
Abstract
Chronic obstructive pulmonary disease (COPD) is a common airway disease, and epithelial mesenchymal transition (EMT) is participated in the pathogenesis of COPD. However, the role of CD147 in COPD remains largely unknown. In order to clarify the role of CD147 in EMT induced by cigarette smoke, we established animal and cell model of EMT by mean of cigarette smoke exposure and detected the expressions of CD147 and EMT markers via PCR, WB and IF. RNA inference was applied to study the role of CD147 in CSE induced EMT in vitro. NAC and H2O2 were used to study oxidative stress signaling pathway in this model. As a result, cigarette smoke exposure upregulated the expressions of CD147, α-SMA, and Vimentin and downregulated the expression of Ecadherin and ZO1 both in vivo and in vitro, which was accompanied by augmented level of oxidative stress. CD147 knockdown would partly inhibit CSE induced EMT, while preincubation of H2O2 could inverse this effect. In conclusion, CD147 promoted EMT in mice and HBE cells induced by cigarette smoke via oxidative stress signaling pathway.
Introduction
Chronic obstructive pulmonary disease (COPD) is a common airway disease with classical symptoms of persistent cough, sputum and breathlessness due to airway inflammation and fibrosis [Citation1]. Nowadays, the regular drugs used for COPD therapy include inhaled corticosteroids (ICS), long-acting β2 adrenergic receptor agonists (LABA), and long-acting muscarinic receptor antagonists (LAMA) etc. [Citation2]. However, a large amount of patients cannot control their daily symptoms even after they received standard therapies. So there is an urgent need for exploring novel drug targets and developing more effective drugs.
As a complex disease, the underlying mechanisms of COPD have not been fully elucidated. Emerging evidences suggested that epithelial mesenchymal transition (EMT) was participated in the pathogenesis of COPD [Citation3]. EMT is a biological process during which epithelial cells lost apical-basal polarity and tight junctions, converted into a different morphology with migration and motile ability. The expressions of biomarkers from epithelial cells decrease, while the expressions of biomarkers from mesenchymal cells increase in this process [Citation4]. In a clinical study performed by Milara et al., the lower expressions of epithelial markers and higher expressions of mesenchymal markers were observed in bronchial epithelial cells derived from COPD patients compared with those from healthy subjects [Citation5]. Sohal et al. found EMT existed in both large and small airways in COPD patients, especially in smoking patients [Citation6].
CD147 is a kind of glycoprotein which belongs to immunoglobulin superfamily. So far, CD147 has been reported to play important roles in cancer, cardiac vascular diseases and autoimmune diseases etc. [Citation7]. Previous studies showed that CD147 regulated EMT in various kinds of cancer cells [Citation8–11], but it remained largely unknown that whether CD147 was participated in EMT process in normal cells, especially in normal airway epithelial cells. There were numerous studies about CD147 and lung cancer [Citation12–16], however, only a small number of studies focused on the relationship between CD147 and COPD [Citation17, Citation18], even if the two diseases shared the same risk factor. Given the fact that EMT was considered as a pivotal link in COPD, we wondered if CD147 could regulate EMT process in COPD. To testify our hypothesis, we carried out a series of experiments both in vitro and in vivo to clarify the role of CD147 in cigarette smoke induced EMT.
Materials and methods
Animals
C57BL/6 mice (male, 8–10 weeks) were purchased from Slac Laboratory Animal Center (Shanghai, China) and raised in the Laboratory Animal Center of Zhejiang Industry University. All experimental protocols were approved by the Ethical Committee for Animal Studies of Hangzhou Medical College.
Cell line and cell culture
Human bronchial epithelial (HBE) cells were purchased from American Type Culture Collection (ATCC), and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) in a 37 °C, 5% CO2 incubator. After reaching more than 90% confluence, cells were in serum-free medium for 12 h to synchronize the cell cycle and to return serum activated signaling pathways to basal levels before exposure to cigarette smoke extract (CSE) or other reagents.
Reagents
Monoclonal antibodies against β-Actin (sc-58673), GAPDH (sc-32233) were from Santa Cruz Biotechnology, Monoclonal antibodies against CD147 (ab108317 for human and ab188190 for mouse), Ecadherin (ab40772) and α-SMA (ab32575) were from Abcam. Polyclonal antibodies against ZO1 (21773-1-AP) and Vimentin (10336-1-AP) were from Proteintech. Small interfering RNA (siRNA) targeted for CD147 were designed and synthesized by GenePharma (Shanghai). N-Acetyl-l-cysteine (NAC, A9165) and Hydrogen peroxide (H2O2) solution (88597) were purchased from sigma.
Cigarette smoke exposure protocol
Twelve mice were divided into two groups: the cigarette smoke-exposed group (CS) and the control group (CON). Each group contained 6 mice. A custom-designed smoking chamber (60 cm × 50 cm × 40 cm) was used for establishing the mouse model. In this study, we made a few adjustments to the smoking protocol referring to the one set up by Li et al. [Citation19]. Briefly, the mice were placed into the chamber and exposed to the smoke of 10 cigarettes (Marlboro, containing 0.8 mg of nicotine and 11 mg of tar per cigarette) twice a day. The total exposure time was near 2 h a day, and the mice were exposed 6 days/week for 12 weeks. The control group was exposed to filtered air. After the last exposure, all the mice were sacrificed. The total particulate matter concentrations measured in the chamber were 160–180 mg/m3.
Cigarette smoke extract preparation
Ten cigarettes (Marlboro) were slowly bubbled through the 50 ml RPMI 1640 medium (Invitrogen) with the help of a peristaltic pump to prepare cigarette smoke solution. The obtained CS solution was defined as 100% CSE. Then the solution was filtered, divided into several aliquots and stored at −80 °C until use.
siRNA studies
After reaching 50% confluence, HBE were transfected with siRNA at 5 nM with GeneMute TM (SignaGen Laboratories), according to the manufacturer’s protocols. After 24 h siRNA transfection, transfected cells were serum-starved for 12 h and then exposed to 10% CSE.
Signal pathway studies
HBE cells were serum-starved for 12 h and treated with NAC at 5 mM or H2O2 at 0.5 mM. After incubation for 30 min, cells were exposed to the 10% CSE for 24 or 48 h. Then the cells were harvested for detect the expressions of EMT markers.
Quantitative real-time PCR analysis
Total RNA from HBE cells and lung tissues was extracted with the Trizol reagent (Invitrogen). Equal amounts of total RNA (1 ug) were reverse transcribed into cDNA PrimerScript RT Reagent Kit (TaKaRa Biotechnology) according to the manufacturer’s instructions. For quantitative real-time PCR (qPCR), SYBR Green PCR Master Mix (TaKaRa Biotechnology) and a CFX96TM quantified PCR system (Bio-Rad) were used according to the manufacturer’s protocol. Relative expression was calculated with the 2−ΔΔCT method. Primers sequences for amplification were listed in .
Table 1. The primers for real-time PCR.
Western blot analysis
The frozen lung tissue samples were homogenized and total cell lysates were obtained in RIPA buffer (Beyotime Biotechnology) supplemented with PMSF (the final concentration was 1 mM), a cocktail of protease inhibitors and phosphatase inhibitors. The samples containing same amounts of protein were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (PVDF membranes, Bio-Rad). Membranes were blocked with 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBST) at room temperature for 60 min followed by incubation with a primary antibody overnight at 4 °C in a specific dilution (1:10,000 for Ecadherin, 1:5000 for ZO1 and 1:2000 for SMA, Vimentin, CD147 and GAPDH). After three washes with TBST, membranes were probing with secondary antibodies (goat anti-rabbit or goat anti-mouse) for 1 h at room temperature. The membrane was reacted with chemiluminescence reagent ECL (Fude Biosciences), and the immune complexes were visualized with by the Western blotting detection system (Bio-Rad).
Immunohistochemistry
Immunohistochemical staining was performed to examine the expressions of Ecadherin, α-SMA, ZO1, Vimentin and 8-OHdG in formalin-fixed, paraffin-embedded sections of each murine lung sample. Briefly, sections were deparaffinized and rehydrated with the use of ethanol solution in a concentration gradient, and followed by an antigen-retrieval step. Endogenous peroxidase activity was blocked by incubating sections with 3% hydrogen peroxide for 15 min. The sections were next blocked with 10% goat serum (ZSGB-BIO, Beijing, China) for 1 h, incubated (overnight, 4 °C) with Ecadherin, α-SMA ZO1, Vimentin and 8-OHdG primary antibodies (1:200–500), and then stained with an anti-mouse IgG secondary antibody (SP-9000 kit; ZSGB-BIO) and 3,3′-diaminobenzidine (DAB; ZSGB-BIO) according to manufacturer’s instructions. Hematoxylin was used for counterstaining.
Immunofluorescence analysis
HBE cells were seeded and cultured on coverslips (Fisherbrand) in 12-well plates. After CD147 siRNA and/or H2O2 treatment, cells were exposed to 10% CSE for 48 h. Washed with PBS, cells were fixed with 4% formaldehyde for 30 min. Cells were blocked with 5% BSA in PBS for 30 min at room temperature, then incubated with primary antibodies overnight at 4 °C in a certain dilution (1:200 for Ecadherin, SMA, ZO1 and Vimentin). The coverslips were then washed and incubated with fluorescein labeled secondary antibody 1 h at room temperature in dark. At last, the cells were counterstained with DAPI and examined through a fluorescence microscope (Axio vert; Carl Zeiss, Jena, Germany).
Reactive oxygen species assay
Intracellular reactive oxygen species (ROS) was detected with the use of Meilun Reactive Oxygen Species Assay Kit (MA0219, Dalian Meilun Biotechnology, CO., Ltd) according to the manufacturer’s instructions. The fluorescence intensity of each sample was measured by microplate reader.
H2O2 assay
The concentration of H2O2 produced by cells was detected with the use of Hydrogen Peroxide Assay Kit (BC3595, Beijing Solarbio Science & Technology Co., Ltd) according to the manufacturer’s instructions. The H2O2 concentration of each sample was measured by microplate reader.
Statistical analysis
Data were expressed as means ± SEM and analyzed with the help of GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA). One-way analysis of variance (ANOVA) was applied to compare the data derived from three or more groups followed by Sidak test for multiple comparisons. Students’ t-test was used in two groups comparison. p < 0.05 was considered statistically significant.
Results
Cigarette smoke upregulated CD147 expression in vivo and in vitro
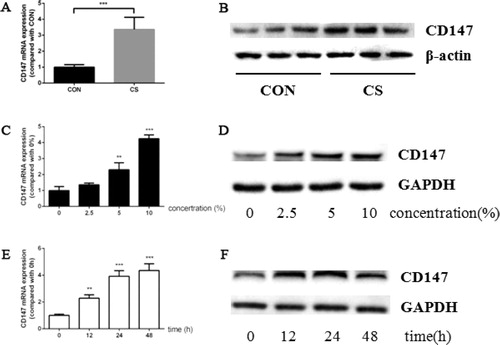
C57Bl/6 mice were exposed to cigarette smoke for 12 weeks. Compared with the mice exposed to filtered air, cigarette smoke increased CD147 expression at both mRNA and protein level (). Moreover, the CD147 expression in HBE cells was also increased after CSE treatment. CSE induced CD147 in HBE cells in a concentration dependent manner. As the concentration of CSE became higher, the expression level of CD147 increased and reached peak value while exposed to 10% CSE (). Then the cells were stimulated by 10% CSE for up to 48 h. At 12 h, 24 h and 48 h, a part of cells were harvested to detect CD147 expression. CD147 mRNA expression increased as the exposure time delayed and reached the highest level at 48 h (). However, CD147 protein expression reached the highest level at 24 h, and decreased at 48 h ().
Figure 1. Cigarette smoke induced CD147 expression in murine lung tissue and HBE cells. (a, b) CD147 mRNA (a) and protein (b) expression in murine lung tissue; (c, d) CD147 mRNA (c) and protein (d) expression in HBE cells treated by CSE ranged from 0–10%; (e, f) CD147 mRNA (e) and protein (f) expression in HBE cells treated by 10% CSE for 0–48 h. (animal study was performed with six replicates in each group, and cell experiments were performed in triplicates, *p < 0.05; **p < 0.01; ***p < 0.001, compared with CON or 0).
Cigarette smoke induced EMT in mice and HBE cells
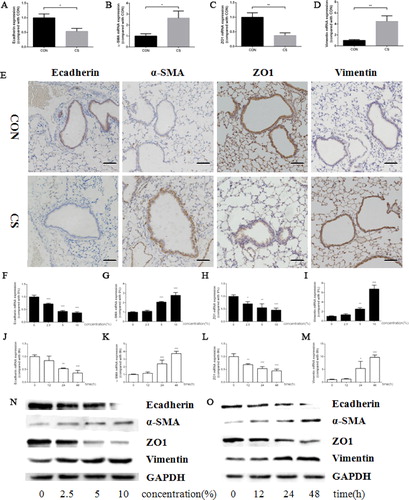
To testify EMT induced by cigarette smoke, Ecadherin, α-SMA, ZO1 and Vimentin were detected as represent markers for epithelial cells and mesenchymal cells, respectively. After 12-week exposure of cigarette smoke, the mRNA expressions of Ecadherin and ZO1 were downregulated while those of α-SMA and Vimentin were upregulated significantly (). In addition, the protein levels of Ecadherin and ZO1 were decreased and those of α-SMA and Vimentin were increased in murine lung tissue, as evidenced by IHC (). Moreover, the alteration of these markers in HBE cells was in line with that in lung tissues, which was confirmed by RT-PCR () and western blot ().
Figure 2. Cigarette smoke upregulated α-SMA, Vimentin expression and downregulated Ecadherin, ZO1 expression both in vivo and in vitro. (a–d) Ecadherin (a), α-SMA (b), ZO1 (c) and Vimentin (d) mRNA expression in murine lung tissue; (e) representative pictures of Ecadherin (left panel), α-SMA (middle left panel), ZO1 (middle right panel) and Vimentin (right panel) protein expression in murine lung tissue by IHC (scale bar, 50 μm); (f–i) Ecadherin (f), α-SMA (g), ZO1 (h) and Vimentin (i) mRNA expression in HBE cells treated by CSE in a series of concentrations; (j–m) Ecadherin (j), α-SMA (k), ZO1 (l) and Vimentin (m) mRNA expression in HBE cells treated by 10% CSE for 0–48 h; (n, o) Ecadherin, α-SMA, ZO1 and Vimentin protein expression in HBE cells treated by CSE in a series of concentrations (n) or 10% CSE for 0–48 h (o). (Animal study was performed with six replicates in each group, and cell experiments were performed in triplicates, *p < 0.05; **p < 0.01; ***p < 0.001, compared with CON or 0).
CD147 was required in cigarette smoke induced EMT
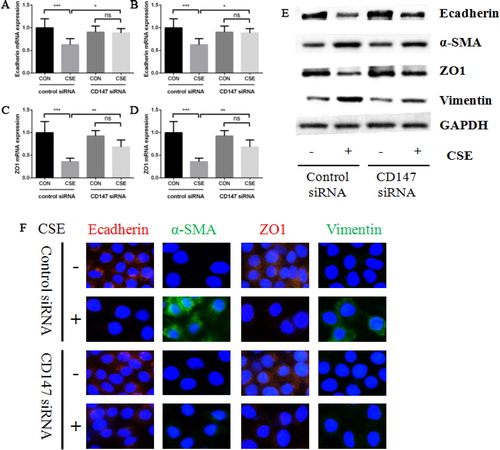
As we observed that cigarette smoke induced EMT and promoted CD147 expression, we next studied the role of CD147 in cigarette smoke induced EMT process. HBE cells were transfected with CD147 siRNA and control siRNA firstly, and treated by 10% CSE for 48 h. As a result, the expressions of Ecadherin and ZO1 were much higher and α-SMA and Vimentin were much lower in CD147 knockdown cells than those in control cells after CSE treatment. This phenomenon was obvious at both mRNA and protein level, as supported by RT-PCR (), western blot () and immunofluorescence ().
Figure 3. The effect of CD147 knockdown on CSE induced EMT markers expression in HBE cells. (a–d) Ecadherin (a), α-SMA (b), ZO1 (c) and Vimentin (d) mRNA expression in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 48 h; (e) Ecadherin, α-SMA, ZO1 and Vimentin protein expression in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 48 h; (f) representative images of Ecadherin (left panel), α-SMA (middle left panel), ZO1 (middle right panel) and Vimentin (right panel) protein expression in HBE cells by IF (All analyses were performed in triplicates, *p < 0.05; **p < 0.01; ***p < 0.001).
Cigarette smoke induced oxidative stress response in vivo and in vitro
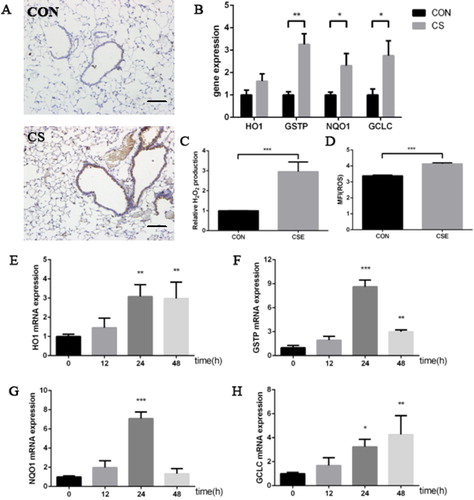
First, the results of IHC showed that 8-OHdG was highly expressed in the lung tissue derived from the mice exposed to cigarette smoke (), which was accompanied by the increasing expression of several anti-oxidative genes, including GSTP, NQO1 and GCLC (). In parallel, intracellular ROS level and the concentration of H2O2 in culture medium increased evidently after 10%CSE treatment for 24 h (). Moreover, the expression levels of anti-oxidative genes were also upregulated after CSE exposure in HBE cells ().
Figure 4. Cigarette smoke induced oxidative stress response in vivo and in vitro. (a) representative pictures of 8-OHdG expression in murine lung tissue by IHC (scale bar, 50 μm); (b) HO1, GSTP, NQO1 and GCLC mRNA expression in murine lung tissue; (c) relative H2O2 production in culture medium in HBE cells treated by 10% CSE for 24 h; (d) mean flurescence intensity (MFI) of intracellular ROS treated by 10% CSE for 24 h; (e–h) HO1 (e), GSTP (f), NQO1 (g) and GCLC (h) mRNA expression in HBE cells treated by 10% CSE (animal study was performed with six replicates in each group, -and cell experiments were performed in triplicates, *p < 0.05; **p < 0.01; ***p < 0.001, compared with CON or 0).
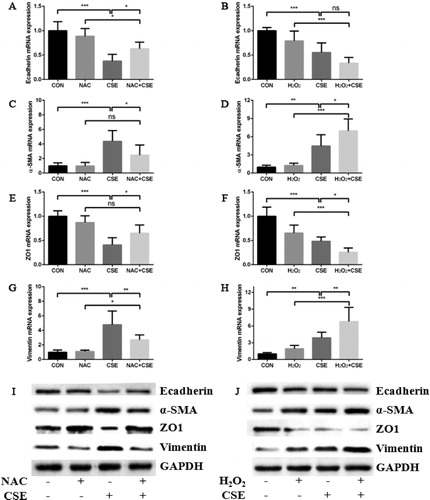
Oxidative stress signaling pathway was involved in cigarette smoke induced EMT
We made use of an inhibitor of oxidative stress signaling pathway, NAC to testify whether oxidative stress was participated in cigarette smoke induced EMT process in HBE cells. As we expected, 5 mM NAC reduced the expressions of α-SMA and Vimentin and augmented the expressions of Ecadherin and ZO1 in CSE induction model (). Conversely, the expressions of α-SMA and Vimentin were further upregulated and the expressions of Ecadherin and ZO1 were further downregulated when HBE cells were pretreated with 0.5 mM H2O2 in the same model ().
Figure 5. Oxidative stress was involved in cigarette smoke induced EMT process. (a, b) the effect of NAC (a) or H2O2 (b) on Ecadherin mRNA expression in HBE cells treated by 10% CSE for 48 h; (c, d) the effect of NAC (c) or H2O2 (d) on α-SMA mRNA expression in HBE cells treated by 10% CSE for 48 h; (e, f) The effect of NAC (e) or H2O2 (f) on ZO1 mRNA expression in HBE cells treated by 10% CSE for 48 h; (g, h) the effect of NAC (g) or H2O2 (h) on Vimentin mRNA expression in HBE cells treated by 10% CSE for 48 h; (i, j) the effect of NAC (i) or H2O2 (j) on Ecadherin, α-SMA, ZO1 and Vimentin protein expression in HBE cells treated by 10% CSE for 48 h. (All analyses were performed in triplicates, *p < 0.05; **p < 0.01; ***p < 0.001).
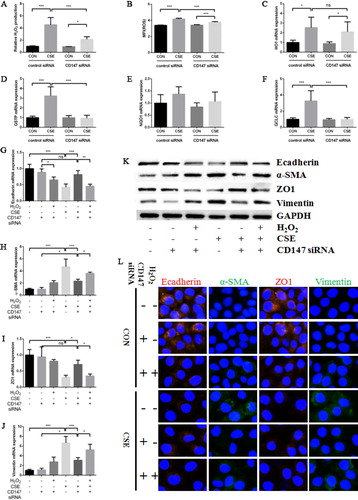
CD147 regulated cigarette smoke induced EMT via oxidative stress signaling pathway
CD147 knockdown did not affect the basal level of intracellular ROS and H2O2 concentration in culture medium. However, the low expression of CD147 significantly reduced the intracellular ROS level and the H2O2 concentration in culture medium increased evidently after CSE treatment (). As mentioned above, knockdown of CD147 inhibited EMT process induced by CSE. In the meanwhile, some anti-oxidative genes were also downregulated in CD147 knockdown cells compared with normal cells after CSE exposure (). Finally, when H2O2 (final concentration was 0.5 mM) was add to CD147 knockdown cells and treated by 10% CSE, the expressions of α-SMA and Vimentin were upregulated and the expressions of Ecadherin and ZO1 were downregulated again, which was confirmed by RT-PCR (), western blot () and immunofluorescence ().
Figure 6. Cigarette smoke promoted EMT in HBE cells via oxidative stress signal pathway. (a) Relative H2O2 production in culture medium in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 24 h; (b) mean flurescence intensity (MFI) of intracellular ROS in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 24 h; (c–f) HO1 (c), GSTP (d), NQO1 (e) and GCLC (f) mRNA expression in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 24 h; (g–j) Ecadherin (g), α-SMA (h), ZO1 (i) and Vimentin (j) mRNA expression in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 48 h with or without preincubation of 0.5 mM H2O2; (k) Ecadherin, α-SMA, ZO1 and Vimentin protein expression in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 48 h with or without preincubation of 0.5 mM H2O2. (l) Representative IF images of Ecadherin (left panel), α-SMA (middle left panel), ZO1 (middle right panel) and Vimentin (right panel) protein expression in HBE cells transfected with control or CD147 siRNA and then treated by 10% CSE for 48 h with or without preincubation of 0.5 mM H2O2. (All analyses were performed in four replicates, *p < 0.05; **p < 0.01; ***p < 0.001).
Discussion
As is known to all, cigarette smoke is the most important risk factor of COPD. The adverse effect of cigarette smoke on organism has not been fully understood partly due to its complex components. EMT is such a morphological and functional change can be induced by cigarette smoke [Citation20]. The declining expressions of epithelial cell markers (including Ecadherin, ZO1, etc.) and the rising expressions of mesenchymal cell markers (including Ncadherin, α-SMA, vimentin, etc.) are major molecular events, which has been observed in different airway epithelial cell lines derived from normal individuals (i.e. HBE, Beas2b) [Citation21–27] or lung cancer patients (i.e. A549) [Citation22, Citation24, Citation28] and primary human bronchial epithelial cells [Citation29–31]. In the meantime, a series of signaling pathways are also activated during this process, including Wnt/β-catenin [Citation27], EGFR/Src/JNK [Citation25], NF-κB [Citation26] and PI3K/Akt [Citation24] etc. Although most studies were limited to cell level, Shen et al. demonstrated that cigarette smoke could induce EMT both in vivo and in vitro, which mediated by TGF-β/Smad, Rac1/PI3K/Akt pathways [Citation32]. In our study, we also observed cigarette smoke upregulated the expressions of α-SMA and Vimentin and downregulated the expressions of Ecadherin and ZO1 both in murine lung tissues and HBE cells, which was in accordance with previous studies.
As cigarette smoke is rich in free radicals, oxidative stress is the direct mechanism that causes injury in cells and tissues. In a previous study, Guan et al. confirmed oxidative stress played an important role in cigarette smoke induced EMT in rats and HBE cells by mean of Ginsenoside Rg1, the main component of a traditional herbal medicine, which had been reported to have anti-oxidative effects [Citation33]. Milara et al. also found ROS mediated CSE induced EMT in primary human bronchial epithelial cells, which could be partly blocked by roflumilast N-oxide, the active metabolite of the PDE4 inhibitor roflumilast [Citation31]. In our study, we directly measured intracellular ROS level and H2O2 concentration in culture medium, and observed an augment of ROS and H2O2 levels after cigarette smoke exposure, which was accompanied by the alteration of EMT markers. NAC, another antioxidant, could partly abrogate the effect, while H2O2 further aggravated this effect. So our work demonstrated that oxidative stress was a key signaling pathway mediating the occurrence of EMT. Continuous stimulation of oxidants will induce antioxidant defense in cells, which is characterized by upregulation of a series of antioxidant and detoxifying enzymes and proteins, including heme oxygenase 1 (HO1), NADP(H): quinone oxidoreductase (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC) and glutathione S-transferases Pi (GSTP) et al. [Citation34]. We also observed an obvious increase of these genes both in animal and cell models induced by cigarette smoke in this study.
Epithelial mesenchymal transition is a common biological process existed in various physiological and pathological conditions. Generally, EMT can be divided into three major types. Type I occurs during embryogenesis, so it is a normal physiological process. Type II, occurring during tissue repair and organ fibrosis, is often caused by persistent stimulation by environmental toxic substance, like cigarette smoke. Type III is characterized by angiogenesis, which is participated in tumor metastasis [Citation35]. As COPD always occurs after chronic cigarette smoke exposure, type II EMT is tightly associated with this disease. In fact, the limitation of airflow in COPD patients is the result of small airway fibrosis and remodeling, which is largely contributed to EMT [Citation3, Citation36]. Many researchers investigated EMT phenomenon in the airway in COPD patients via detecting effective molecules, including S100A4, vimentin, α-SMA, Ecadherin, ZO-1, etc. [Citation5, Citation6, Citation37, Citation38]. However, the relationship between the expression level of EMT markers and the severity or prognosis of this disease was hardly discussed in these papers, which might be obtained more concern in the future.
CD147 was originally discovered in a cell line derived from lung cancer. There were numerous studies about the role of CD147 in lung cancer. Most studies indicated that CD147 enhanced the ability of invasion and promoted tumor metastasis. The high expression of CD147 meant a poor prognosis for lung cancer patients [Citation12–16]. So far, several researchers have focused their attention on the relationship between CD147 and EMT. Rodriguez-Teja et al. reported that CD147 suppressed EMT in prostate gland cancer [Citation10]. On the contrary, other studies indicated that CD147 promoted EMT in liver cancer [Citation8], colorectal cancer [Citation9] and tongue cancer [Citation11]. However, to our knowledge, there were few research discussing whether CD147 could regulate EMT in COPD. In our study, we first reported the high expression of CD147 in an animal model by cigarette smoke exposure and observed the upregulation of CD147 in HBE cells in concentration dependent manner after CSE treatment. Then we demonstrated that CD147 promoted EMT both in vivo and in vitro induced by cigarette smoke. Moreover, our work also suggested oxidative stress signaling pathway was involved in CD147 mediated EMT process. This novel finding would be a step forward to our understanding about the function of CD147.
It was also worth to note that CD147 expression was not always upregulated in HBE cells by CSE treatment. In fact, the protein level of CD147 began to decrease after 10% CSE treatment for more than 24 h, despite that CD147 mRNA expression maintained at high level. The inconsistency between mRNA and protein expressions of CD147 suggested the synthesis of CD147 protein is inhibited or the degradation of CD147 protein was enhanced after CSE stimulation for a long time, which might be associated with an unknown mechanism of post-transcriptional control. On the other hand, we detected an increase of CD147 protein expression in animal model in which mice were exposed to cigarette smoke for 12 weeks, but we have not construct an animal model with longer exposure time (i.e. 24 weeks). If we extend the exposure time, the variation of CD147 expression level might be similar to that we observed in cell experiments. The kinetic change of CD147 in long-term CS exposure model and underlying mechanisms will be further investigated in future studies.
In conclusion, our study first demonstrated that CD147 promoted EMT in mice and HBE cells induced by cigarette smoke via oxidative stress signaling pathway. As CD147 is involved in COPD, further investigating the underlying mechanism may contribute to new therapy targets to this disease.
Author contributions
H. Zhou contributed to establishing cell model, molecular biology detection by PCR and WB, analysis and interpretation of data and drafting manuscript. Y. Liu was responsible for IHC of animal tissues and IF of cell samples. Z. Wang, Y. Yang, D. Yuan and M. Li took part in animal treating and collecting various kinds of samples. X. Zhang carried out statistical analysis and revised manuscript. Y. Li designed this study and revised manuscript. All authors reviewed the manuscript.
Disclosure Statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9.
- Higham A, Quinn AM, Cancado JED, et al. The pathology of small airways disease in COPD: historical aspects and future directions. Respir Res. 2019;20(1):49. doi:10.1186/s12931-019-1017-y.
- Courtney JM, Spafford PL. The role of epithelial-mesenchymal transition in chronic obstructive pulmonary disease. Cells Tissues Organs. 2017;203(2):99–104. doi:10.1159/000450919.
- Das V, Bhattacharya S, Chikkaputtaiah C, et al. The basics of epithelial–mesenchymal transition (EMT): A study from a structure, dynamics, and functional perspective. J Cell Physiol. 2019;234(9):14535–14555. doi:10.1002/jcp.28160.
- Milara J, Peiro T, Serrano A, et al. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–420. doi:10.1136/thoraxjnl-2012-201761.
- Sohal SS, Reid D, Soltani A, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15(6):930–938. doi:10.1111/j.1440-1843.2010.01808.x.
- Xiong L, Edwards CK, 3rd, Zhou L. The biological function and clinical utilization of CD147 in human diseases: A review of the current scientific literature. Int J Mol Sci. 2014;15(10):17411–17441. doi:10.3390/ijms151017411.
- Wu J, Ru NY, Zhang Y, et al. HAb18G/CD147 promotes epithelial-mesenchymal transition through TGF-beta signaling and is transcriptionally regulated by Slug. Oncogene. 2011;30(43):4410–4427. doi:10.1038/onc.2011.149.
- Xu T, Zhou M, Peng L, et al. Upregulation of CD147 promotes cell invasion, epithelial-to-mesenchymal transition and activates MAPK/ERK signaling pathway in colorectal cancer. Int J Clin Exp Pathol. 2014;7(11):7432–7441.
- Rodriguez-Teja M, Gronau JH, Minamidate A, et al. Survival outcome and EMT suppression mediated by a lectin domain interaction of Endo180 and CD147. Mol Cancer Res. 2015;13(3):538–547.
- Suzuki S, Toyoma S, Tsuji T, et al. CD147 mediates transforming growth factor-beta1-induced epithelial-mesenchymal transition and cell invasion in squamous cell carcinoma of the tongue. Exp Ther Med. 2019;17(4):2855–2860. doi:10.3892/etm.2019.7230.
- Zhong X, Li M, Nie B, et al. Overexpressions of RACK1 and CD147 associated with poor prognosis in stage T1 pulmonary adenocarcinoma. Ann Surg Oncol. 2013;20(3):1044–1052. doi:10.1245/s10434-012-2377-4.
- Xu XY, Lin N, Li YM, et al. Expression of HAb18G/CD147 and its localization correlate with the progression and poor prognosis of non-small cell lung cancer. Pathol Res Pract. 2013;209(6):345–352. doi:10.1016/j.prp.2013.02.015.
- Fei F, Li X, Xu L, et al. CD147-CD98hc complex contributes to poor prognosis of non-small cell lung cancer patients through promoting cell proliferation via the PI3K/Akt signaling pathway. Ann Surg Oncol. 2014;21(13):4359–4368. doi:10.1245/s10434-014-3816-1.
- Matsumoto T, Nagashio R, Ryuge S, et al. Basigin expression as a prognostic indicator in stage I pulmonary adenocarcinoma. Pathol Int. 2018;68(4):232–240. doi:10.1111/pin.12646.
- Liu B, Wan ZH, Sheng BW, et al. Overexpression of EMMPRIN is associated with lymph node metastasis and advanced stage of non-small cell lung cancer: a retrospective study. BMC Pulm Med. 2017;17(1):214. doi:10.1186/s12890-017-0540-1.
- Jouneau S, Khorasani N, de Souza P, et al. EMMPRIN (CD147) regulation of MMP-9 in bronchial epithelial cells in COPD. Respirology. 2011;16(4):705–712. doi:10.1111/j.1440-1843.2011.01960.x.
- Berg J, Halvorsen AR, Bengtson MB, et al. Levels and prognostic impact of circulating markers of inflammation, endothelial activation and extracellular matrix remodelling in patients with lung cancer and chronic obstructive pulmonary disease. BMC Cancer. 2018;18(1):739. doi:10.1186/s12885-018-4659-0.
- Li Y, Gu C, Xu W, et al. Therapeutic effects of amniotic fluid-derived mesenchymal stromal cells on lung injury in rats with emphysema. Respir Res. 2014;15(1):120. doi:10.1186/s12931-014-0120-3.
- Vu T, Jin L, Datta PK. Effect of cigarette smoking on epithelial to mesenchymal transition (EMT) in lung cancer. J Clin Med. 2016;5(4):44.
- Veljkovic E, Jiricny J, Menigatti M, et al. Chronic exposure to cigarette smoke condensate in vitro induces epithelial to mesenchymal transition-like changes in human bronchial epithelial cells, BEAS-2B. Toxicol In Vitro. 2011;25(2):446–453. doi:10.1016/j.tiv.2010.11.011.
- Eurlings IM, Reynaert NL, van den Beucken T, et al. Cigarette smoke extract induces a phenotypic shift in epithelial cells; involvement of HIF1alpha in mesenchymal transition. PLoS One. 2014;9(10):e107757. doi:10.1371/journal.pone.0107757.
- Liu Y, Luo F, Xu Y, et al. Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015;282(1):9–19. doi:10.1016/j.taap.2014.10.022.
- Wang Q, Wang H, Zhang Y, et al. Activation of uPAR is required for cigarette smoke extract-induced epithelial-mesenchymal transition in lung epithelial cells. Oncol Res. 2014;21(6):295–305. doi:10.3727/096504014X13946388749036.
- Zhang L, Gallup M, Zlock L, et al. Cigarette smoke disrupts the integrity of airway adherens junctions through the aberrant interaction of p120-catenin with the cytoplasmic tail of MUC1. J Pathol. 2013;229(1):74–86. doi:10.1002/path.4070.
- Zhao Y, Xu Y, Li Y, et al. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol Sci. 2013;135(2):265–276. doi:10.1093/toxsci/kft150.
- Zou W, Zou Y, Zhao Z, et al. Nicotine-induced epithelial-mesenchymal transition via Wnt/beta-catenin signaling in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;304(4):L199–209. doi:10.1152/ajplung.00094.2012.
- Zhang H, Liu H, Borok Z, et al. Cigarette smoke extract stimulates epithelial-mesenchymal transition through Src activation. Free Radic Biol Med. 2012;52(8):1437–1442. doi:10.1016/j.freeradbiomed.2012.01.024.
- Milara J, Peiro T, Serrano A, et al. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther. 2014;28(2):138–148. doi:10.1016/j.pupt.2014.02.001.
- Li E, Xu Z, Liu F, et al. Continual exposure to cigarette smoke extracts induces tumor-like transformation of human nontumor bronchial epithelial cells in a microfluidic chip. J Thorac Oncol. 2014;9(8):1091–1100. doi:10.1097/JTO.0000000000000219.
- Milara J, Peiro T, Serrano A, et al. Simvastatin increases the ability of roflumilast n-oxide to inhibit cigarette smoke-induced epithelial to mesenchymal transition in well-differentiated human bronchial epithelial cells in vitro. COPD. 2015;12(3):320–331. doi:10.3109/15412555.2014.948995.
- Shen HJ, Sun YH, Zhang SJ, et al. Cigarette smoke-induced alveolar epithelial-mesenchymal transition is mediated by Rac1 activation. Biochim Biophys Acta. 2014;1840(6):1838–1849. doi:10.1016/j.bbagen.2014.01.033.
- Guan S, Xu W, Han F, et al. Ginsenoside Rg1 attenuates cigarette smoke-induced pulmonary epithelial-mesenchymal transition via inhibition of the TGF-beta1/Smad pathway. Biomed Res Int. 2017;2017:1–12. doi:10.1155/2017/7171404.
- Zhao H, Eguchi S, Alam A, et al. The role of nuclear factor-erythroid 2 related factor 2 (Nrf-2) in the protection against lung injury. Am J Physiol Lung Cell Mol Physiol. 2017;312(2):L155–L62. doi:10.1152/ajplung.00449.2016.
- Stone RC, Pastar I, Ojeh N, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016;365(3):495–506. doi:10.1007/s00441-016-2464-0.
- Sohal SS. Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir Invest. 2017;55(2):104–113. doi:10.1016/j.resinv.2016.11.006.
- Sohal SS, Reid D, Soltani A, et al. Evaluation of epithelial mesenchymal transition in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12(1):130. doi:10.1186/1465-9921-12-130.
- Mahmood MQ, Sohal SS, Shukla SD, et al. Epithelial mesenchymal transition in smokers: large versus small airways and relation to airflow obstruction. Int J Chron Obstruct Pulmon Dis. 2015;10:1515–1524.