
Abstract
This Phase II, randomized, parallel group study was conducted as part of US regulatory requirements to identify the most appropriate dose of the long-acting muscarinic antagonist glycopyrronium bromide (GB) for use in a single-inhaler triple-therapy combination with the inhaled corticosteroid beclomethasone dipropionate plus the long-acting β2-agonist formoterol fumarate. Eligible subjects were adults with COPD and post-bronchodilator forced expiratory volume in 1 s (FEV1) 40–80% predicted. Subjects were randomized to receive inhaled double-blind GB 6.25, 12.5, 25 or 50 µg or placebo, all twice daily (BID), or open-label tiotropium 18 µg once daily for six weeks. The primary objective was to evaluate the efficacy of GB versus placebo in terms of FEV1 area under the curve between 0 and 12 h at Week 6. Of 733 subjects randomized, 682 (93.0%) completed the study. For the primary endpoint, all GB doses were superior to placebo (p < 0.05), with a dose-response up to 25 µg BID, and 25 and 50 µg BID both superior to 6.25 µg BID (p < 0.05). Results for the secondary spirometry endpoints were consistent with the primary endpoint. Overall, the efficacy of GB 25 and 50 µg BID was broadly consistent with that of tiotropium. The incidence of adverse events, both overall and for the most common preferred terms, was low and similar in all treatment groups, including placebo (overall, 22.3–29.3%). Based on the totality of the efficacy and safety data, the optimal GB dose is 25 µg BID.
Introduction
Given the progressive nature of chronic obstructive pulmonary disease (COPD), many patients require additional pharmacotherapy over time to control symptoms or prevent exacerbations [Citation1]. Those with the most severe disease may require inhaled triple therapy, with an inhaled corticosteroid (ICS), a long-acting β2-agonist (LABA) and a long-acting muscarinic antagonist (LAMA) [Citation2–10]. Although such triple therapy can be achieved using a combination of inhalers, complex regimens, especially those that involve multiple different inhalers, may adversely impact adherence, with associated negative impacts on clinical outcomes and healthcare utilization [Citation11,Citation12]. As a consequence, a number of single-inhaler triple-therapy combinations are becoming available.
This manuscript reports the results of a dose-finding study that was conducted as part of US regulatory requirements to identify the most appropriate dose of the LAMA glycopyrronium bromide (GB) for use in an extrafine formulation (i.e. with mass median aerodynamic diameter <2 µm [Citation13]) single-inhaler triple-therapy combination with the ICS beclomethasone dipropionate (BDP) plus the LABA formoterol fumarate (FF). Such extrafine particles are more able to consistently reach small airways than non-extrafine particles [Citation14–16], with resulting enhanced drug delivery to these areas, improved overall lung deposition, and lower deposition in the oropharynx [Citation17].
Materials and methods
Trial design
This was a Phase II, multicenter, randomized, double-blind, placebo- and active-controlled, parallel group, dose-ranging study. After an initial screening visit (after which non-permitted COPD maintenance medication was discontinued) and a 14-day run-in period, subjects were randomized equally to one of six treatment groups, to inhaled GB 6.25, 12.5, 25 or 50 µg or placebo, all twice daily (BID) via pressurized metered-dose inhaler (pMDI), or tiotropium 18 µg once daily (QD) via the manufacturer’s proprietary single-dose dry-powder inhaler. Over the six-week treatment period, subjects then attended visits at Weeks 3 and 6. Albuterol (via pMDI) was permitted as rescue medication throughout the study (including during the run-in period), but not within 6 h prior to the start of a visit. At the screening visit, any subject receiving an ICS had this medication switched to the equivalent dose of BDP pMDI; this was maintained at a stable dose throughout the study (including the run-in period). LABAs, LAMAs, short-acting β2-agonists (other than rescue medication), and short-acting muscarinic antagonists were not permitted starting from the screening visit and for the duration of the study.
Pre-dose and serial post-dose (up to 12 h) spirometry (for forced expiratory volume in 1 s [FEV1] and forced vital capacity [FVC]) were conducted at the randomization visit and after 6 weeks; at Week 3, only pre-dose spirometry was conducted. Inspiratory capacity (IC) was assessed using a slow vital capacity maneuver pre-dose at randomization and Weeks 3 and 6. Results from the Baseline Dyspnea Index (BDI) questionnaire were recorded at the screening and randomization visits, and from the Transition Dyspnea Index (TDI) questionnaire at the Weeks 3 and 6 visits. Subjects recorded their symptoms (using the Evaluating Respiratory Symptoms in COPD [E-RS] questionnaire) and rescue medication use daily in an electronic diary. Adverse events were captured throughout the study, with 12-lead electrocardiogram (ECG) and vital signs data recorded pre-dose at each visit and post-dose at the randomization and Week 6 visits. Hematology and blood chemistry evaluations were performed at the screening and Week 6 visits, with vital signs evaluations at each visit and 24-h Holter data recorded from the day prior to the randomization and Week 6 visits.
The study was approved by the independent ethics committees for each institution, and was performed in accordance with the principles of the Declaration of Helsinki, and the International Conference on Harmonization Notes for Guidance on Good Clinical Practice (ICH/CPMP/135/95). The protocol was amended twice, with the first amendment completed prior to recruitment of any subject. The main changes in the second amendment were to widen the lung function inclusion criterion (the lower limit was reduced from FEV1 ≥50% to ≥40% of predicted normal), and the list of permissible prior COPD medication was expanded (to include inhaled LABA, LABA/LAMA and ICS/LABA/LAMA). The study is registered in ClinicalTrials.gov (NCT03084796).
Participants
The main inclusion criteria were: ≥40 years of age; current or ex-smokers; a diagnosis of COPD, with post-bronchodilator (ipratropium 84 µg) FEV1 ≥40% and <80% of the predicted normal value, FEV1/FVC ratio <0.7; a bronchodilator response to ipratropium (≥5% increase in FEV1); symptomatic at screening, with COPD Assessment Test total score ≥10 and BDI focal score ≤10 (both confirmed at randomization); and the use for at least two months prior to screening of LAMA, ICS/LABA (fixed or free combination), ICS + LAMA, LABA, LABA/LAMA (fixed or free combination), or ICS/LABA/LAMA (fixed or free combination). All subjects provided written informed consent prior to any study-related procedure.
The key exclusion criteria were: a current diagnosis of asthma or asthma-COPD overlap; a moderate-or-severe exacerbation that had not resolved within 14 days prior to screening; clinically significant cardiovascular conditions or laboratory abnormalities; or unstable concurrent disease that may have impacted efficacy or safety (as judged by the investigator). The full inclusion and exclusion criteria are listed in the supplement.
Randomization and masking
Subjects were allocated by the interactive response technology provider to treatment arms according to a central randomization list (generated by an independent third party), stratified by region. Subjects, investigators, site staff and sponsor personnel were blinded to GB (and matching placebo) assignment for the duration of the study. Tiotropium was administered open-label.
Objectives and related assessments
The primary objective was to evaluate the efficacy of GB by comparison with placebo in terms of change from baseline in FEV1 area under the curve between 0 and 12 h (AUC0–12h) normalized by time at Week 6. The secondary objectives were to compare the effect of GB versus placebo on: change from baseline in FEV1 AUC0-12h at Day 1, and FEV1 AUC0-4h and peak0-4h at Day 1 and Week 6; time to onset of action (change from baseline in post-dose FEV1 ≥100 mL) at Day 1; change from baseline in pre-dose morning FEV1 at Weeks 3 and 6, FVC AUC0-12h, AUC0-4h and peak0-4h at Day 1 and Week 6, and pre-dose morning IC at Weeks 3 and 6; TDI focal score and response (TDI focal score ≥1) at Weeks 3 and 6; and change from baseline in percentage of rescue medication-free days, average use of rescue medication, and average E-RS total score in each inter-visit period and over the entire treatment period. The safety and tolerability of the study treatments were assessed throughout the study.
Sample size and statistical methods
We estimated that a total of 594 evaluable subjects (99 per group) would provide 80% power to detect a mean difference of 120 mL between each dose of GB and placebo at a two-sided significance level of 0.0125 (taking the Bonferroni adjustment into account, since four dose levels were tested: 0.0125 = 0.05/4), assuming an SD of 250 mL [Citation18,Citation19]. Considering a non-evaluable rate of 15%, 702 subjects (117 per group) were planned to be randomized.
The primary efficacy endpoint was analyzed using a linear mixed model for repeated measures including treatment, visit, treatment by visit interaction, region, and smoking status at screening as fixed effects, and the baseline value (average of the pre-dose FEV1 measurements on Day 1) and baseline by visit interaction as covariates. An unstructured covariance matrix was assumed. Comparisons between each GB dose and placebo were adjusted for multiplicity, based on the parametric simulation method by Edwards and Berry [Citation20].
The majority of the secondary efficacy endpoints were analyzed using a similar model to the primary endpoint, although without any adjustment for multiplicity. TDI response (i.e. TDI focal score ≥1) at Weeks 3 and 6 was analyzed using a multiple logistic regression model that included treatment, region, and smoking status as fixed effects and the BDI as a covariate. GB and tiotropium were not compared statistically.
The intention-to-treat (ITT) population was defined as all randomized subjects who received at least one dose of study treatment and with at least one available evaluation of efficacy (primary or secondary efficacy variables) after baseline. The safety population was defined as all randomized subjects who received at least one dose of study treatment.
Results
Participants
The first subject entered the study on 28 July 2017, with the last subject completing on 6 June 2018. The study enrolled at 109 investigative sites, all in the US. Of 733 subjects randomized, 682 (93.0%) completed the study (). The ITT and safety populations were the same. The baseline demographics and disease characteristics of the subjects in the ITT population are shown in . Overall, 76.1% of subjects received concomitant ICS throughout the study (75.2, 75.6, 71.9, 81.3, 76.9 and 75.6% in the GB 6.25, 12.5, 25 and 50 µg BID, placebo, and tiotropium groups, respectively).
Figure 1. Subject disposition.
*The protocol permitted subjects to be screened multiple times. †One subject randomized to GB 25 µg BID withdrew prior to receiving study medication, and so was excluded from the safety and ITT populations. GB, glycopyrronium bromide; BID, twice daily; QD, once daily; ITT, intention-to-treat.
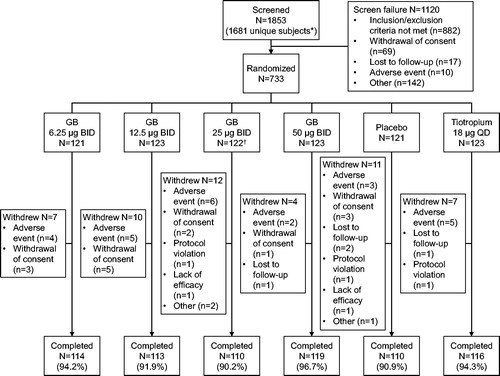
Table 1. Subject demographics and disease characteristics at screening (ITT population).
Outcomes
Primary endpoint
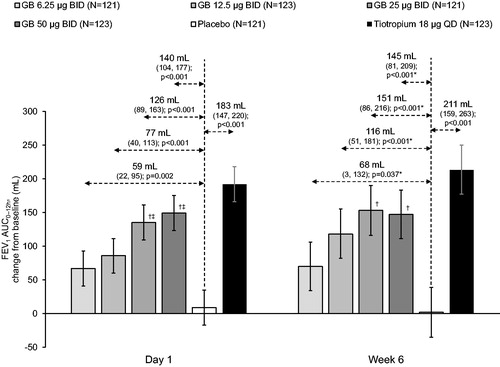
For the primary endpoint (FEV1 AUC0–12h at Week 6), all GB doses were superior to placebo, with differences of 68, 116, 151 and 145 mL for GB 6.25, 12.5, 25 and 50 µg BID, respectively (all p < 0.05). There was a dose-response for GB doses up to 25 µg BID, with GB 25 and 50 µg BID both superior to GB 6.25 µg BID (p < 0.05; ). Tiotropium was also superior to placebo for this endpoint (difference 211 mL; p < 0.001), with a tiotropium-placebo difference numerically higher than with any of the GB doses. We also conducted two post-hoc analyses: one with subjects subgrouped by lung function at screening, and one with subjects subgrouped by reversibility to ipratropium. Patients with severe airflow obstruction at screening (post-bronchodilator FEV1 ≥30 to <50% predicted) responded better to all active treatments than those with moderate obstruction, with higher adjusted mean treatment-placebo differences (although with wider confidence intervals, perhaps due to the smaller sample size; ). In contrast, the extent of bronchodilator response to ipratropium at screening did not consistently impact the treatment-placebo differences ().
Figure 2. FEV1 AUC0–12h on Day 1 and at Week 6 (ITT population).
Bars are adjusted mean changes from baseline, with 95% confidence intervals (*95% CI and p values adjusted for multiplicity). Values are adjusted mean treatment–placebo differences, 95% confidence intervals and p values. †p < 0.05 vs GB 6.25 µg BID; ‡p < 0.05 vs GB 12.5 µg BID. Tiotropium–GB differences were not analyzed. ITT, intention-to-treat; GB, glycopyrronium bromide; BID, twice daily; QD, once daily; FEV1, forced expiratory volume in 1 second; AUC, area under the curve.
Secondary endpoints
For FEV1 AUC0–12h on Day 1 there was a dose-response across the GB doses, with significant differences of 59, 77, 126 and 140 mL for GB 6.25, 12.5, 25 and 50 µg BID, respectively (all p < 0.01), and significant differences for GB 25 and 50 µg BID vs GB 6.25 and 12.5 µg BID (p < 0.05; ). Tiotropium was also superior to placebo (p < 0.001), with a tiotropium-placebo difference (183 mL) that was numerically higher than with GB. The results for FEV1 AUC0–4h and FEV1 peak0–4h were consistent with those for FEV1 AUC0–12h, with a dose-response across all GB doses on Day 1 and up to 25 µg BID at Week 6 (). For these endpoints, GB 25 and 50 µg BID were again superior to GB 6.25 and 12.5 µg BID on Day 1, and to GB 6.25 µg BID at Week 6 (p < 0.05). Tiotropium was superior to placebo (p < 0.001), with tiotropium-placebo differences numerically higher than with GB at Week 6, but similar to the two higher GB doses on Day 1. Similarly, for pre-dose morning FEV1 there was a dose-response for GB doses up to 25 µg at Week 3, and for all doses at Week 6, with the efficacy of the two higher GB doses (treatment–placebo differences of 122 and 111 mL for GB 25 and 50 µg BID, respectively, at Week 3 and 119 and 142 mL at Week 6) similar to that of tiotropium (122 and 124 mL at Weeks 3 and 6, respectively; ). There was a dose-response across GB doses for time to onset of action on Day 1, with onset faster than placebo with all GB doses (median 27.3–45.1 min; p ≤ 0.002) and with tiotropium (28.1 min; p < 0.001; ). Onset was faster with GB 25 and 50 µg BID than with GB 6.25 µg BID (p < 0.001), and with GB 50 µg BID than with GB 12.5 µg BID (p = 0.013). Results for the FVC endpoints were consistent with those for the FEV1 endpoints, with a dose-response across GB doses on Day 1, and up to 25 µg BID at Week 6, and with the two higher doses consistently superior to GB 6.25 µg BID (Supplementary Figures 5–7).
Figure 3. Pre-dose morning FEV1 at Weeks 3 and 6 (ITT population).
Bars are adjusted mean changes from baseline, with 95% confidence intervals. Values are adjusted mean treatment–placebo differences, 95% confidence intervals and p values. †p < 0.05 vs GB 6.25 µg BID. Tiotropium–GB differences were not analyzed. ITT, intention-to-treat; GB, glycopyrronium bromide; BID, twice daily; QD, once daily; FEV1, forced expiratory volume in 1 second.
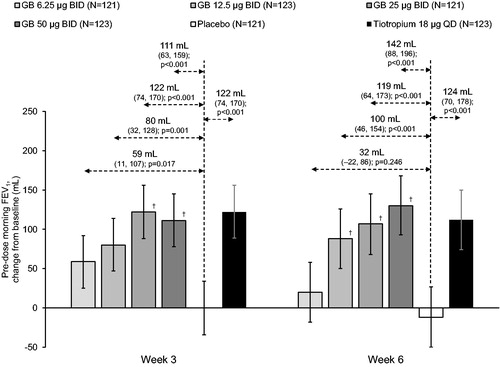
Figure 4. TDI focal score at Weeks 3 and 6 (ITT population).
Bars are adjusted mean, with 95% confidence intervals. Values are adjusted mean treatment–placebo differences, 95% confidence intervals and p values. †p < 0.05 vs GB 6.25 µg BID. Tiotropium–GB differences were not analyzed. ITT, intention-to-treat; GB, glycopyrronium bromide; BID, twice daily; QD, once daily; TDI, Transition Dyspnea Index.
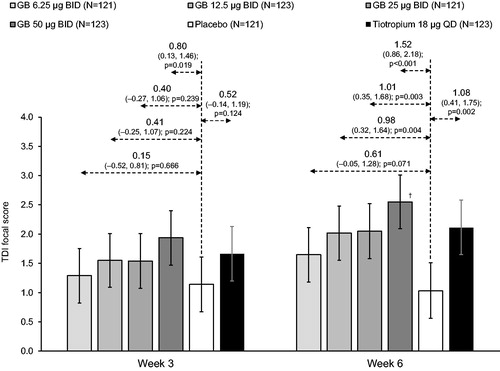
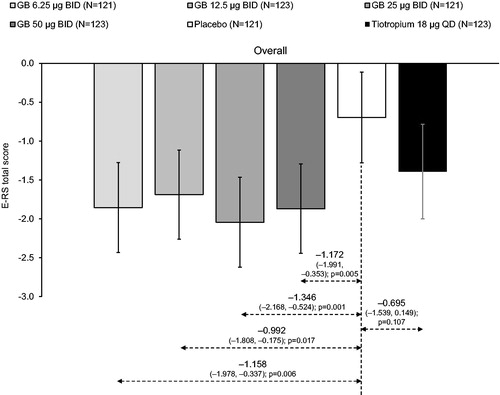
Figure 5. E-RS total score, analyzed across the study duration (ITT population).
There were no significant differences between GB doses. Tiotropium–GB differences were not analyzed. ITT, intention-to-treat; GB, glycopyrronium bromide; BID, twice daily; QD, once daily; E-RS, Evaluating Respiratory Symptoms questionnaire.
None of the GB doses or tiotropium had consistently significant effects versus placebo on pre-dose morning IC, with high variability around the mean, as indicated by the wide confidence intervals, and a larger change from baseline at Week 3 than at Week 6 for most treatments, including placebo (Supplementary Figure 8). At Week 6, GB 25 µg BID increased mean IC by 111 mL over placebo (p < 0.05), versus a non-significant 74 mL increase with tiotropium.
For TDI focal score at Week 3, only GB 50 µg BID differed significantly from placebo (difference 0.80; p = 0.019); by the Week 6 assessment, the three higher GB doses and tiotropium were significantly different vs placebo (differences of 0.98, 1.01, 1.52 and 1.08 for GB 12.5, 25 and 50 µg BID and tiotropium, respectively; all p < 0.01), with the treatment-placebo difference for GB 25 and 50 µg BID and tiotropium exceeding the 1 unit threshold for clinical relevance (). A high proportion of subjects in all treatment groups (including placebo) were TDI responders at both timepoints (). At Week 3, only GB 50 µg BID demonstrated significantly greater response compared to placebo (68.3% vs 56.3%; odds ratio 1.74; p = 0.042). At Week 6, significantly more subjects responded to all GB doses and tiotropium than placebo (62.7–74.6% vs. 47.8%; all p < 0.05), with a dose-response across the GB doses, and a higher proportion of responders to GB 50 µg BID than GB 6.25 µg BID.
Table 2. TDI focal score responder analysis at Weeks 3 and 6 (ITT population).
Overall, use of rescue medication during the run-in period was low (both the percentage of rescue medication-free days and average use; ). Despite this, use during the treatment period reduced from baseline in all arms. When analyzed overall, there were significant reductions versus placebo for all four GB groups in terms of both rescue-free days and average use; differences versus placebo were not significant for the tiotropium group.
Symptoms, as assessed with the E-RS, decreased (i.e. improved) from baseline in all groups, with a significantly greater reduction in all GB groups than placebo overall and across the two inter-visit periods ( and ). Although symptoms also decreased in the tiotropium group, none of the differences versus placebo were significant and the improvements in the tiotropium group were numerically lower than in all GB groups.
Safety
The incidence of adverse events, both overall and for the most common preferred terms, was low and similar in all treatment groups (including placebo; 24.8–29.3% with GB, 22.3% with placebo, and 26.0% with tiotropium; ). Few subjects had adverse events considered related to treatment (2.4–7.4% with GB, 4.1% with placebo, and 2.4% with tiotropium); the only preferred term related to treatment that occurred in at least three subjects in any group was dyspnea (in three subjects in the GB 6.25 µg BID group). The majority of adverse events were mild or moderate in intensity. The only severe event related to treatment was constipation, in a subject receiving GB 6.25 µg BID; this was also classified as serious, and resulted in treatment discontinuation. Overall, there was no correlation between adverse events leading to treatment discontinuation and GB dose. Six subjects had cardiac adverse events that were assessed as related to the study treatment (two [1.7%] subjects in the GB 6.25 µg BID group, and one [0.8%] subject each in the GB 12.5 and 25 µg BID, placebo and tiotropium groups). All were mild or moderate in intensity.
Table 3. Adverse events, overall and most common preferred terms (≥2% of subjects in any group; safety population).
Few subjects had clinically relevant changes in clinical laboratory or vital signs parameters (including systolic and diastolic blood pressure and heart rate), with no relationship to GB dose. The incidence of notable QTc (Fridericia’s correction) interval values and notable changes was low in all groups, with no relation to GB dose ().
Table 4. QTc (Fridericia’s correction) interval data at any post-dose timepoint (Safety population).
Discussion
In this Phase II dose-ranging study, for the FEV1 and FVC endpoints there was overall a dose-response across GB doses up to 25 µg BID, with GB 50 µg BID not consistently providing additional efficacy, and no statistically significant differences between these two doses. The 25 µg BID dose was also the lowest dose to consistently demonstrate FEV1 improvements of at least 100 mL compared with placebo – an improvement that has been described as a difference that patients can perceive [Citation21].
The effect of the active treatments on IC was more variable, as illustrated by broad confidence intervals around the means, highlighting the difficulty of conducting a multi-site study with IC as a secondary endpoint. Despite this, there was some evidence of a dose-response in the Week 6 evaluations for GB doses up to 25 µg BID, consistent with the forced spirometry evaluations. For the symptom endpoints (TDI and E-RS), all active treatments had a greater effect toward the end of the study (Week 6 for TDI, and Weeks 3 to 6 for E-RS). All subjects had to be symptomatic on study entry, so these data are consistent with a gradual reduction in symptoms, despite a low baseline use of rescue medication during the run-in period. In the Week 6 TDI focal score evaluation, the treatment–placebo differences for GB 25 and 50 µg BID and tiotropium exceeded the 1 unit difference considered clinically meaningful [Citation22].
The overall efficacy of the two higher GB doses was broadly consistent with that of tiotropium. There were no formal statistical comparisons pre-specified or performed between GB and tiotropium; however, for the FEV1 and FVC endpoints the efficacy of tiotropium was generally numerically greater than GB. This was most marked for the AUC0–12h assessments, so is likely due to differences in dosing intervals (with GB dosed twice daily, whereas tiotropium was dosed once daily) – and in this context it is notable that the efficacy of GB 25 and 50 µg BID were similar to tiotropium for pre-dose morning FEV1, a timepoint that assesses treatment effect at the end of the dosing interval equally for both molecules. Lung function was not assessed over the period from 12 to 24 h post-morning dose in this study, but given GB is dosed BID it is likely that the bronchodilator efficacy of GB would be greater than tiotropium over this period, consistent with data from a previous study that compared QD and BID dosing of another formulation of GB [Citation23].
The efficacy results of the current study are consistent with those of a previous dose-response study that also identified the most appropriate GB dose of this extrafine formulation to be 25 µg BID [Citation24], with the efficacy of this dose similar to tiotropium. This is also consistent with data for a dry powder formulation of glycopyrronium, in which a total daily dose of 50 µg was comparable to tiotropium in terms of lung function, dyspnea and health status [Citation25]. The bronchodilator effects of inhaled glycopyrronium have been studied using both once and twice a day administration, with one study showing little difference between these regimens when the same daily dose is administered entirely in the morning or divided between morning and evening [Citation23]. However, there is a suggestion that BID administration of LAMAs may be particularly useful in the management of nighttime and early morning symptoms [Citation26].
Importantly, there was no dose-response across GB doses for any of the safety parameters, with the overall incidence of adverse events (especially those considered related to treatment) being low. In particular, the incidence of cardiovascular events was low, with no relationship to GB dose of systolic and diastolic blood pressure, heart rate or notable QTcF interval values or changes.
Given this study was conducted to support dose selection of GB for use in an inhaled triple ICS/LABA/LAMA formulation, one limitation is that only three-quarters of subjects were receiving concomitant ICS, and concomitant LABA was not permitted. However, if subjects had been permitted other bronchodilators as concomitant therapy, this could potentially blunt the dose-response to GB, making interpretation of the results challenging. As strengths, rescue short-acting bronchodilator use (albuterol) was not permitted from 6 h prior to each visit, with centralized spirometry reading using standardized equipment used to optimize the quality of the lung function data, and with heart rate and QTc (Fridericia’s correction) data collected from 24 h Holter analyses.
In conclusion, based on the totality of the efficacy and safety data, the optimal GB dose is 25 µg BID. This dose has been utilized in the extrafine formulation single-inhaler triple-therapy combination BDP/FF/GB, which has demonstrated efficacy in both COPD and asthma [Citation7–9,Citation27].
Declarations of interest
EK is employee and founder of Altitude Clinical Consulting, Medford OR, USA, and former medical director of Clinical Research Institute, Medford USA during 1997-2020. He has performed consulting, served on advisory boards, or received travel reimbursement from Amphastar, AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Mylan, Novartis, Sunovion, Theravance, and Connect Biopharmaceuticals, and has conducted multicenter clinical research trials for some 40 pharmaceutical companies.
GF has no relevant conflicts of interest to disclose.
JP has no relevant conflicts of interest to disclose.
LDLC has no relevant conflicts of interest to disclose.
ME, CB and GG are employees of Chiesi, the sponsor of the study.
Supplemental Material
Download PDF (430.1 KB)Acknowledgments
The authors would like to thank the investigators and subjects at the investigative sites for their support of this study. Writing support was provided by David Young of Young Medical Communications and Consulting Ltd. This support was funded by Chiesi Farmaceutici S.p.A.
Data availability
Chiesi commits to sharing with qualified scientific and medical Researchers, conducting legitimate research, patient-level data, study-level data, the clinical protocol and the full clinical study report of Chiesi Farmaceutici S.p.A.-sponsored interventional clinical trials in patients for medicines and indications approved by the European Medicines Agency and/or the US Food and Drug Administration after 1st January 2015, following the approval of any received research proposal and the signature of a Data Sharing Agreement. Chiesi provides access to clinical trial information consistently with the principle of safeguarding commercially confidential information and patient privacy. To date, the current study is out of scope of the Chiesi policy on Clinical Data Sharing. Other information on Chiesi’s data sharing commitment, access and research request’s approval process are available in the Clinical Trial Transparency section of http://www.chiesi.com/en/research-and-development/.
Additional information
Funding
References
- Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease [Internet]. 2020. [cited 2020 Sep 5]. Available from: https://goldcopd.org/gold-reports/.
- Brusselle G, Price D, Gruffydd-Jones K, et al. The inevitable drift to triple therapy in COPD: an analysis of prescribing pathways in the UK. Int J Chron Obstruct Pulmon Dis. 2015;10:2207–2217.
- Worth H, Buhl R, Criée C-P, et al. GOLD 2017 treatment pathways in 'real life': An analysis of the DACCORD observational study. Respir Med. 2017;131:77–84. DOI:https://doi.org/10.1016/j.rmed.2017.08.008
- Vestbo J, Vogelmeier C, Small M, et al. Understanding the GOLD 2011 Strategy as applied to a real-world COPD population. Respir Med. 2014;108(5):729–736. DOI:https://doi.org/10.1016/j.rmed.2014.03.002
- Lipson DA, Barnacle H, Birk R, et al. FULFIL Trial: Once-daily triple therapy for patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;196(4):438–446. DOI:https://doi.org/10.1164/rccm.201703-0449OC
- Lipson DA, Barnhart F, Brealey N, et al. Once-daily single-inhaler triple versus dual therapy in patients with COPD. N Engl J Med. 2018;378(18):1671–1680. DOI:https://doi.org/10.1056/NEJMoa1713901
- Papi A, Vestbo J, Fabbri L, et al. Extrafine inhaled triple therapy versus dual bronchodilator therapy in chronic obstructive pulmonary disease (TRIBUTE): a double-blind, parallel group, randomised controlled trial. Lancet. 2018;391(10125):1076–1084. DOI:https://doi.org/10.1016/S0140-6736(18)30206-X
- Singh D, Papi A, Corradi M, et al. Single inhaler triple therapy versus inhaled corticosteroid plus long-acting β2-agonist therapy for chronic obstructive pulmonary disease (TRILOGY): a double-blind, parallel group, randomised controlled trial. Lancet. 2016;388(10048):963–973. DOI:https://doi.org/10.1016/S0140-6736(16)31354-X
- Vestbo J, Papi A, Corradi M, et al. Single inhaler extrafine triple therapy versus long-acting muscarinic antagonist therapy for chronic obstructive pulmonary disease (TRINITY): a double-blind, parallel group, randomised controlled trial. Lancet. 2017;389(10082):1919–1929. DOI:https://doi.org/10.1016/S0140-6736(17)30188-5
- Ferguson GT, Rabe KF, Martinez FJ, et al. Triple therapy with budesonide/glycopyrrolate/formoterol fumarate with co-suspension delivery technology versus dual therapies in chronic obstructive pulmonary disease (KRONOS): a double-blind, parallel-group, multicentre, ph 3 randomised controlled trial. Lancet Respir Med. 2018;6(10):747–758. DOI:https://doi.org/10.1016/S2213-2600(18)30327-8
- Yu AP, Guérin A, de Leon DP, et al. Therapy persistence and adherence in patients with chronic obstructive pulmonary disease: multiple versus single long-acting maintenance inhalers. J Med Econ. 2011;14(4):486–496. DOI:https://doi.org/10.3111/13696998.2011.594123
- Yu AP, Guérin A, de Leon DP, et al. Clinical and economic outcomes of multiple versus single long-acting inhalers in COPD. Respir Med. 2011;105(12):1861–1871. DOI:https://doi.org/10.1016/j.rmed.2011.07.001
- Hillyer EV, Price DB, Chrystyn H, on behalf of the Respiratory Effectiveness Group, Small Airways Study Group, et al. Harmonizing the nomenclature for therapeutic aerosol particle size: a proposal. J Aerosol Med Pulm Drug Deliv. 2018;31(2):111–113. DOI:https://doi.org/10.1089/jamp.2017.1396
- Braido F, Scichilone N, Lavorini F, et al. Manifesto on small airway involvement and management in asthma and chronic obstructive pulmonary disease: an Interasma (Global Asthma Association - GAA) and World Allergy Organization (WAO) document. World Allergy Organ J. 2016;9:1–6.
- Lavorini F, Pedersen S, Usmani OS, et al. Dilemmas, confusion, and misconceptions related to small airways directed therapy. Chest. 2017;151(6):1345–1355. DOI:https://doi.org/10.1016/j.chest.2016.07.035
- Usmani OS, Barnes PJ. Assessing and treating small airways disease in asthma and chronic obstructive pulmonary disease. Ann Med. 2012;44(2):146–156. DOI:https://doi.org/10.3109/07853890.2011.585656
- Usmani OS, Biddiscombe MF, Barnes PJ. Regional lung deposition and bronchodilator response as a function of beta2-agonist particle size. Am J Respir Crit Care Med. 2005;172(12):1497–1504. DOI:https://doi.org/10.1164/rccm.200410-1414OC
- LaForce C, Feldman G, Spangenthal S, et al. Efficacy and safety of twice-daily glycopyrrolate in patients with stable, symptomatic COPD with moderate-to-severe airflow limitation: the GEMI study. Int J Chron Obstruct Pulmon Dis. 2016;11:1233–1243.
- Kerwin E, Stiler TM, Korenblat P, et al. Efficacy and safety of twice-daily glycopyrrolate versus placebo in patients with COPD: The GEM2 Study. J Copd F. 2016;3(2):549–559. DOI:https://doi.org/10.15326/jcopdf.3.2.2015.0157
- Edwards D, Berry JJ. The efficiency of simulation-based multiple comparisons. Biometrics. 1987;43(4):913–928.
- Donohue JF. Minimal clinically important differences in COPD lung function. COPD. 2005;2(1):111–124. DOI:https://doi.org/10.1081/copd-200053377
- Mahler DA, Witek TJ. The MCID of the Transition Dyspnea Index is a total score of one unit. COPD. 2005;2(1):99–103. DOI:https://doi.org/10.1081/copd-200050666
- Arievich H, Overend T, Renard D, et al. A novel model-based approach for dose determination of glycopyrronium bromide in COPD. BMC Pulm Med. 2012;12:74.
- Singh D, Scuri M, Collarini S, et al. Bronchodilator efficacy of extrafine glycopyrronium bromide: the Glyco 2 study. COPD. 2017;12:2001–2014. DOI:https://doi.org/10.2147/COPD.S137659
- Kerwin E, Hébert J, Gallagher N, et al. Efficacy and safety of NVA237 versus placebo and tiotropium in patients with COPD: the GLOW2 study. Eur Respir J. 2012;40(5):1106–1114. DOI:https://doi.org/10.1183/09031936.00040712
- McGarvey L, Morice AH, Smith JA, et al. Effect of aclidinium bromide on cough and sputum symptoms in moderate-to-severe COPD in three phase III trials. BMJ Open Resp Res. 2016;3(1):e000148. DOI:https://doi.org/10.1136/bmjresp-2016-000148
- Virchow JC, Kuna P, Paggiaro P, et al. Single inhaler extrafine triple therapy in uncontrolled asthma (TRIMARAN and TRIGGER): two double-blind, parallel-group, randomised, controlled phase 3 trials. Lancet. 2019;394(10210):1737–1749. DOI:https://doi.org/10.1016/S0140-6736(19)32215-9