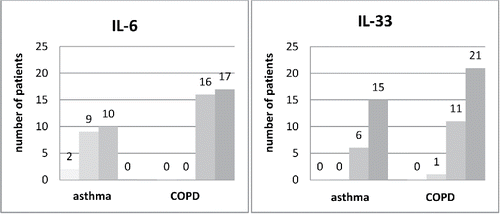
Bronchial tissue samples of adequate quality were obtained in 21 asthma and 33 COPD patients. The degree of IL-6 expression in bronchial mucosa samples differed significantly in asthmatics and COPD patients, p < 0.001. In 17 (51.5%) and 16 (48.5%) COPD patients mucosal expression of IL-6 protein was rated as 3+ and 2+, respectively. In contrast, none of the asthmatics showed mucosal IL-6 protein expression of 3+ and 11/21 (52%) patients presented IL-6 expression of 0 or 1+. High mucosal IL-33 expression was found in a significant percentage of asthma and COPD patients. In 15 (71.5%) and 6 (28.5%) of asthma patients and in 21 (64%) and 11 (33%) of COPD patients it was rated as 3+ and 2+, respectively ().
Figure 3. Distribution of tissue IL-6 and IL-33 protein expression in bronchial mucosa biopsy samples in patients with asthma (n = 21) and chronic obstructive pulmonary disease (n = 33). The X-axis presents the intensity of the immunostainings (■ 0, ■1+,■ 2+,■ 3+) separately for asthma and COPD groups.
In asthma, a significant positive correlation between IL-6 and IL-33 protein tissue expression was observed (r = 0.49, p < 0.05). In COPD patients, mucosal IL-6 protein expression correlated with serum IL-6 level (r = −0.42, p < 0.05), and with the number of mucosal CD8+ cells (r = 0.4, p < 0.05). No other significant correlations between the tissue expression of both cytokines and their concentration in the investigated materials were found.
IL-33 and IL-6 mRNA expression in induced sputum cells
There were no significant differences in IS mRNA expression of IL-33 and IL-6 between asthma and COPD ().
A strong correlation between tissue IL-6 protein expression and IS mRNA for IL-6 in the COPD group was found (r = 0.75, p < 0.05). In COPD, a correlation between IS IL-33 mRNA expression and lung tissue CD4+ and CD8+ lymphocyte count was found (r = −0.69 and r = −0.68, respectively, p < 0.05 for both).
Relationships between IL-33 and IL-6 levels with peripheral/airway eosinophil and neutrophil count, tobacco exposure and pulmonary function
In COPD, serum IL-6 concentration correlated inversely with IS eosinophilia (r = −0.36 and r = −0.49 for eosinophil absolute count and percentage, respectively, p < 0.05). We failed to show any relationship between IL-33 concentration/expression and eosinophils in COPD patients (). In asthma, a negative correlation between EBC IL-6 concentration and sputum eosinophil percentage was found and IS IL-33 correlated positively with tissue eosinophilia ()
In COPD patients, IS neutrophil count was significantly higher than in asthma patients and correlated positively with FEV1 (r = 0.45, p < 0.05) and FVC (r = 0.41, p < 0.05). In asthma patients, peripheral and airway neutrophil count correlated negatively with peripheral/ airway eosinophils (r = −0.59 and r = −0.5, p < 0.05 for both). There were no correlations between serum and IS neutrophil count and IL-6 and IL-33 levels in both investigated groups.
We found a positive correlation between sputum IL-6 level and the number of pack-years (r = 0.4, p < 0.05) in COPD patients. No relationship between IL-33 level and smoking history was found in both groups of patients.
We did not demonstrate any significant relationships between the concentration/expression of both investigated cytokines and pulmonary function in separate analyses for asthma and COPD (this refers to all the spirometrical as well as lung volume indices). However, when both groups were analyzed together, serum IL-6 correlated with FEV1 (r = −0.3, p < 0.05), FVC (r = −0.28, p < 0.05), FEV1/FVC (r = −0.38, p < 0.05) and RV (r = 0.41, p < 0.05). Also, there was a correlation between IS IL-6 and FEV1/FVC (r = −0.39, p < 0.05) as well as RV (r = 0.27, p < 0.05). No such correlations between pulmonary function and IL-33 were found.
No differences in IL-6 and IL-33 were found between mild and moderate stages of both asthma and COPD.
Discussion
The present study demonstrated that IL-33 is detectable in serum, IS and EBC not only in patients with asthma, but also in patients with COPD. IL-33 and IL-6 concentrations in IS were significantly higher than those in serum and EBC. Interestingly, IL-33 expression in bronchial samples was high not only in asthmatics, but also in COPD patients. Thus, we believe our study provides some new data on the immunopathological and inflammatory phenotyping of asthma and COPD. To our knowledge, this is the first study comparing IL-33 expression in bronchial walls with IL-33 concentration in EBC and IS. This is also the first study directly comparing IL-33 expression/concentration in patients with asthma and COPD. As we were not able to find any other data on IL-33 concentrations in IS and EBC in COPD patients, we believe this is the first study addressing this issue. We found only one recent study that evaluated IL-33 and TSLP concentrations in EBC from asthmatics (Citation36). It should also be emphasized that our study was performed in patients with the mild-to-moderate stage of the two diseases and not treated with steroids for at least 6 weeks. This is in contrast with the majority of previous studies that had been performed in patients with severe/refractory asthma or COPD receiving steroids or other anti-inflammatory agents.
IL-33 is mainly expressed in the nuclei of various cell types, including epithelial and endothelial cells, and fibroblasts that respond to specific stimuli (Citation37). IL-33 may act as a nuclear alarmin for innate immunity after damage, or infection of epithelial barriers (Citation38). Some authors postulated that the expression of membrane-bound IL-33 receptor (IL-33R) is restricted to hematopoietic cells, particularly Th2 lymphocytes, mast cells, eosinophils and basophils (Citation39). IL-33 promotes Th2 immunity and systemic inflammation (Citation12,Citation40) and is associated with various inflammatory diseases, such as rheumatoid arthritis, atopic allergy, asthma, and cardiovascular diseases (Citation41). Much less data on the role of IL-33 in COPD are available, especially in the mild-to-moderate stage. Some authors suggested that IL-33 would be cleaved by caspase-1; however, this hypothesis was based on studies in murine models and site is not conserved between humans and mice (Citation42). Moreover, the cleavage products are biologically less active than the precursor (Citation43). Therefore, the translation of the results obtained in experiments on mice might be difficult.
Earlier human studies demonstrated that bronchial epithelium and smooth muscle are an important IL-33 reservoir in the lung and that the expression of IL-33 is elevated in asthma (Citation13,Citation14,Citation20). Histologic observations supported the intracellular and nuclear localization of IL-33 in epithelial cells (Citation44). The role of IL-33 has been widely investigated in asthma, contrary to COPD. Recent studies have demonstrated that IL-33 level in sputum, serum as well as its tissue expression correlated with asthma severity (Citation12,Citation45,Citation46). We were not able to confirm this relationship, either in asthma or in COPD as we failed to find any correlations between IL-33 and FEV1 or disease severity criteria (as defined in GINA and GOLD). In contrast to the previous studies, our study involved only patients with mild and moderate, but not severe disease. We cannot exclude that some correlations between IL-33 and disease severity would have emerged if we had also investigated patients in severe stages of asthma and COPD. It should be emphasized that the majority of earlier measurements were performed in serum and biopsy material, only a few studies evaluated induced sputum (Citation45), and even less assessed EBC (Citation36). Gluck et al. showed that in asthmatics not only IL-33 level, but also TSLP was increased in EBC (Citation36). Hamzaoie et al. demonstrated that sputum IL-33 concentration and mRNA expression was significantly higher in children with mild-to-moderate asthma compared with healthy controls (Citation45).
There are only few reports on IL-33 concentration in COPD. Recent human and murine studies suggest that IL-33 also plays a role in this disease. Xia et al. reported elevated IL-33 expression in peripheral blood and airways of patients with COPD, compared with controls (Citation37). Byers et al. have demonstrated that lung levels of IL-33 were increased in mice with chronic obstructive lung disease and in humans with very severe COPD (Citation20). Several studies have demonstrated that IL-33 is highly expressed in epithelial cells from many tissues, including airways (Citation37,Citation47) in response to contact with allergens, pathogens, tobacco smoke, and other common environmental agents (Citation14,Citation44). We found no relationship between IL-33 level and smoking history in our patients. This observation may at least partially be explained by the fact that 74.5% of the investigated patients were ex-smokers and IL-33 may not have been markedly elevated because they were not currently exposed to cigarette smoke.
The current study provided the first data directly comparing the concentration of IL-33 in asthma and COPD patients. Serum, IS, EBC concentrations of IL-33 were comparable in asthma and COPD. This could be due to the fact that IL-33 is not only associated with Th2-type immune response, but can also regulate Th1-type cytokine involvement in the immune response. This was shown by Bourgeois et al. (Citation48), who reported the ability of IL-33 to promote a Th1 cytokine profile in a mouse model. Our results also indicate that IL-33 concentration in all tested materials and mRNA expression in IS cells may not differentiate between asthma and COPD, in contrast to IL-6. This is confirmed by the similar IL-33 expression in IS cells from patients with asthma and COPD and is consistent with the results of our previous studies (Citation3,Citation49).
One of the easily measurable clinical features of obstructive lung diseases is the increased number of inflammatory cells, mainly eosinophils. In our study the number of eosinophils in all materials was statistically higher in asthma than in COPD group, and in the asthma group, tissue eosinophilia positively correlated with IL-33 level in IS. We cannot exclude that IL-33 contributes to eosinophil chemotaxis to the airways and lung tissue what may induce a vicious circle due to the further production of inflammatory mediators by eosinophils. We failed to show any relationship between IL-33 concentration/expression and eosinophils in patients with COPD. Kim et al. showed a positive correlation between peripheral eosinophil count and plasma IL-33 level in COPD patients (Citation50), this has also been reported in asthma animal models (Citation10).
The second cytokine evaluated in our study was IL-6. There were at least two arguments supporting the rationale for combining measurements of IL-33 with those of IL-6. In contrast to IL-33, which, according to the literature, seemed to be associated with asthma, IL-6 is a pleiotropic proinflammatory cytokine, which plays an important role in the pathogenesis of COPD. One of our previous studies demonstrated significantly increased IL-6 levels in induced sputum from COPD patients when compared to asthmatics (Citation32). In a different study, elevated levels of IL-6 in serum and sputum of COPD patients were associated with the degree of lung function impairment (Citation51). He et al. found high levels of IL-6 and 8-iso-prostaglandin in EBC and serum from patients with COPD and hypothesized that this phenomenon could be related to pulmonary hypertension (Citation52). Thus, performing the study on IL-33 in asthma and COPD, we wanted to have comparative data on one reference cytokine that is typical for the inflammatory process in COPD. The second argument was that some data suggested the relationship between IL-33 and IL-6. The study by Moulin et al. showed that IL-33 stimulated the production of pro-inflammatory mediators, including IL-6, IL-1b, TNF-a, MCP-1 and PGD2 by mouse mast cells (Citation53). The effect of IL-33 appeared to be independent of mast cell degranulation; although it cannot be excluded that IL-33 could modulate degranulation in a more physiological context, such as, for instance, after sensitization by IgE. In the current study, we found a positive correlation between IS IL-6 and IL-33 in the asthma group. In the COPD group, we found a negative correlation between IL-6 and IL-33 levels in EBC, in contrast to the other tested materials. Of note, the concentration of IL-33 did not differ between asthma and COPD. It may be hypothesized in COPD, IL-33 secretion may be activated in a different pathway which, in contrast to the pathway in asthma, is not eosinophil-dependent. However, these considerations are purely assumptive and based only on the lack of correlations between eosinophils and IL-33 levels/expression in our study. It must be emphasized that the investigated materials reflect different compartments and EBC content may not fully correspond with the cellular composition of IS and peripheral blood.
We realize several limitations of our study. First, it would be more interesting to assess the expression of TSLP and IL-25 in addition to IL-33 and IL-6, which would allow for a broader view on the role of epithelial derived cytokines in obstructive lung diseases. We plan to perform IL-25 and TSLP tissue expression assays in the biopsy materials in the near future. Second, all enrolled participants were diagnosed with mild-to-moderate degree of asthma or COPD, which was due to the fact that in severe stages of both diseases, sputum induction would not have been possible because of the risk of complications related with the procedure. It is possible that in patients with severe asthma or COPD, the expression level of the studied cytokines and its correlation with the investigated factors could be different. Another limitation is that the immunohistochemical staining with monoclonal antibodies is a semi-quantitative method, which allows the tissue expression of cytokines to be defined only at a low-high level, based on the degree of immunostaining intensity.
Conclusions
IL-33 and IL-6 are detectable in serum, IS and EBC not only in asthma patients, but also in patients with COPD. In the COPD group, serum and IS IL-6 concentrations were statistically higher than in the asthma group. The tissue expression of IL-33 and IL-33 concentrations in the investigated biological samples were on comparable level in both obstructive airway diseases. Our findings may suggest that IL-33 activation is a common pathway in asthma and COPD.
Declaration of interest statement
The authors declare no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Author contributions
KG, PNG and RK designed the study. KG and PNG were responsible for literature search, patient recruitment, data analysis and wrote the first draft. PK and RK performed bronchoscopy procedures. PNG carried out the cytological and cytokines concentration evaluations. MSP performed histological studies in bronchial biopsy specimens and was involved in data analysis and interpretation. MPG did the preliminary study in ELISA and PCR measurements and performed the induced sputum processing. All authors critically reviewed the manuscript and contributed to the final version.
Additional information
Funding
References
- Culpitt SV, Rogers DF, Traves SL, Barnes PJ, Donnelly LE. Sputum matrix metalloproteases: comparison between chronic obstructive pulmonary disease and asthma. Respir Med 2005;99(6):703–10. https://doi.org/10.1016/j.rmed.2004.10.022.
- Fabbri LM, Romagnoli M, Corbetta L, Casoni G, Busljetic K, Turato G, Ligabue G, Ciaccia A, Saetta M, Papi A. Differences in airway inflammation in patients with fixed airflow obstruction due to asthma or chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003;167(3):418–24. https://doi.org/10.1164/rccm.200203-183OC.
- Górska K, Paplińska-Goryca M, Nejman-Gryz P, Goryca K, Krenke R. Eosinophilic and neutrophilic airway inflammation in the phenotyping of mild-to-moderate asthma and chronic obstructive pulmonary disease. Journal of Chronic Obstructive Pulmonary Disease. 2017; 14 (2);181–189, DOI:10.1080/15412555.2016.1260539 COPD.
- Agusti A, Bel E, Thomas M, Vogelmeier C, Brusselle G, Holgate S, Humbert M, Jones P, Gibson PG, Vestbo J. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J 2016;47(2):410–9. https://doi.org/10.1183/13993003.01359-2015.
- Ghebre MA, Bafadhel, M, Desai Dhananjay, Cohen, Suzanne E. Biological clustering supports both “Dutch” and “British” hypotheses of asthma and chronic obstructive pulmonary disease – Journal of Allergy and Clinical Immunology. The Journal of Allergy and Clinical Immunology. 2015;135(1):63–72.e10. doi:10.1016/j.jaci.2014.06.035.
- Bafadhel M, McKenna S, Terry S, Mistry V, Pancholi M, Venge P, Lomas DA, Barer MR, Johnston SL, Pavord ID. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med 2012;186(1):48–55. https://doi.org/10.1164/rccm.201108-1553OC.
- Mirchandani AS, Salmond RJ, Liew FY. Interleukin-33 and the function of innate lymphoid cells. Trends Immunol 2012;33(8):389–96. https://doi.org/10.1016/j.it.2012.04.005.
- Konya V, Mjösberg J. Lipid mediators as regulators of human ILC2 function in allergic diseases. Immunol Lett 2016;179:36–42. https://doi.org/10.1016/j.imlet.2016.07.006.
- Kaur D, Gomez E, Doe C, Berair R, Woodman L, Saunders R, Hollins F, Rose FR, Amrani Y, May R. IL-33 drives airway hyper-responsiveness through IL-13-mediated mast cell: Airway smooth muscle crosstalk. Allergy 2015;70(5):556–67. https://doi.org/10.1111/all.12593.
- Lee HY, Rhee CK, Kang JY, Byun JH, Choi JY, Kim SJ, Kim YK, Kwon SS, Lee SY. Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Exp Lung Res 2014;40(2):66–76. https://doi.org/10.3109/01902148.2013.870261.
- Togbe D, Fauconnier L, Madouri F, Marchiol T, Chenuet P, Rouxel N, Ledru A, Erard F, Quesniaux V, Ryffel B. Thymic stromal lymphopoietin enhances th2/th22 and reduces il-17a in protease-allergen-induced airways inflammation. Allergy 2013; Article ID 971036, 14 pages https://doi.org/10.1155/2013/971036 ISRN Allergy 2013.
- Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol 2009;183(8):5094–103.
- Préfontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, Martin JG, Hamid Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol 2010;125(3):752–4. https://doi.org/10.1016/j.jaci.2009.12.935.
- Shang J, Zhao J, Wu X, Xu Y, Xie J, Zhao J. Interleukin-33 promotes inflammatory cytokine production in chronic airway inflammation. Biochem Cell Biol 2015;93(4):359–66. https://doi.org/10.1139/bcb-2014-0163.
- Neveu WA, Allard JL, Raymond DM, Bourassa LM, Burns SM, Bunn JY, Irvin CG, Kaminsky DA, Rincon M. Elevation of IL-6 in the allergic asthmatic airway is independent of inflammation but associates with loss of central airway function. Respir Res 2010;11:28. https://doi.org/10.1186/1465-9921-11-28.
- Naik SP, P A M, B S J, Madhunapantula SV, Jahromi SR, Yadav MK. Evaluation of inflammatory markers interleukin-6 (IL-6) and matrix metalloproteinase-9 (MMP-9) in asthma. J Asthma 2016; 54(6):584–593. doi: 10.1080/02770903.2016.1244828. 1–10.
- Sakthivel P, Breithaupt A, Gereke M, Copland DA, Schulz C, Gruber AD, et al. Soluble CD200 correlates with interleukin-6 levels in sera of COPD patients: potential implication of the cd200/cd200r axis in the disease course. Lung 2016;195(1):59–68. doi: 10.1007/s00408-016-9962-4.
- Rincon M, Irvin CG. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci 2012;8(9):1281–90. https://doi.org/10.7150/ijbs.4874.
- Mazur W, Stark H, Sovijärvi A, Myllärniemi M, Kinnula VL. Comparison of 8-isoprostane and interleukin-8 in induced sputum and exhaled breath condensate from asymptomatic and symptomatic smokers. Respiration 2009;78(2):209–16.
- Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, Wu K, You Y, Alevy Y, Girard JP. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest 2013;123(9):3967–82. https://doi.org/10.1172/JCI65570.
- Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, Mori M, Pham TH, Ward CK, Criner GJ. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 2015;42(3):566–79. https://doi.org/10.1016/j.immuni.2015.02.011.
- GINA_Report_2010_1.pdf. February 12, 2016. Available from: http://www.ginasthma.org/local/uploads/files/GINA_Report_2010_1.pdf
- GOLDReport_April112011.pdf. February 12, 2016. Available from: http://www.goldcopd.org/uploads/users/files/GOLDReport_April-112011.pdf
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P. Standardisation of spirometry. Eur Respir J 2005;26(2):319–38. https://doi.org/10.1183/09031936.05.00034805.
- Crapo RO, Casaburi R, Coates AL, Enright PL, Hankinson JL, Irvin CG, et al. Guidelines for methacholine and exercise challenge testing-1999. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med 2000;161(1):309–29.
- Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, Coates A, van der Grinten CP, Gustafsson P, Hankinson J. Interpretative strategies for lung function tests. Eur Respir J 2005;26(5):948–68. https://doi.org/10.1183/09031936.05.00035205.
- Vignola AM, Rennar SI, Hargreave FE, Fah JV, Bonsignore MR, Djukanović R, et al. Standardised methodology of sputum induction and processing. Future directions. Eur Respir J Suppl 2002;37:51s–55s.
- Djukanović R, Sterk PJ, Fahy JV, Hargreave FE. Standardised methodology of sputum induction and processing. Eur Respir J Suppl 2002;37:1s–2s. https://doi.org/10.1183/09031936.02.00000102.
- Horváth I, Hunt J, Barnes PJ, Alving K, Antczak A, Baraldi E, Becher G, van Beurden WJ, Corradi M, Dekhuijzen R. Exhaled breath condensate: methodological recommendations and unresolved questions. Eur Respir J 2005;26(3):523–48. https://doi.org/10.1183/09031936.05.00029705.
- Górska K, Maskey-Warzęchowska M, Nejman-Gryz P, Korczyński P, Prochorec-Sobieszek M, Krenke R. Comparative study of periostin expression in different respiratory samples in patients with asthma and chronic obstructive pulmonary disease. Pol Arch Med Wewn 2016;126(3):124–37.
- Gorska K, Korczynski P, Mierzejewski M, Kosciuch J, Zukowska M, Maskey-Warzechowska M, Krenke R. Comparison of endobronchial ultrasound and high resolution computed tomography as tools for airway wall imaging in asthma and chronic obstructive pulmonary disease. Respir Med 2016;117:131–8. https://doi.org/10.1016/j.rmed.2016.06.011.
- Grubek-Jaworska H, Paplińska M, Hermanowicz-Salamon J, Białek-Gosk K, Dąbrowska M, Grabczak E, et al. IL-6 and IL-13 in induced sputum of COPD and asthma patients: correlation with respiratory tests. Respiration 2012;84(2):101–7.
- Hastie AT, Moore WC, Li H, Rector BM, Ortega VE, Pascual RM, Peters SP, Meyers DA, Bleecker ER, National Heart. Biomarker surrogates do not accurately predict sputum eosinophil and neutrophil percentages in asthmatic subjects. J Allergy Clin Immunol 2013;132(1):72–80. https://doi.org/10.1016/j.jaci.2013.03.044.
- Berry M, Morgan A, Shaw DE, Parker D, Green R, Brightling C, Bradding P, Wardlaw AJ, Pavord ID. Pathological features and inhaled corticosteroid response of eosinophilic and non-eosinophilic asthma. Thorax 2007;62(12):1043–9. https://doi.org/10.1136/thx.2006.073429.
- Taylor CR, Levenson RM. Quantification of immunohistochemistry–issues concerning methods, utility and semiquantitative assessment II. Histopathology 2006;49(4):411–24. https://doi.org/10.1111/j.1365-2559.2006.02513.x.
- Glück J, Rymarczyk B, Kasprzak M, Rogala B. Increased levels of interleukin-33 and thymic stromal lymphopoietin in exhaled breath condensate in chronic bronchial asthma. Int Arch Allergy Immunol 2016;169(1):51–6. https://doi.org/10.1159/000444017.
- Xia J, Zhao J, Shang J, Li M, Zeng Z, Zhao J, Wang J, Xu Y, Xie J. Increased IL-33 expression in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 2015;308(7):L619–627. https://doi.org/10.1152/ajplung.00305.2014.
- Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol 1950. 2012;188(7):3488–95.
- Drube S, Heink S, Walter S, Löhn T, Grusser M, Gerbaulet A, et al. The receptor tyrosine kinase c-Kit controls IL-33 receptor signaling in mast cells. Blood 2010;115(19):3899–906. https://doi.org/10.1182/blood-2009-10-247411.
- Hayakawa H, Hayakawa M, Kume A, Tominaga S. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem 2007;282(36):26369–80. https://doi.org/10.1074/jbc.M704916200.
- Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol 2010;10(2):103–10. https://doi.org/10.1038/nri2692.
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005;23(5):479–90. https://doi.org/10.1016/j.immuni.2005.09.015.
- Cayrol C, Girard J-P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA 2009;106(22):9021–6. https://doi.org/10.1073/pnas.0812690106.
- Moussion C, Ortega N, Girard J-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PloS One 2008;3(10):e3331. https://doi.org/10.1371/journal.pone.0003331.
- Hamzaoui A, Berraies A, Kaabachi W, Haifa M, Ammar J, Kamel H. Induced sputum levels of IL-33 and soluble ST2 in young asthmatic children. J Asthma 2013;50(8):803–9. https://doi.org/10.3109/02770903.2013.816317.
- Guo Z, Wu J, Zhao J, Liu F, Chen Y, Bi L, Liu S, Dong L. IL-33 promotes airway remodeling and is a marker of asthma disease severity. J Asthma 2014;51(8):863–9. https://doi.org/10.3109/02770903.2014.921196.
- Kurokawa M, Matsukura S, Kawaguchi M, Ieki K, Suzuki S, Odaka M, Watanabe S, Homma T, Sato M, Yamaguchi M. Expression and effects of IL-33 and ST2 in allergic bronchial asthma: IL-33 induces eotaxin production in lung fibroblasts. Int Arch Allergy Immunol 2011;155 Suppl 1:12–20. https://doi.org/10.1159/000327259.
- Bourgeois E, Van LP, Samson M, Diem S, Barra A, Roga S, Gombert JM, Schneider E, Dy M, Gourdy P. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol 2009;39(4):1046–55. https://doi.org/10.1002/eji.200838575.
- Paplińska-Goryca M, Nejman-Gryz P, Górska K, Białek-Gosk K, Hermanowicz-Salamon J, Krenke R. Expression of inflammatory mediators in induced sputum: comparative study in asthma and COPD. Advs Exp. Medicine, Biology - Neuroscience and Respiration 2016;1–13. DOI 10.1007/5584_2016_165
- Kim SW, Rhee CK, Kim KU, Lee SH, Hwang HG, Kim YI, Kim DK, Lee SD, Oh YM, Yoon HK. Factors associated with plasma IL-33 levels in patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2017;12:395–402. https://doi.org/10.2147/COPD.S120445.
- Donaldson GC, Seemungal TAR, Patel IS, Bhowmik A, Wilkinson TMA, Hurst JR, Maccallum PK, Wedzicha JA. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest 2005;128(4):1995–2004. https://doi.org/10.1378/chest.128.4.1995.
- He H, Tao Y, Chen X, Qiu H, Zhu J, Zhang J, et al. High levels of interleukin-6 and 8-iso-prostaglandin in the exhaled breath condensate and serum of patients with chronic obstructive pulmonary disease related pulmonary hypertension. Chin Med J (Engl) 2014;127(9):1608–12.
- Moulin D, Donzé O, Talabot-Ayer D, Mézin F, Palmer G, Gabay C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007;40(3):216–25. https://doi.org/10.1016/j.cyto.2007.09.013.